- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunogold Electron Microscopy of the Autophagosome Marker LC3
Published: Vol 7, Iss 24, Dec 20, 2017 DOI: 10.21769/BioProtoc.2648 Views: 13480
Reviewed by: Vikash VermaAswad KhadilkarMartin SachseAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Even though autophagy was firstly observed by transmission electron microscopy already in the 1950s (reviewed in Eskelinen et al., 2011), nowadays this technique remains one of the most powerful systems to monitor autophagic processes. The autophagosome, an LC3-positive double membrane structures enclosing cellular materials, represents the key organelle in autophagy and its simple visualization and/or numeration allow to draw important conclusions about the autophagic flux. Therefore, the accurate identification of autophagosomes is crucial for a comprehensive and detailed dissection of autophagy. Here we present a simple protocol to identify autophagosomes by transmission electron microscopy coupled to immunogold labeling of LC3 starting from a relatively low cell number, which we recently developed to follow the autophagic pathway during viral-mediated human carcinogenesis.
Keywords: AutophagyBackground
The autophagosome represents the key structure in macroautophagy, a catabolic degradation system for cellular cytosolic constituents. Macroautophagy (or simply autophagy) is initiated by the formation of the phagophore, a membrane able to expand itself engulfing organelles and proteins that finally closes around sequestered components forming an organelle known as autophagosome. Next, during the maturation process, the autophagosome can fuse with lysosomes to form autolysosomes where trapped materials are degraded and recycled back by pumps located in the lysosomal limiting membrane (Glick et al., 2010).
Since malfunction of autophagy has been linked to a variety of human disease, viral infection, neurodegeneration, immune function, and cancer (Schneider and Cuervo, 2014), a more comprehensive and detailed dissection of autophagy is increasingly required. Indeed, numerous methodologies to study autophagy have been developed so far (Klionsky et al., 2016), and most of them need to be simultaneously performed in order to correctly define how autophagy is working in a particular experimental system. Among them, transmission electron microscopy (TEM) coupled with specific immunogold labeling (immunogold electron microscopy-IEM) remains one of the most accurate methods for autophagy detection because of its exquisite ability to provide fine details of specific ultracellular structures. Indeed, in addition to their peculiar morphology (Yla-Anttila et al., 2009), autophagosomes are also characterized by the presence of microtubule-associated protein 1 light chain 3, or LC3, which is generally recruited on autophagic membranes after lipidation with phosphatidylethanolamine (LC3-II) during autophagic activation (Kabeya et al., 2000). Therefore, TEM coupled with labeling of cells with anti-LC3 antibody and gold probes conjugated to a secondary antibody allows the unequivocal identification of the autophagosome, and indirectly, permits to monitor autophagic flux.
Several approaches are commonly used for the immunogold labelling of cultured cells: pre-embedding (immunolabeling before resin embedding) and post-embedding methods (immunolabeling after resin embedding) including also Tokuyasu cryosections, where cryoprotected embedded specimens are firstly frozen by immersion in liquid nitrogen and then cut with cryo-ultramicrotome at -120 °C before immunolabelling (Tokuyasu, 1980; Eskelinen et al., 2002; Jager et al., 2004). However, both techniques require at least 3 x 106 cells to obtain an adequate pellet for sample preparation, inferring that initial cell number is a critical factor to consider during planning of IEM experiments.
Therefore, we describe herein a pre-embedding protocol for autophagosome detection starting from small cell numbers (less than 0.1 x 106 cells), which allows performing IEM analysis in primary cells characterized by low replicative potential. We recently used this protocol to detect LC3-positive autophagosomes on populations of primary human keratinocytes transduced with retroviral vectors relevant to viral carcinogenesis (Mattoscio et al., 2017). In principle, this methodology could be applied to detect other antigens of interest, since we used the same protocol also to study the subcellular localization of the SUMO-conjugating enzyme UBC9 (Mattoscio et al., 2017), and may be used with different cellular species for which the initial cell number is also a limiting factor.
Materials and Reagents
- 13 mm Nunc Thermanox coverslips (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 150067 )
- 24-well tissue culture-treated plates (Corning, Costar®, catalog number: 3524 )
- Eppendorf® Safe-Lock microcentrifuge tubes (Sigma-Aldrich, catalog number: T9661)
Manufacturer: Eppendorf, catalog number: 022363204 . - Edge razor blade (Sigma-Aldrich, catalog number: Z740504 )
- 200 mesh grids (Sigma-Aldrich, catalog number: G5151 )
- Chang Monolayer Molds (Electron Microscopy Sciences, catalog number: 70920 )
- Ethanol BioUltra, for molecular biology (Sigma-Aldrich, catalog number: 51976 )
- Dulbecco’s phosphate-buffered saline, calcium, magnesium (Sigma-Aldrich, catalog number: D8662 )
- Paraformaldehyde (Sigma-Aldrich, catalog number: P6148 )
Note: To prepare 4% paraformaldehyde (PFA) solution, in a fume hood dissolve 4 g of PFA in 90 ml of water at 60 °C and few drops of 1 N NaOH (until the solution appears without PFA precipitate), then remove from heating, add 10 ml 10x PBS, fill up with water up to 100 ml, and cool. Do not heat solution above 70 °C since PFA will break down at higher temperatures. Use the solution immediately or store for 1 week at 4 °C or at -20 °C for a longer time. - Glutaraldehyde (Sigma-Aldrich, catalog number: G7776 )
- Quenching solution: 50 mM glycine (Sigma-Aldrich, catalog number: 410225 ) in PBS
- Primary antibody: rabbit polyclonal anti-LC3 (Novus Biologicals, catalog number: NB100-2331 )
- Washing buffer (0.1% BSA, 0.1% saponin in PBS)
- Secondary antibody conjugated with gold: 1.4 nm Nanogold IgG goat anti rabbit IgG (Nanoprobes, catalog number: 2003 )
- Gold enhancement solution (GoldEnhanceTM EM) (Nanoprobes, catalog number: 2113 ), made by four different components: A (enhancer), B (activator), C (initiator) and D (buffer)
- Uranyl acetate (Electron Microscopy Sciences, catalog number: 22400 )
Note: Uranyl acetate is mildly reactive and highly toxic by ingestion. Please dispose of waste according to your institutional policy. - Embed-812 resin kit (Electron Microscopy Sciences, catalog number: 14120 )
Notes:- This product has been discontinued and replaced by EMbed 812.
- For a better preservation, mix Embed-812 (32 g) with Dodecenyl succinie anhydride (DDSA) (16 g) and NMA (Nadic Methyl Anhydride) (17.4 g). Mix well, add 0.8 g of the accelerator DMP30 (2,4,6-tri (dimethylaminomethyl) phenol), mix and store in 20 ml syringe at -20 °C. Warm to room temperature before use.
- This product has been discontinued and replaced by EMbed 812.
- Hydrofluoric acid (Fisher Scientific, catalog number: 14-650-236)
Manufacturer: Avantor Performance Materials, J.T. Baker, catalog number: 9560-01 . - Blocking buffer (see Recipes)
- Bovine serum albumin (BSA, IgG-Free and Protease-Free) (Jackson ImmunoResearch, catalog number: 001-000-162 )
- Normal goat serum (Jackson ImmunoResearch, catalog number: 005-000-121 )
- Ammonium chloride (Sigma-Aldrich, catalog number: A9434 )
- Saponin (Sigma-Aldrich, catalog number: 47036 )
- Phosphate buffer (Sigma-Aldrich, catalog number: P3619 )
- Sodium chloride (Sigma-Aldrich, catalog number: S7653 )
- Bovine serum albumin (BSA, IgG-Free and Protease-Free) (Jackson ImmunoResearch, catalog number: 001-000-162 )
- Reduced osmium tetroxide (see Recipes)
- Sato lead solution (see Recipes)
- Lead citrate tribasic (Sigma-Aldrich, catalog number: 15326 )
- Lead nitrate (Merck, catalog number: 1073980100 )
- Sodium citrate tribasic dehydrate (Sigma-Aldrich, catalog number: S4641 )
- Lead citrate tribasic (Sigma-Aldrich, catalog number: 15326 )
Equipment
- Fume hood
- 60 °C Oven (Memmert, model: UN30 )
- Ultramicrotome (Leica Microsystem, model: Leica EM UC7 )
- Transmission Electron microscope (Carl Zeiss, model: Leo 912AB ) equipped with a slow-scan Proscan camera (ProScan)
- Rocking shaker
Procedure
- Fixation
- Sterilize the coverslips with ethanol, wash three times with PBS, and accommodate one slide for each well of the 24-well plate.
- Plate the cells to be tested on these slides in appropriate medium to obtain a 60-70% confluency on the following day.
Note: To obtain enough technical replicates, we usually prepare 6 slides for each condition to be tested. In our study, we used primary keratinocytes expressing empty-control vector or a plasmid for the overexpression of the HPV oncoviral proteins E6/E7 (seeded about 1 x 105 cells/well) (Mattoscio et al., 2017). In addition, to unequivocally identify autophagosomes, appropriate positive and negative controls of autophagosome formation should be prepared. For example, consider activating/inhibiting autophagy with rapamycin 10-500 nM (to increase autophagosomes formation) or with class III PI3K inhibitors (such as 1-10 mM 3-methyladenine or 10-100 nM wortmannin to inhibit autophagosomes accumulation), respectively (Mizushima et al., 2010). - The next day, remove the medium, wash 2 times with PBS, and fix cells in 4% freshly-prepared PFA in 0.1 M phosphate buffer pH 7.4 for 1 h at room temperature.
Note: Since glutaraldehyde lowers protein antigenicity, although it provides best structural preservation, we prefer to initially fix cells with PFA alone, and then re-fix with glutaraldehyde after antigen labeling. - Remove PFA, wash 2 times with PBS. Store fixed cells at 4 °C.
Note: Since PFA fixation is reversible, for long time storage keep cells in storage solution (1% PFA in PBS) instead of PBS alone.
- Sterilize the coverslips with ethanol, wash three times with PBS, and accommodate one slide for each well of the 24-well plate.
- Immunolabeling
- Remove fixative or storage solution, wash wells three times for 5 min each in PBS, and treat with quenching solution for 10 min at room temperature to quench the free aldehydes of the primary fixative.
- Permeabilize cells by incubation with 0.25% saponin, 0.1% BSA in PBS for 10 min at room temperature.
- Incubate cells for 30 min at room temperature with blocking buffer (0.2% BSA, 5% normal goat serum, 50 mM NH4Cl, 0.1% saponin, 20 mM phosphate buffer, 150 mM NaCl, see Recipes) to block non-specific staining.
- Remove blocking buffer and incubate coverslips with anti-LC3 primary antibody (1:150 dilution) in blocking buffer, for 2 h at room temperature.
- Wash cells with washing buffer (0.1% BSA, 0.1% saponin in PBS) 4 times for 5 min each.
- Incubate coverslips with 1.4 nm Nanogold goat anti rabbit secondary antibody conjugated with nanogold (1:200 dilution) in blocking buffer for 1 h at RT.
- Wash slides 3 times with washing buffer and wash three times in PBS.
- Re-fix stained cells with 1% glutaraldehyde in PBS for 30 min at room temperature to preserve ultrastructure. Rinse in PBS and leave in PBS at 4 °C until next step.
- Remove fixative or storage solution, wash wells three times for 5 min each in PBS, and treat with quenching solution for 10 min at room temperature to quench the free aldehydes of the primary fixative.
- Gold enhancement
- Equilibrate all the components of gold enhancement solution (A, B, C, D) at room temperature before use.
- Wash 3 x 5 min with 50 mM glycine in PBS.
- Wash 3 x 5 min with 1% BSA, 0.05% Tween in PBS.
- Rinse 3 times in PBS and in the meanwhile activate gold enhancement solution by adding in an Eppendorf tube one part each of component (A-enhancer and B-activator), mix well and incubate for 5 min.
Note: Gold enhancement solution should be prepared immediately before use. Increasing the amount of component B might help in reducing nonspecific signals. - Add to the gold enhancement mix one part of components C (initiator) and D (buffer), respectively, rinse cells in distilled water and add few drops of the solution to the coverslip. Prepare about 40 µl of gold enhancement solution per coverslip.
- Check the appearance of a dark purple/black precipitate (Figure 1). Incubation time with the gold enhancement solution may vary. Usually, it takes between 1 to 3 min for the full development. Always compare with the negative control (without primary antibody).
Note: The goal of this step is to enlarge gold particles associated to the secondary antibody. To improve penetration and density labeling, gold particles of small size are preferable (1.4 nm). Since these nanogold particles will be hardly visible in samples, this step is mandatory. However, as an alternative to gold, a silver enhancement solution could be used. Silver enhancement is both commercially available (Nanoprobes, catalog number 2013) or can be prepared from single components (Stierhof et al., 1991). Silver enhancement is light sensitive and can be damaged by osmium tetroxide, therefore gold enhancement solution is usually preferred for its simplicity of use. - Wash three times for 5 min each with distilled water.

Figure 1. Gold enhancement. Put a drop of gold enhancement solution on the coverslips and incubate until a dark precipitate appears (visible by eye). Longer incubations may cause darker signals. Check under a bright field microscope or a stereomicroscope for a fine control of gold development. Note that the dark precipitate will not appear in the negative control (CTRL: no primary antibody). Bottom: magnification of coverslips at Time = 0 and at the end of gold enhancement incubation (T = 3 min). Please note that the dark precipitate appeared only where the primary antibody is present.
- Equilibrate all the components of gold enhancement solution (A, B, C, D) at room temperature before use.
- Contrast enhancement
- Post-fix samples with reduced osmium tetroxide (see Recipes) for 1 h at RT. Wash 3 times for 5 min in 0.1 M sodium cacodylate and wash three times in distilled water.
Note: The use of reduced osmium fixation step preserves membrane lipids. The addition of potassium ferrocyanide reduces the osmium making it more reactive to membranes, improving their contrast. - En bloc stain with 1% uranyl acetate water solution overnight at 4 °C.
Note: Do not expose to light. Uranyl acetate improves image contrast due to the high affinity of uranyl and lead ions for proteins, nucleic acids, and hydroxyl groups in carbohydrates and RNA, respectively. It is important to use water instead of alcoholic solution to better preserve cellular structures.
- Post-fix samples with reduced osmium tetroxide (see Recipes) for 1 h at RT. Wash 3 times for 5 min in 0.1 M sodium cacodylate and wash three times in distilled water.
- Embedding
- Dehydrate specimens in an ascending ethanol series 30%, 50%, 70%, 80%, 90%, 96% for 5 min in each of the ascending series. Finally, dehydrate 3 times in 100% ethanol for 10 min every time.
- Replace absolute ethanol with a 1:1 solution of ethanol-embedding medium, on a rocking shaker for 2 h at room temperature.
- Remove coverslips from the ethanol-resin mix, absorb the excess of resin with a paper tissue and place the coverslips on top of a drop of resin. After 1 h, move the coverslips to a new drop of resin (Figure 2A).

Figure 2. Embedding. Move the coverslip on drop of resin and incubate for one hour (A). Place a drop of Epon on a Chang Mold and lay the coverslips (B). - Fill the cavities of a Chang monolayer Mold with few drops of resin and lay the coverslips (make sure to clean the top side of the coverslip from the excess of resin) on top of the cavity (Figure 2B).
- Place in the oven at 45 °C overnight, then increase the temperature to 60 °C and bake for further 24 h.
- Dehydrate specimens in an ascending ethanol series 30%, 50%, 70%, 80%, 90%, 96% for 5 min in each of the ascending series. Finally, dehydrate 3 times in 100% ethanol for 10 min every time.
- Sectioning
- To detach Epon from slide, take the Epon block out of the oven and let it cool down. Then, immerse block in liquid nitrogen for 30 sec and then in hot water. This will detach resin containing embedded cells from the coverslip where cells were initially grown.
Note: Alternatively, put the coverslips in a plastic multiwell, cover with a solution of concentrated hydrofluoric acid (30%), and shake for 5-10 min until the glass is completely dissolved. - Wash the samples several times with distilled water, soak the samples in 0.1 M cacodylate buffer for 30 min, then wash with distilled water and dry.
- With a single edge razor blade remove a small portion of the resin block and stick with a cyanoacrylate glue on top of an empty resin block (Figure 3).

Figure 3. Sectioning. After glass coverslips have been removed, cut with a razor blade a small portion of the resin block and glue on top of an empty resin block (plasticine, blue material in this image). After 1-2 h, the sample can be placed in an ultramicrotome for sectioning. - Cut ultrathin sections of 60-90 nm thick and collect sections onto 200 mesh grids with square or hexagonal openings.
- Stain grids with 1% uranyl acetate water solution for 5 min and then Sato’s lead solution (see Recipes) for 2 min.
- Observe under an electron microscope and take digital micrographs.
Note: To visualize autophagosomes, specimens should be observed at a magnification of 4,000 (for an overview) to 10,000x (to finely observe ultrastructure) looking for double-membraned organelles positive for LC3 gold particles, and containing undigested cytoplasmic contents (Figure 4A).
- To detach Epon from slide, take the Epon block out of the oven and let it cool down. Then, immerse block in liquid nitrogen for 30 sec and then in hot water. This will detach resin containing embedded cells from the coverslip where cells were initially grown.
Data analysis
In this protocol, we applied a pre-embedding technique (immunolabeling before samples were embedded in resin) to detect LC3 in low-abundant cells preparation. We recently used this method to image both UBC9 and LC3-positive autophagosomes in primary human keratinocytes expressing either an Empty vector or HPV E6/E7 oncoviral proteins (Mattoscio et al., 2017). Indeed, TEM analysis showed the presence of UBC9 inside double-membranes intracellular structures that we identified as autophagic compartments by LC3-immunogold TEM, suggesting an autophagic dependent route for UBC9 degradation.
Although TEM is one of the best technique to evaluate the autophagic process, the fact that autophagy is a dynamic process makes it difficult to extrapolate meaningful data from a single ultrastructural snapshot. Moreover, some late autophagic structures are difficult to identify due to the degradative process that takes place upon fusion to lysosomes. In Figure 4 we reported the morphology of the autophagic structures (indicated by arrows) in human keratinocytes comparing side by side the classical EM preparation (left) with LC3 immunolabeling (right), illustrating typical results obtained following our strategy. The combination of the initial mild fixation with PFA alone and the pre-embedding staining allowed a better antigen preservation despite preserving detailed information on the cell structure (Figure 4) and the labeling of cells with anti-LC3 antibody allows the unequivocal identification of the autophagic vacuoles. Moreover, cell fixation directly on microscopy slides without the need to scrape and pellet cells allows to work with a relatively low cell number and to maintain cell ultrastructure. Therefore, we believe that this protocol could be easily applied to detect additional low-abundant antigens in rare cell populations. However, a limitation of this approach is that it does not enable colocalization studies, which can be achieved only with post-embedding EM using gold probes of different sizes.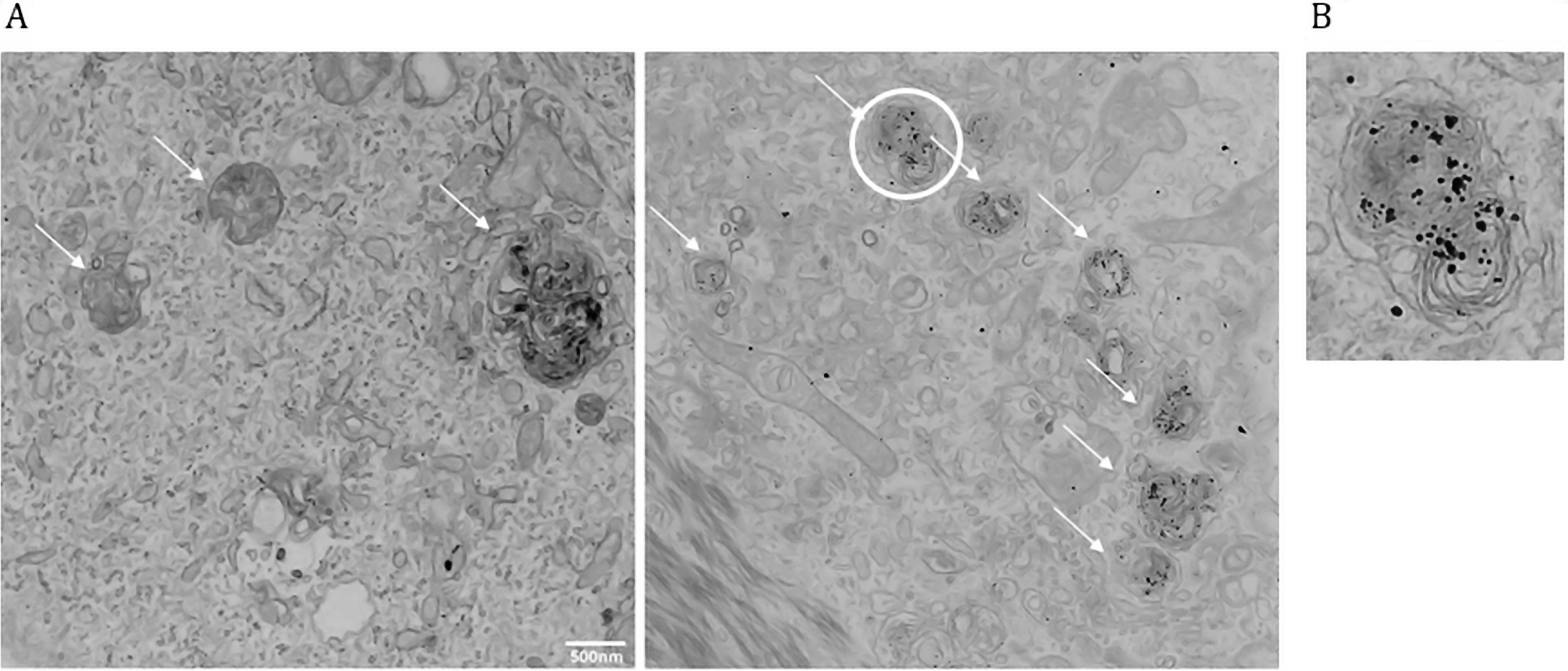
Figure 4. Autophagosomes identification using TEM and LC3 immunostaining. A. Left: TEM image of primary human keratinocytes. Right: TEM coupled with LC3 immunogold labeling. Arrows indicate autophagic vacuoles. Note that gold particles (LC3) are selectively enriched in autophagic structures. B. Magnification of the autophagic vacuole circled in Figure 4A (right). Note the membrane structure, LC3 positivity, and fusion with a lysosome (darker body).
Recipes
- Blocking buffer
0.2% bovine serum albumin (BSA, IgG-Free and Protease-Free)
5% normal goat serum
50 mM ammonium chloride
0.1% saponin
20 mM phosphate buffer
150 mM sodium chloride - Reduced osmium tetroxide
1% osmium tetroxide (OsO4)
1.5% potassium ferrocyanide
0.1 M cacodylate buffer pH 7.4 (from sodium cacodylate)
Note: Protect OsO4 from direct exposure to light during storage and use. OsO4 is highly toxic, handling and disposal in a fume hood are required. - Sato lead solution
0.21 g of lead citrate tribasic
0.15 g lead nitrate
0.15 g lead acetate trihydrate
1 g of sodium citrate tribasic dehydrate
Mix the chemicals and dissolve in 41 ml of distilled water. This makes a milky solution. Then, add 9 ml 4% NaOH and the solution will become clear. Filter the solution and store in a dark glass bottle at room temperature
Acknowledgments
Work in S.C. lab related to these topics is supported by Associazione Italiana per la Ricerca sul Cancro (A.I.R.C.) (IG 2015 Id.16721) and from European Commission-FP7-HPVAHEAD. DM was a Fondazione Italiana per la Ricerca sul Cancro (F.I.R.C.) fellow. The protocol is adapted from Mattoscio et al., 2017, and has been developed with the aid of the San Raffaele Institute Microscopy Facility ‘Alembic’. The authors declare no conflicts of interest.
References
- Eskelinen, E. L., Reggiori, F., Baba, M., Kovacs, A. L. and Seglen, P. O. (2011). Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy 7(9): 935-956.
- Eskelinen, E. L., Prescott, A. R., Cooper, J., Brachmann, S. M., Wang, L., Tang, X., Backer, J. M. and Lucocq, J. M. (2002). Inhibition of autophagy in mitotic animal cells. Traffic 3(12): 878-893.
- Glick, D., Barth, S. and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J Pathol 221(1): 3-12.
- Jäger, S., Bucci, C., Tanida, I., Ueno, T., Kominami, E., Saftig, P. and Eskelinen, E. L. (2014). Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117(Pt 20): 4837-48.
- Kabaeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., Kominami, E., Ohsumi, Y., and Yoshimori, T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19(21): 5720-8.
- Klionsky, D. J., Abdalla, F. C., Abeliovich, H., Abraham, R. T., Acevedo-Arozena, A., Adeli, K., Agholme, L., Agnello, M., Agostinis, P., Aguirre-Ghiso, J. A., Ahn, H. J., Ait-Mohamed, O., Ait-Si-Ali, S, Akematsu, T. and Akira, S., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12(1): 1-222.
- Mattoscio, D., Casadio, C., Miccolo, C., Maffini, F., Raimondi, A., Tacchetti, C., Gheit, T., Tagliabue, M., Galimberti, V. E., De Lorenzi, F., Pawlita, M., Chiesa, F., Ansarin, M., Tommasino, M. and Chiocca, S. (2017). Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog 13(3): e1006262.
- Mizushima, N., Yoshimori, T. and Levine, B. (2010). Methods in mammalian autophagy research. Cell 140: 313-326.
- Schneider, J. L. and Cuervo, A. M. (2014). Autophagy and human disease: emerging themes. Curr Opin Genet Dev 26: 16-23.
- Stierhof YD, Humbel BM, Schwarz H. (1991). Suitability of different silver enhancement methods applied to 1 nm colloidal gold particles: an immunoelectron microscopic study. J Electron Microsc Tech 17(3): 336-43.
- Tokuyasu, K. T. (1980). Immunochemistry on ultrathin frozen sections. Histochem J 12(4): 381-403.
- Yla-Anttila, P., Vihinen, H., Jokitalo, E. and Eskelinen, E. L. (2009). Monitoring autophagy by electron microscopy in mammalian cells. Methods Enzymol 452: 143-164.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mattoscio, D., Raimondi, A., Tacchetti, C. and Chiocca, S. (2017). Immunogold Electron Microscopy of the Autophagosome Marker LC3. Bio-protocol 7(24): e2648. DOI: 10.21769/BioProtoc.2648.
Category
Cell Biology > Cell staining > Protein
Immunology > Host defense > General
Cell Biology > Cell imaging > Electron microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.