- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

MicroScale Thermophoresis as a Tool to Study Protein-peptide Interactions in the Context of Large Eukaryotic Protein Complexes
Published: Vol 7, Iss 23, Dec 5, 2017 DOI: 10.21769/BioProtoc.2632 Views: 24890
Reviewed by: Tie LiuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Protein-peptide interactions are part of many physiological processes, for example, epigenetics where peptide regions of histone complexes are crucial for regulation of chromatin structure. Short peptides are often also used as alternatives to small molecule drugs to target protein complexes. Studying the interactions between proteins and peptides is thus an important task in systems biology, cell biology, biochemistry, and drug design. However, this task is often hampered by the drawbacks of classical biophysical methods for analysis of molecular interactions like surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC), which require immobilization of the interaction partners or very high sample concentrations. MicroScale Thermophoresis (MST) is an innovative method that offers the possibility to determine the important parameters of a molecular interaction, such as dissociation constant, stoichiometry, and thermodynamics. Moreover, it does so in a rapid and precise manner, with free choice of buffers or biological liquids, no need for sample immobilization, and very low sample consumption. Here we describe two MST assays in detail, which analyze (i) the interactions between certain peptide stretches of the eukaryotic RNA polymerase II and a protein subunit of the eukaryotic transcription elongation complex and (ii) interactions between N-terminal histone tail peptides and epigenetic reader proteins. These experiments show that MST is able to characterize protein-peptide interactions that are triggered by only minor changes in the peptide, for example, only one phosphorylation at a specific serine residue.
Keywords: MicroScale ThermophoresisBackground
Biological Background: Protein-protein interactions (PPIs) are essential for almost all cellular processes, ranging from DNA replication, transcription, and translation over the formation of metabolic enzyme complexes and large cellular machineries, to the sensing and relay of biological signals. Given the physiological importance of PPIs, protein complexes are considered high-value targets in drug development (Milroy et al., 2014; Nevola and Giralt, 2015). However, traditional, small-molecule based targeting approaches often fail to produce potent inhibitors, mostly due to the large and often flat protein interfaces (Sperandio et al., 2010). Alternative strategies involve peptide-based inhibitors that often mimic part of the binding surface of an interaction partner (Boersma et al., 2012; Azzarito et al., 2013; Arkin et al., 2014; Pelay-Gimeno et al., 2015). Such interactions between proteins and peptides of typically 5-20 amino acids are frequently found in nature, for example with antibodies, kinases, phosphatases, MHC proteins, or epigenetic reader proteins (Stanfield and Wilson, 1995). Moreover, the specific targeting of PPIs with small peptides can help to elucidate composition and function of large and sophisticated protein complexes like the eukaryotic RNA polymerase II elongation complex (Antosz et al., 2017) or the specificity of epigenetic reader proteins (Josling et al., 2015).
Technical Background: In order to precisely and comprehensively characterize protein-protein and protein-peptide interactions with respect to affinity, kinetics, and thermodynamics, an array of biophysical techniques and methods have been developed. Well established technologies are isothermal titration calorimetry (ITC) (Pierce et al., 1999; Ghai et al., 2012), surface plasmon resonance (SPR)-based assays (Pattnaik, 2005; Ishii et al., 2013), native mass spectrometry (Heck, 2008; Yan et al., 2017), biolayer interferometry (Abdiche et al., 2008; Concepcion et al., 2009), static or dynamic light scattering (Wen et al., 1996; Kameyama and Minton, 2006; Hanlon et al., 2010), as well as fluorescence spectroscopy (Lundblad et al., 1996; Lin et al., 2014; Raines, 2015). Although some of these methods uniquely provide specific information like on- and off-rates or the precise complex stoichiometry, they are often lab-intense and complex, are often problematic due to high sample consumption, low-sensitivity, surface immobilization, mass transport limitations, as well as buffer restrictions or fail to quantify interactions involving small peptides. The innovative MicroScale Thermophoresis technology (MST) is a powerful technique that overcomes such limitations and allows for the fast, precise, cost-efficient, and quality-controlled characterization of molecular interactions with very low sample consumption, no need of sample immobilization, and a free choice of buffers or bioliquids. MST enables researchers to determine the important parameters of molecular interactions, such as binding affinity (from pico-molar o milli-molar), binding stoichiometry, and interaction thermodynamics (Wienken et al., 2010; Jerabek-Willemsen et al., 2011). Importantly, MST does not impose any limitations regarding molecular weight of the interacting molecules and thus quantifying interactions between proteins of several hundred kDa in size and short peptides with molecular weights below 1 kDa is straightforward and precise (Antosz et al., 2017).
The physical phenomenon of thermophoresis: The physical phenomenon of ‘thermophoresis’ describes the movement of molecules along a temperature gradient. This movement is dependent on the size, charge, and hydration shell of the molecules (Braun and Libchaber, 2002; Duhr and Braun, 2006). Interactions between a protein and a ligand (other protein, peptide, DNA, RNA, small molecule) alter at least one of these parameters resulting in a different thermophoretic mobility along the temperature gradient (Jerabek-Willemsen et al., 2014). Thus interactions between large proteins and small ligands, such as short peptides, which only entail negligible changes in size and charge, will significantly influence the thermophoresis due to changes in the hydration shell.
MicroScale Thermophoresis: MicroScale Thermophoresis is an innovative technology that utilizes the thermophoretic principle to study molecular interactions in solution (Duhr et al., 2004; Baaske et al., 2010; Jerabek-Willemsen et al., 2011). The thermophoretic movement of molecules is induced by generating a microscopic temperature gradient inside a very thin glass capillary using an infrared (IR) laser. The corresponding technical setup of an MST device (Monolith NT.115, NanoTemper Technologies GmbH, Munich, Germany) is shown in Figure 1A. This gradient is focused in a diameter of around 50 µM and comprises a temperature difference ∆T of 2-6 °C. In the same area, the thermophoretic movement of the molecules is tracked by fluorescence optics using either the intrinsic fluorescence of tryptophans in proteins or peptides (Seidel et al., 2012) or the fluorescence signal of an extrinsic fluorophore that is coupled to one of the interacting partners (Schubert et al., 2012; Zillner et al., 2012). Upon heating, molecules either deplete or accumulate in the center of the temperature gradient and the fluorescence optics, which can be quantified by the Soret coefficient ST, with chot and ccold representing the concentration in the heated and non-heated regions of the capillary and ∆T the temperature difference along the gradient (Duhr and Braun, 2006):![]()
A typical MST measurement is depicted in Figure 1B. In the beginning, the fluorescence in the capillary is measured at the fixed starting temperature for usually five seconds. Then the temperature gradient is induced by the IR laser, which results in a steep drop of the measured fluorescence signal, the so-called temperature- or T-jump. This signal jump results from temperature-dependent changes in the quantum yield of the used fluorophore. After that, a slower, thermophoresis-driven depletion of fluorescent molecules in the optical focus corresponding to the Soret coefficient results in an exponential decrease of the fluorescence signal. After a measurement time of typically 20-30 sec, the IR laser is turned off again, which results in a thermophoretic back-diffusion of fluorescent molecules into the optical focus and a concomitant reverse T-jump.
In order to determine the equilibrium dissociation constant (KD) of the complex under study, a series of MST measurements with a fixed amount of fluorescent binding partner and increasing amounts of non-fluorescent partner are recorded (Figure 1C). The 16 MST traces are normalized with respect to their initial fluorescence. The concentration range of the non-fluorescent partner is chosen in a way that the lowest concentration results in a practically fully ‘unbound’ fluorescent partner and the highest concentration results in a fully ‘bound’ fluorescent partner. Usually 16 dilutions of the non-fluorescent partner are prepared ranging from about 10-fold above to the 10-fold below the projected KD, supplemented with the same amount of fluorescent partner and loaded into 16 individual capillaries. The concentration of the fluorescent partner is usually kept below the projected KD in order to allow for a precise determination of the true equilibrium affinity. The differences between the ‘cold’ and ‘hot’ states of each of the 16 MST traces is then used to determine the change in fluorescence for each trace:![]()
Plotting these values against the ligand concentration finally results in a typical binding isotherm, which yields the KD value of the interaction (Figure 1D).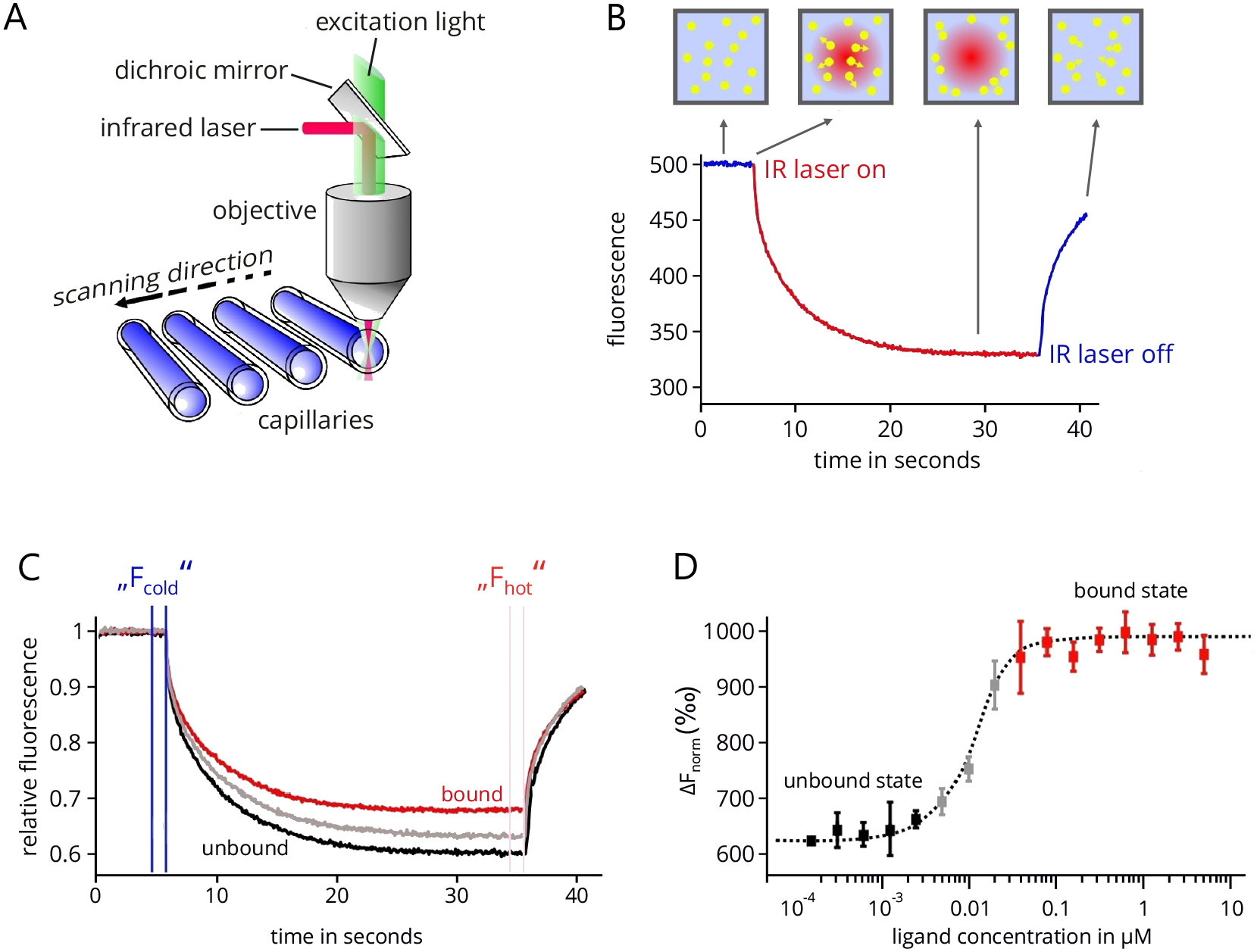
Figure 1. MicroScale Thermophoresis. A. Technical setup of an MST device. The thermophoretic movement of molecules inside the glass capillary is induced by an infrared laser that is focused on a 50 µM wide area. In the same area fluorescence emission is used to track this movement. In total, 16 capillaries can be analyzed subsequently in one device. B. An example of MST trace. After an initial delay of five seconds, the IR laser is turned on to establish a temperature gradient. Following the T-jump phase, the thermophoretic movement leads to an exponential decrease of fluorescent molecules in the optics focus. After a measurement time of typically 20-30 sec, the laser is turned off. C. Combined traces of a typical MST experiment in which 16 capillaries are analyzed that contain the same concentration of the fluorescent interaction partner but increasing concentrations of the non-fluorescent partner. All traces are normalized to the same initial relative fluorescence value of 1. D. Final binding isotherm resulting from plotting the difference in normalized fluorescence against the concentration of the non-fluorescent binding partner (ligand). Figure modified from Entzian and Schubert, 2016; originally kindly provided by NanoTemper Technologies, Munich.
Advantages and drawbacks of MST: MST offers several integral advantages over other biophysical methods for characterizing protein-protein and protein-peptide interactions. First, the MST traces and lateral scans of the filled capillaries provide a straight-forward sample quality control as they allow for the easy detection of aggregation, precipitation, and adsorption effects. This enables the researcher to quickly alter and optimize technical and buffer conditions to increase sample stability and data quality. Importantly, these controls take less than two minutes for 16 capillaries. Also the full required measurement to get an equilibrium KD takes less than 15 min. Moreover, MST is immobilization-free and thus allows for the determination of affinities in practically all buffers and even in complex bioliquids like lysates and sera (Wienken et al., 2010; Seidel et al., 2013). Also, MST excels by very low sample consumption, which can be a crucial issue when working with hard-to-produce eukaryotic proteins. Usually, the fluorescent interaction partner is assayed at very low nanomolar concentrations (5-20 nM) and the non-fluorescent partner usually–depending on the KD–in the nanomolar to low micromolar range. Finally, the high dynamic range of detectable affinities (pM to mM) and non-existing size limitations allow for the characterization of a wide variety of different molecular interactions involving proteins, peptides, small molecules, DNA, and RNA, just to name a few. However, MST does not allow for determining on and off-rates of molecular interactions. Nevertheless, binding parameters derived from MST experiments agree well with established state-of-the-art methods like SPR or ITC (Ramakrishnan et al., 2012; Chen et al., 2015; Stoltenburg et al., 2015; Wan et al., 2015; Harazi et al., 2017). Finally, it has to be stated that for most MST applications one interaction partner has to be modified with a fluorophore. Thus, if neither a peptide, nor a protein can be labeled with a fluorophore (e.g., because neither contains lysine or cysteine residues for fluorophore coupling), standard MST cannot be applied. In such cases, however, label-free MST analysis can be an alternative, due to the read-out of intrinsic tyrosine and tryptophan fluorescence of the protein or the peptide.
Materials and Reagents
- Low volume, low binding reaction tubes (e.g., Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 90410 )
- Eppendorf tubes (1.5 ml) (Sigma-Aldrich, catalog number: T9661)
Manufacturer: Eppendorf, catalog number: 022363204 . - PCR tubes (200 µl, NanoTemper Technologies GmbH, Munich, Germany)
- Pipette tips (10 μl, 100 μl, 1,000 μl) (STARLAB INTERNATIONAL, catalog number: S1111 )
- FITC-labelled peptides of the CTD of the A. thaliana RNA polymerase II (Biomatik, Wilmington, Delaware, USA)
Note: ‘pS’ denotes a phosphorylated serine; peptides are labeled with Fluorescein isothiocyanate [FITC, CAS number: 27072-45-3] at the N-terminus.- CTD-noP (sequence: PSYSPTSPSYSP)
- CTD-Ser2P (sequence: TSPSY(pS)PTSPSY)
- CTD-Ser5P (sequence: SYSPT(pS)PSYSPT)
- CTD-noP (sequence: PSYSPTSPSYSP)
- Protein SPT6L (Phe1218-Asp1412)
Note: Expressed in Escherichia coli with a GST-tag and purified by glutathione-sepharose affinity chromatography as described in (Kammel et al., 2013). - Protein HP1 from Plasmodium falciparum (P. falciparum)
Note: Expressed in E. coli BL21-CodonPlus(DE3)-RIL cells with a hexahistidine tag and purified by Ni2+-IMAC chromatography (Josling et al., 2015). - Protein HP1 from Drosophila melanogaster (D. melanogaster)
Note: Expressed in E. coli BL21-CodonPlus(DE3)-RIL cells with a hexahistidine tag and purified by Ni2+-IMAC chromatography (Josling et al., 2015). - Histone peptides
All histone peptides were purchased from ANASPEC, Kaneka Eurogentec (Liege, Belgium) - Pluronic® F-127 (CAS number: 9003-11-6) (Sigma-Aldrich, catalog number: P2443 )
- Sodium dodecyl sulfate, sodium salt (SDS; CAS number: 151-21-3) (Sigma-Aldrich, catalog number: L3771 )
- Sodium phosphate (CAS number: 7601-54-9) (Sigma-Aldrich, catalog number: 342483 )
- Ethylenediaminetetraacetic acid (EDTA, CAS number: 60-00-4) (Sigma-Aldrich, catalog number: E9884 )
- DL-Dithiothreitol (DTT, CAS number: 3483-12-3) (Sigma-Aldrich, catalog number: 43815 )
- Phenylmethanesulfonyl fluoride (PMSF, CAS number: 329-98-6) (Sigma-Aldrich, catalog number: 78830 )
- Tris(hydroxymethyl)aminomethane (Tris, CAS number: 77-86-1) (Sigma-Aldrich, catalog number: T1503 )
- Sodium chloride (NaCl, CAS number: 7647-14-5) (Sigma-Aldrich, catalog number: S7653 )
- Magnesium chloride (MgCl2, CAS number: 7786-30-3) (Sigma-Aldrich, catalog number: M8266 )
- Tween® 20 (CAS number: 9005-64-5) (Sigma-Aldrich, catalog number: P1379 )
- NaP buffer (see Recipes)
- MST-T buffer (see Recipes)
Equipment
- Pipettes (Eppendorf Reference® 20, 100, 200, 1,000) (Eppendorf, catalog numbers: 4920000032 , 4920000059 , 4920000067 , 4920000083 )
- MiscroScale Thermophoresis instrument Monolith NT.115 (NanoTemper Technologies, model: Monolith NT.115 )
- MiscroScale Thermophoresis instrument Monolith NT.LabelFree (NanoTemper Technologies, model: Monolith NT.LabelFree )
- Monolith NTTM capillaries ‘Premium coated’ (NanoTemper Technologies, Munich, Germany)
- Incubator (Eppendorf, model: ThermoMixer® comfort , catalog number: 5360000011)
- Centrifuge (Eppendorf, model: 5424 R , catalog number: 5404000219)
Software
- MO.Control (NanoTemper Technologies, Munich, Germany)
- MO.Affinity Analysis (NanoTemper Technologies, Munich, Germany)
Procedure
- Solution preparation
Notes: Buffers- For the interactions between SPT6L(Phe1218-Asp1412) and the CTD peptides of the A. thaliana RNA polymerase II, the assay buffer is NaP buffer (see Recipes).
- For the interactions between the HP1 proteins and the histone peptides, the assay buffer is MST-T buffer (see Recipes).
- Preparation of the working solutions of the targets
- Peptides CTD-noP, CDT-Ser2P, and CDT-Ser5P: These peptides serve as targets for binding of SPT6L(Phe1218-Asp1412).
- Follow the manufacturers’ instructions and dissolve the peptides.
- Prepare the target working solution by diluting the peptide stocks to 80 nM with MST-T buffer.
- Follow the manufacturers’ instructions and dissolve the peptides.
- HP1 proteins from P. falciparum and D. melanogaster. These proteins serve as the targets for binding of the different histone peptides. Labeling of the targets is not necessary in this context, because their intrinsic tryptophan fluorescence can be used with the Monolith NT.LabelFree device.
Prepare the target working solution by diluting the protein stocks to 3.5 µM in MST-T buffer.
- Peptides CTD-noP, CDT-Ser2P, and CDT-Ser5P: These peptides serve as targets for binding of SPT6L(Phe1218-Asp1412).
- Preparation of the working solution of the ligand
- SPT6L(Phe1218-Asp1412) protein: This protein serves as the ligand for binding of the CTD-peptides.
- Dilute the ligand stock with MST-T buffer to 4.3 mM. Ideally, the concentration of the working solution of the ligand should be around 50-times higher than the projected KD value.
Note: Centrifugation of the ligand stock for 5 min at 14,000 x g at 4 °C may help remove aggregates. Low volume, low binding reaction tubes (e.g., Thermo Fisher Scientific) are recommended to avoid adsorption of molecules to the tube walls. - When preparing the ligand working solution it has to be considered that–depending on the dilution factor–a certain amount of ligand stock buffer can still be present in the working solution. If this exceeds 1%, we recommend adjusting the final MST buffer accordingly.
- Dilute the ligand stock with MST-T buffer to 4.3 mM. Ideally, the concentration of the working solution of the ligand should be around 50-times higher than the projected KD value.
- Histone peptides: These peptides serve as the ligands for the measurements with the two HP1 protein targets.
- Follow the manufacturers’ instructions and dissolve the peptides.
- Prepare the ligand working stock by diluting the peptide stocks to 20 mM in NaP buffer.
- Follow the manufacturers’ instructions and dissolve the peptides.
- SPT6L(Phe1218-Asp1412) protein: This protein serves as the ligand for binding of the CTD-peptides.
- Preparation of the ligand dilution series
- The ligand is diluted in 16 serial steps in 200 µl PCR tubes. Dilute the ligand by 50% (1:1 dilution) in each of the sixteen steps.
Note: The concentration finder tool implemented in the control software simulates binding data and helps with finding the right concentration range for the dilution series. - Add 10 µl of assay buffer into PCR tubes 2 to 16.
- Fill 20 µl of the ligand working solution in PCR tube 1.
- Transfer 10 µl from PCR tube 1 into PCR tube 2 and mix properly by pipetting up and down several times.
Note: Avoid vortexing or heavy shaking in order to prevent protein denaturation. - Transfer 10 µl from PCR tube 2 into PCR tube 3 and mix. Repeat this process for the remaining dilution steps.
- Discard the excess 10 µl from the last tube. All 16 PCR tubes should now contain a volume of 10 µl.
Note: It is important to avoid any buffer dilution effects. All 16 PCR tubes should contain the exact same buffer composition. If the ligand stock solution is in a different buffer than the MST assay buffer, the best practice is to prepare an aliquot of the MST assay buffer that features the exact same composition as the buffer of the ligand working solution. This buffer is then used for pipetting the serial dilution series. For example, if the ligand stock solution is in buffer A + 2 mM DTT and is diluted 10-fold in buffer A to yield the ligand working solution, the DTT concentration in the working solution is 0.2 mM. Thus use buffer A + 0.2 mM DTT for preparing the serial dilution series.
- The ligand is diluted in 16 serial steps in 200 µl PCR tubes. Dilute the ligand by 50% (1:1 dilution) in each of the sixteen steps.
- Preparation of the final MST mix
- Although a volume of 4 µl is sufficient to fill the MST capillaries, it is advised to prepare at least 20 µl of the final MST mix in order to minimize pipetting errors.
- Add 10 µl of the target working solution to each 10 µl ligand dilution step and mix properly by pipetting up and down several times.
Note: Avoid vortexing or heavy shaking in order to prevent protein denaturation. - Incubate at room temperature for five minutes to achieve binding equilibrium. Longer incubation times or different incubation temperatures may be required depending on the specific targets and ligands. The specific incubation time and temperature have to be chosen with a priori knowledge about the interaction to be analyzed. As a general suggestion, we recommend to incubate for between 5 and 20 min at room temperature.
- Fill 16 capillaries with the 16 MST mixes by dipping the capillaries into the sample.
Note: Do not touch the capillaries in the middle section where the optical measurement will take place. - Place the capillaries onto the capillary tray and start the MST device.
- Although a volume of 4 µl is sufficient to fill the MST capillaries, it is advised to prepare at least 20 µl of the final MST mix in order to minimize pipetting errors.
- For the interactions between SPT6L(Phe1218-Asp1412) and the CTD peptides of the A. thaliana RNA polymerase II, the assay buffer is NaP buffer (see Recipes).
- MST measurement
- Starting the MST device
- Start the MST control software and adjust the desired temperature by enabling temperature control. Usually measurements are performed at 25 °C. Wait for the temperature to reach the predefined value.
Note: MST instruments can be temperature-controlled from 22 to 45 °C. - Place the capillary tray in the MST device.
- Set the LED channel (fluorescence excitation) to ‘blue’ for FITC fluorophore labels and set the LED power to gain a fluorescence signal of 300 to 1,000 units at an MST device with a standard sensor and 6,000 to 18,000 on a device with a high-sensitivity sensor.
Note: Other fluorescent labels may require the ‘red’ LED setting. Check the excitation wavelengths of the used fluorophore.
- Start the MST control software and adjust the desired temperature by enabling temperature control. Usually measurements are performed at 25 °C. Wait for the temperature to reach the predefined value.
- Capillary scan
- Perform a capillary scan in order to check different quality aspects of the sample.
- Check the capillary scan to see if the maximum fluorescence signal obtained falls in the ranges described above.
- Check the capillary scan for sticking effects of labeled target to the glass walls of the capillaries. This leads to U-shaped or flattened peaks.
- Check the capillary scan for pipetting errors. These lead to inconsistent fluorescence values across the 16 MST mixes.
- Check the capillary scan for ligand-dependent fluorescence enhancement or quenching effects. These lead to increasing or decreasing fluorescence values with increasing ligand concentration in the 16 MST mixes.
- Perform a capillary scan in order to check different quality aspects of the sample.
- MST measurement
- Before starting an MST measurement, make sure to exclude any sticking effects, pipetting errors, enhancing/quenching effects, and ensure a sufficient fluorescence signal.
- Assign each of the 16 MST mixes with the respective final ligand concentration in the control software. In newer control software versions, only the highest ligand concentration (usually in MST mix 1) is set and the dilution type (e.g., 1:1 or 1:2) is chosen.
Note: The ligand concentration that has to be entered at this point is half the concentration of the ligand dilution series described above, because the dilution samples were mixed 1:1 with target solution. - Enter the fixed concentration of the fluorescently labeled target.
Note: This concentration is also half the concentration of the target working solution due to the preparation of the MST mixes described above. - For most applications, the default settings (initial fluorescence for 5 sec, recording of thermophoresis for 30 sec, and after-thermophoresis fluorescence for a further 5 sec) are sufficient.
- Adjust the MST-power to 20%.
Notes:- In order to receive the best signal-to-noise ratio and to avoid unspecific effects, a laser power of 20-40% is recommended. In some cases, a higher laser power may be required to get good separation of unbound and bound molecules.
- In newer control software versions only the MST power settings ‘low’, ‘medium’, and ‘high’ are available. ‘Medium’ is recommended for most applications.
- In order to receive the best signal-to-noise ratio and to avoid unspecific effects, a laser power of 20-40% is recommended. In some cases, a higher laser power may be required to get good separation of unbound and bound molecules.
- Enter a destination folder path where the experiment file will be saved. The experiment will be saved as a .ntp file.
- Start the MST measurement. The measurement will take between 10-15 min, depending on the times set.
- Repeat the MST measurement at least twice for a more reliable determination of the equilibrium binding affinity.
Note: In order to test the technical reproducibility of the measurement, the same capillaries can be used for several MST measurements.
- Before starting an MST measurement, make sure to exclude any sticking effects, pipetting errors, enhancing/quenching effects, and ensure a sufficient fluorescence signal.
- Starting the MST device
Data analysis
- Data analysis
- Start the MST analysis software (MO.Affinity Analysis) and load the previously saved .ntp file from the destination folder.
- Select the ‘MST’ analysis type.
Note: If the initial fluorescence in the capillary scan shows signs of ligand-dependent fluorescence effects, it is also possible to choose the ‘initial fluorescence’ analysis type. However, possible ligand-dependent fluorescence effects should be carefully investigated with an SD-test (see Notes). - Add the respective technical or biological repeat runs to the MST analysis by drag-and-drop or by clicking the ‘+’ button below the individual repeat runs.
- In order to get information on raw data, MST traces, capillary scans, capillary shape profiles, initial fluorescence, and bleaching rate, click the ‘information’ button below the respective repeat runs.
Note: It is also possible to inspect these raw data during later steps of the analysis. - Check the automated inspection of the data for sticking effects, as well as aggregation and/or precipitation effects. Also visually inspect the data for these effects (e.g., U-shaped capillary shape peaks, bumps and spikes in the MST traces).
Note: A detailed description of this quality control step is described in the Notes section and in Figure 2.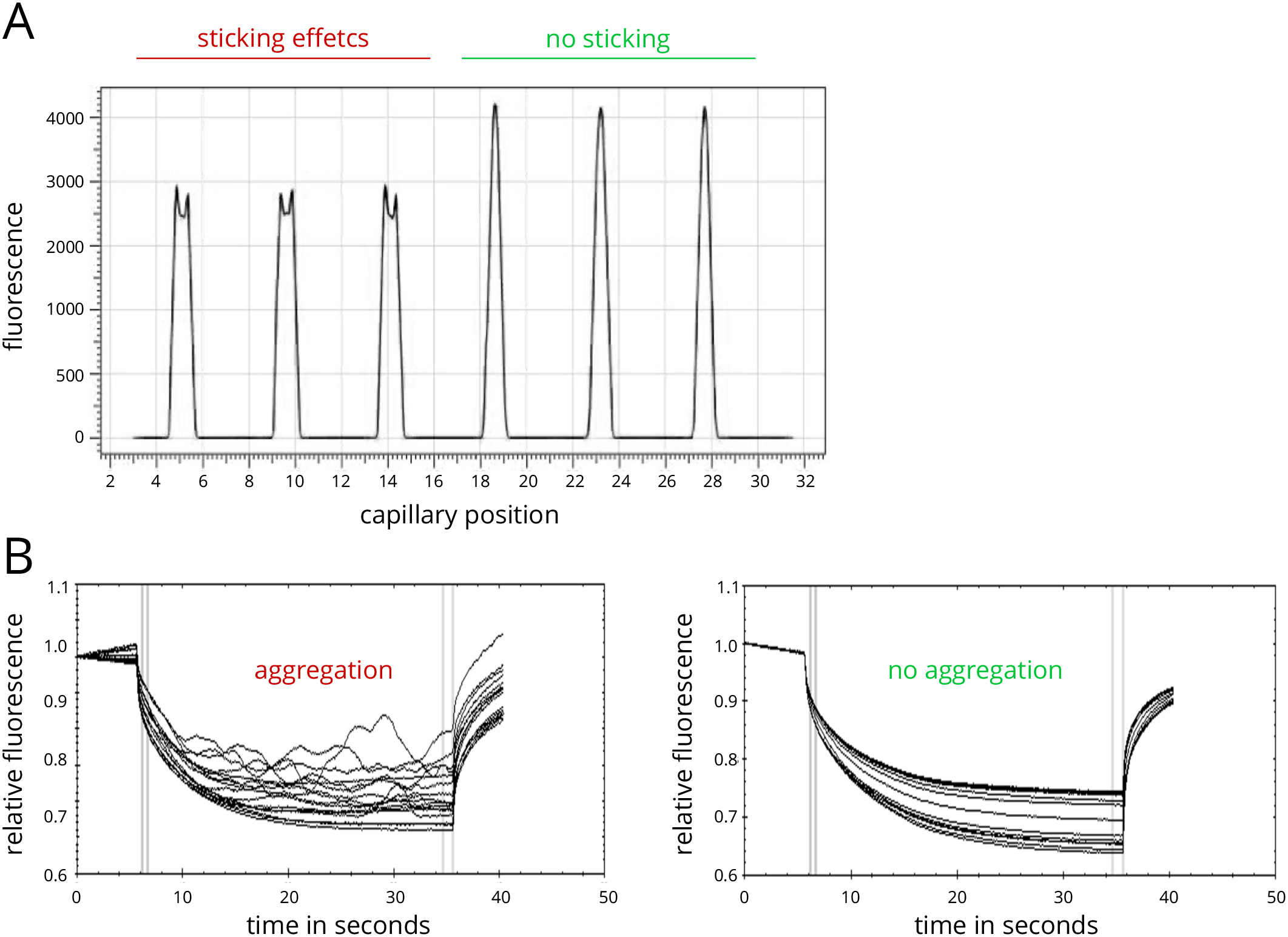
Figure 2. Assay optimization for MST experiments. A. Example of capillary scan with adsorption/sticking effects in capillaries 1-3 and high-quality samples in capillaries 4-6. B. Example of MST traces showing sample aggregation (left panel) and high quality data (right panel). - Evaluate the data by switching to the dose-response panel.
Note: Normally, the standard evaluation modes, which automatically adjust the time ranges for determining Fhot and Fcold in order to guarantee an optimal signal-to-noise ratio, are sufficient for most experiments. However, the user is also free to change the evaluation mode to ‘expert’, where it is possible to manually adjust the time ranges used for evaluation of the data. - For determining the KD value of an interaction, select the ‘KD’ model.
Note: Although the ‘Hill’ model may provide a better fit for the data in many cases, it is advised to choose this model only, when it is justified by known properties of the studied interaction. This could, for example, be a non-1:1 stoichiometry of the interaction or cooperative binding. - For a better comparison of different MST experiments, binding isotherms can be normalized to the fraction of bound molecules (FB) by the following equation:
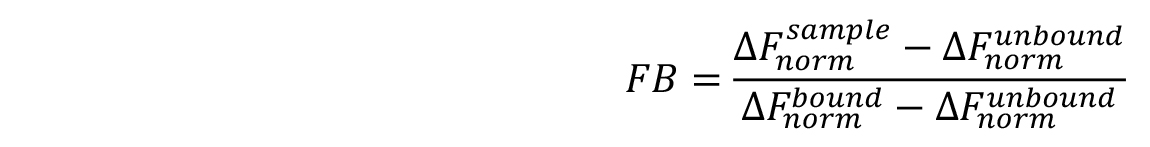
where,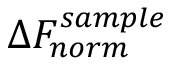 is the value of an individual MST mix,
is the value of an individual MST mix, 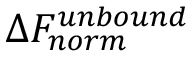 is the value of the unbound state, i.e., the MST mix with the lowest concentration of the ligand, and
is the value of the unbound state, i.e., the MST mix with the lowest concentration of the ligand, and 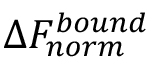 is the value of the bound state, i.e., the MST mix with the highest concentration of the ligand.
is the value of the bound state, i.e., the MST mix with the highest concentration of the ligand.
- Start the MST analysis software (MO.Affinity Analysis) and load the previously saved .ntp file from the destination folder.
- Example results
- Interaction between short repeat peptides of the C-terminal domain of the A. thaliana RNA polymerase II and SPT6L
- The synthesis of mRNAs (and other RNAs) by the 12-subunit RNA polymerase II (RNAPII) is of fundamental importance for eukaryotic organisms. Accordingly, transcription by RNAPII is regulated at various steps of the transcription cycle, including polymerase recruitment as well as transcriptional initiation and elongation (Kornberg, 2007). Importantly, the carboxy-terminal domain (CTD) of the largest subunit of RNAPII is differentially modified during the transcription cycle (Buratowski, 2009; Jeronimo et al., 2016). Phosphorylation of the Ser5 residue of the CTD heptapeptide repeats occurs, for instance, during early elongation, while Ser2 is increasingly phosphorylated when elongation proceeds. The differential modification of the RNAPII-CTD is critical for the timely and coordinated recruitment of factors that modulate transcript elongation on chromatin templates as well as of factors that are involved in the processing of mRNAs (5’end capping, splicing, 3’end polyadenylation) (Moore and Proudfoot, 2009; Bentley, 2014). Also in the plant model Arabidopsis thaliana a variety of so-called transcript elongation factors were identified that facilitate RNAPII transcription of repressive chromatin (Van Lijsebettens and Grasser, 2014). One subgroup of these transcript elongation factors is histone chaperones that assist RNAPII by disassembling nucleosomes in the path of the polymerase, thus promoting polymerase progression (Zhou et al., 2015). Examples of these histone chaperones involved in transcriptional elongation are the H2A/H2B chaperone FACT and the H3/H4 chaperone SPT6, both of which are encoded by essential genes in Arabidopsis (Lolas et al., 2010; Gu et al., 2012). SPT6 occurs in Arabidopsis in two variants termed SPT6L and SPT6 (Gu et al., 2012). In a recent study that analyzed the composition of the RNAPII transcript elongation complex by affinity purification from Arabidopsis cells in combination with mass spectrometry, SPT6L–but not SPT6–was identified as a constituent of the complex (Antosz et al., 2017). Since SPT6 from other organisms binds directly to the RNAPII-CTD as studied by fluorescence anisotropy and NMR (Sun et al., 2010; Liu et al., 2011), it was examined whether Arabidopsis SPT6L also interacts with the RNAPII-CTD.
- Using MST, binding of the putative SPT6L interaction domain (Phe1218-Asp1412) was analyzed with differentially phosphorylated, synthetic, N-terminally FITC-labelled RNAPII CTD repeat peptides (Antosz et al., 2017). The targets in this experiment were the peptides, which were synthesized with either no phosphorylation, or a phosphoserine at position 2 or 5 and labeled with FITC at their N-terminal residue. The sequences of the peptides were: CTD-noP (N-PSYSPTSPSYSP-C), CTD-Ser2P (N-TSPSY(pS)PTSPSY-C) and CTD-Ser5P (N-SYSPT(pS)PSYSPT-C), with (pS) representing a phosphorylated Ser residue. The ligand in this experiment was the STP6L interaction domain, which was expressed with a GST-tag in E. coli and purified by glutathione-sepharose affinity chromatography (Antosz et al., 2017). The experiments were performed on a Monolith NT.115 device with an MST-power of 40% and an LED-power of 80% at 25 °C.
- The MST assay showed that SPT6L(Phe1218-Asp1412) is indeed able to interact with the synthetic CTD peptides and that it has a significantly higher affinity for the peptide with a phosphorylated Ser2 residue. The KD value of this interaction was determined as 134.8 ± 26.6 µM and was highly reproducible over several independent repeats (Figure 3). Experiments with the non-phosphorylated peptide or the Ser5-phosphorylated peptide showed much weaker binding and a reliable determination of the KD values was not possible (estimated KD around 1 mM). It is important to note that this is not an intrinsic issue of the MST technique, but rather a consequence from the concentration of the ligand stock, which was too low to analyze MST mixtures with higher ligand concentrations (highest possible final ligand concentration in an MST mix was 2.15 mM). Thus, the MST assay showed that the interaction domain of SPT6L from A. thaliana also directly binds to the RNAPII-CTD, as has been shown previously for SPT6L proteins from other organisms. Moreover, the assay revealed that the affinity of SPT6L for Ser2-phosphorylated CTD repeat peptides of the RNAPII is at least 10-fold higher than for non-phosphorylated or Ser5-phosphorylated CTDs. These experiments demonstrate how MST can be successfully applied to study interactions between the subunits of sophisticated eukaryotic protein complexes.
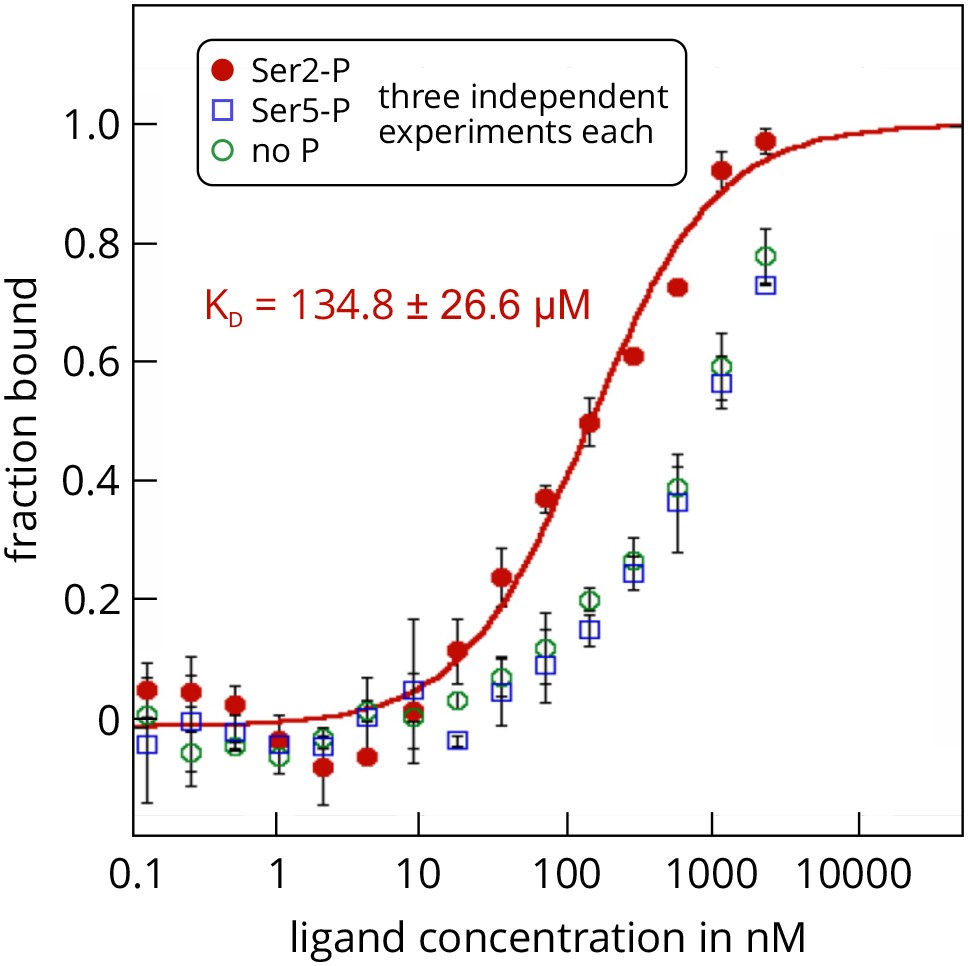
Figure 3. MST analysis of the interactions between GST-SPT6L(Phe1218-Asp1412) and three repeat peptides of the atRNAPII-CTD. The protein SPT6L(Phe1218-Asp1412) served as the unlabelled ligand in this example and was added in increasing concentrations to the three FITC-labelled peptides that served as targets. The peptides resemble specific regions of the C-terminal domain of the A. thaliana RNA polymerase II and were either non-phosphorylated (green), or phosphorylated at Ser2 (red) or Ser5 (blue). Raw fluorescence data were normalized to the fraction of bound target. Error bars represent standard deviations from three individual repeat measurements. The KD was determined from a fit with the ‘KD’ model to the law of mass action (R2 = 0.98). The figure is modified from Antosz et al., 2017 (www.plantcell.org, Copyright American Society of Plant Biologists).
- The synthesis of mRNAs (and other RNAs) by the 12-subunit RNA polymerase II (RNAPII) is of fundamental importance for eukaryotic organisms. Accordingly, transcription by RNAPII is regulated at various steps of the transcription cycle, including polymerase recruitment as well as transcriptional initiation and elongation (Kornberg, 2007). Importantly, the carboxy-terminal domain (CTD) of the largest subunit of RNAPII is differentially modified during the transcription cycle (Buratowski, 2009; Jeronimo et al., 2016). Phosphorylation of the Ser5 residue of the CTD heptapeptide repeats occurs, for instance, during early elongation, while Ser2 is increasingly phosphorylated when elongation proceeds. The differential modification of the RNAPII-CTD is critical for the timely and coordinated recruitment of factors that modulate transcript elongation on chromatin templates as well as of factors that are involved in the processing of mRNAs (5’end capping, splicing, 3’end polyadenylation) (Moore and Proudfoot, 2009; Bentley, 2014). Also in the plant model Arabidopsis thaliana a variety of so-called transcript elongation factors were identified that facilitate RNAPII transcription of repressive chromatin (Van Lijsebettens and Grasser, 2014). One subgroup of these transcript elongation factors is histone chaperones that assist RNAPII by disassembling nucleosomes in the path of the polymerase, thus promoting polymerase progression (Zhou et al., 2015). Examples of these histone chaperones involved in transcriptional elongation are the H2A/H2B chaperone FACT and the H3/H4 chaperone SPT6, both of which are encoded by essential genes in Arabidopsis (Lolas et al., 2010; Gu et al., 2012). SPT6 occurs in Arabidopsis in two variants termed SPT6L and SPT6 (Gu et al., 2012). In a recent study that analyzed the composition of the RNAPII transcript elongation complex by affinity purification from Arabidopsis cells in combination with mass spectrometry, SPT6L–but not SPT6–was identified as a constituent of the complex (Antosz et al., 2017). Since SPT6 from other organisms binds directly to the RNAPII-CTD as studied by fluorescence anisotropy and NMR (Sun et al., 2010; Liu et al., 2011), it was examined whether Arabidopsis SPT6L also interacts with the RNAPII-CTD.
- Interactions between histone tail peptides and HP1 proteins
- Eukaryotic genomes are usually organized as chromatin, a compact storage form of DNA wound around scaffold proteins. The lowest level of chromatin are nucleosomes, which are octameric complexes of histone proteins circled by a DNA stretch of roughly 150 nucleotides (Kornberg, 1974; Luger et al., 1997). The histone monomers possess flexible, N-terminal tails that are the site of frequent post-translational modifications like acetylations, methylations, or phosphorylations. The frequency and nature of these modifications are important regulators of chromatin structure and DNA accessibility (Jenuwein and Allis, 2001): Modified histone tails can influence interactions between neighboring nucleosomes and can recruit other proteins that influence chromatin structure (Shogren-Knaak et al., 2006). One such ‘reader’ protein is the heterochromatin protein 1 (HP1), a small (around 30 kDa), highly conserved protein that specifically binds to methylated histones, for example to histone tail peptides of histone H3 that feature a bi- or tri-methylation at Lys9 (H3K9met) (Eskeland et al., 2007). Binding of HP1 to methylated histone tails is an important factor in epigenetic regulations and the formation of heterochromatin, i.e., regions of chromatin with low transcriptional activity and high compacting ratio (Kwon and Workman, 2011).
- MST enables the researcher to determine the typical interaction parameters of the interactions between HP1 proteins and histone tails in-solution (i.e., without any immobilization) and label-free (i.e., without the need for attaching a fluorescent label to one of the interaction partners). It thus provides the possibility to study these interactions in a system that is very close to the physiological conditions of the interactions. This is possible, because the short histone tail peptides do not contain fluorescent amino acids and at the same time the HP1 proteins we studied contain enough Trp and Tyr residues in order to generate a reliable intrinsic fluorescence signal that can be measured with the Monolith NT.LabelFree MST device. We analyzed the interactions between two HP1 homologs (from Plasmodium falciparum and Drosophila melanogaster) with four different tail peptides of histone H3 (20 amino acids): one unmodified peptide, two di-methylated peptides (at Lys4 and Lys9), and one acetylated peptide (at Lys9).
- For each experiment, the HP1 protein served as the target and was used at a final, constant concentration of 3.5 µM. The peptides served as ligands and were assayed at a final concentration, ranging from 10 nm to 10 mM. The Monolith NT.LabelFree was set to an LED power of 20% and an MST power of 40%, with a thermophoresis time interval of 30 sec. Raw fluorescence data were normalized to the fraction of bound ligand and fitted with the ‘KD’ model describing the law of mass action. HP1 from P. falciparum (pfHP1) binds to the di-methylated peptide H3K9(Me2) with a KD value of 4.36 ± 0.14 µM (Figure 4A). Additional experiments showed that this interaction is highly specific, because pfHP1 does not bind to another di-methylated peptide that differs only in the position of the methylated lysine residue (H3K4(Me2)), nor to an acetylated peptide (H3K9(Ac)) or the unmodified peptide (H3) (Figure 4B). We could also show that this interaction specificity is conserved, because also HP1 from D. melanogaster (dmHP1) displays the same binding preferences (Figures 4C and 4D). However, the KD of the interaction with H3K9(Me2) is about 7-fold higher (31.12 ± 0.48 µM).
- These experiments also demonstrate that binding isotherms determined with MST are highly reliable. The standard deviations from several individual repeats of a binding experiment are low and the determined KD values are highly reproducible (Figures 4A and 4C). Moreover, the KD value for the interaction between pfHP1 und H3K9(Me2) (4.36 µM) is in good agreement with a value of 7 µM determined by ITC (Jacobs and Khorasanizadeh, 2002). This shows that MST is well suited to study interactions in epigenetic systems. Importantly, the measurements can be done in solution, with very low sample consumption (an important point to consider when working with eukaryotic proteins or synthetic peptides), and also in bioliquids. The technique thus allows for the fast, easy, and flexible characterization of various histone-reader protein interactions.
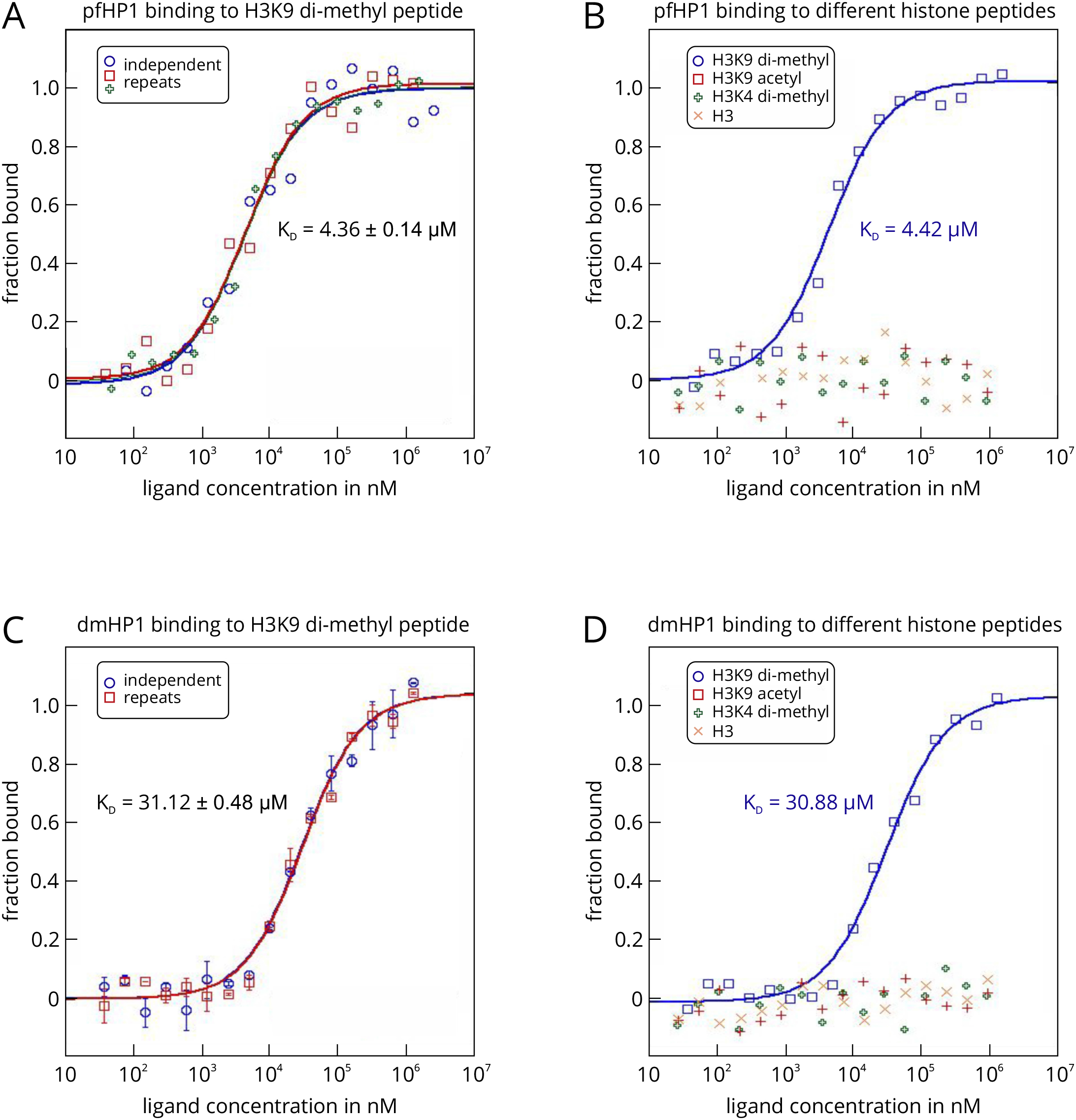
Figure 4. MST analysis of the interactions between P. falciparum and D. melanogaster HP1 and different histone peptides using the label-free MST technology. The HP1 proteins from P. falciparum (pfHP1) and D. melanogaster (dmHP1) served as the targets in this example. In this example, it was not necessary to attach a fluorescent label to the targets, because both proteins featured high enough molar extinction coefficients to analyze the interactions on a Monolith NT.LabelFree device. A prerequisite for this was also that the histone peptides did not contain tryptophan and tyrosine residues (no fluorescence above 300 nm).
- Eukaryotic genomes are usually organized as chromatin, a compact storage form of DNA wound around scaffold proteins. The lowest level of chromatin are nucleosomes, which are octameric complexes of histone proteins circled by a DNA stretch of roughly 150 nucleotides (Kornberg, 1974; Luger et al., 1997). The histone monomers possess flexible, N-terminal tails that are the site of frequent post-translational modifications like acetylations, methylations, or phosphorylations. The frequency and nature of these modifications are important regulators of chromatin structure and DNA accessibility (Jenuwein and Allis, 2001): Modified histone tails can influence interactions between neighboring nucleosomes and can recruit other proteins that influence chromatin structure (Shogren-Knaak et al., 2006). One such ‘reader’ protein is the heterochromatin protein 1 (HP1), a small (around 30 kDa), highly conserved protein that specifically binds to methylated histones, for example to histone tail peptides of histone H3 that feature a bi- or tri-methylation at Lys9 (H3K9met) (Eskeland et al., 2007). Binding of HP1 to methylated histone tails is an important factor in epigenetic regulations and the formation of heterochromatin, i.e., regions of chromatin with low transcriptional activity and high compacting ratio (Kwon and Workman, 2011).
- Interaction between short repeat peptides of the C-terminal domain of the A. thaliana RNA polymerase II and SPT6L
Notes
- Detection of adsorption and capillary sticking effects and troubleshooting
The capillary scan can detect sticking or adsorption effects that occur between the targets and the glass surface of the capillaries. Irregular peak shapes (Figure 2A, capillaries 1-3), such as U-shaped or flattened peaks, indicate such sticking or adsorption effects. In many cases a change of capillary type (standard treated, premium coated, or hydrophobic) can prevent them, resulting in regularly shaped peaks (Figure 2A, capillaries 4-6). If a change of capillary type does not enhance the capillary scan peak shapes, a change in buffer conditions or the addition of detergents like Tween (0.005-0.1%) or Pluronic F-127 (0.01-0.1%) may prevent sticking and adsorption. Generally, a capillary test should be performed with one MST mix prior to the measurement of the full 16 MST mixes in order to find conditions where no sticking and adsorption occurs. This stage of an MST experiment is essential for high data quality later on. - Detection of fluorescence effects and troubleshooting
- The initial capillary scan also provides information on possible pipetting errors or a ligand-dependent change in fluorescence emission. Pipetting errors are reflected in random height differences of the capillary peaks. A high pipetting accuracy is mandatory to avoid such errors. Also, buffer dilution effects should be avoided, i.e., the buffer used for preparing the dilution steps should be exactly the same buffer as the one of the ligand working solution. Large differences in the fluorescence readout between individual capillaries may also be a sign of sample aggregation. Aggregates are characterized by a much higher fluorescence density and thus increase the fluorescence readout when coming into the optical readout focus.
- Systematic changes in the fluorescence signal that correlate with the ligand concentration, however, hint at a possible fluorescence enhancement or quenching effect by the ligand. In order to exclude other possibilities, like unspecific fluorescence decrease, for these systematic changes, an SD-test is carried out: If the systematic change of fluorescence is really ligand-dependent, this effect should not be detectable under denaturing conditions and the fluorescence signal should become identical for all capillaries. If, however, the fluorescence signal still shows deviations under denaturing conditions, the reason is probably a loss of fluorescently labeled molecule due to adsorption, sticking, or aggregation effects. A standard SD-test is performed as follows: 10 µl of the first MST mix and 10 µl of the last MST mix are each transferred to a fresh PCR tube containing 10 µl of a 2x SD mix (4% SDS, 40 mM DTT). After mixing, the samples are incubated at 95 °C for 5 min to ensure denaturation. Tow fresh capillaries are filled and the fluorescence intensities are recorded at the same settings as before.
- The initial capillary scan also provides information on possible pipetting errors or a ligand-dependent change in fluorescence emission. Pipetting errors are reflected in random height differences of the capillary peaks. A high pipetting accuracy is mandatory to avoid such errors. Also, buffer dilution effects should be avoided, i.e., the buffer used for preparing the dilution steps should be exactly the same buffer as the one of the ligand working solution. Large differences in the fluorescence readout between individual capillaries may also be a sign of sample aggregation. Aggregates are characterized by a much higher fluorescence density and thus increase the fluorescence readout when coming into the optical readout focus.
- Detection of aggregation effects and troubleshooting
Sample aggregation and/or precipitation can easily be detected in the MST traces as bumps and spikes (Figure 2B, left panel). Samples with no aggregation usually display smooth MST traces (Figure 2B, right panel). If aggregation occurs in a sample, a change of capillary type, buffer conditions and additives such as detergents (0.005-0.1% Tween-20, 0.01-0.1% Pluronic F-127, or similar) or BSA (> 0.5 mg/ml), pH conditions, and salt concentrations may optimize solubility of the molecules. Large aggregates may be removed prior to the measurement by centrifugation (at least 10 min at 14,000 x g). In order to ensure optimal data quality, the MST traces should resemble the ones shown in the right panel of Figure 2B. - Data analysis and curve fitting
- In principle, MST traces can be evaluated with respect to the different underlying physical phenomena. The temperature jump (or T-jump) describes the sudden change if fluorescence emission upon temperature change, which is often characteristic for different fluorophores. The T-jump is highly affected by binding of the ligand in close proximity to the fluorophore. The slower thermophoresis phase of an MST trace is influenced by the movement of the molecules along the induced temperature gradient and is thus highly sensitive to changes in size, charge, and hydration shell of the molecules under study.
- In the ‘expert’ analysis mode of the MO.Affinity Analysis software the user can vary the time points chosen for either T-jump or thermophoresis evaluation. If aggregation occurs in the samples and the instructions described above do not resolve these issues, it may be possible to use the early parts of the thermophoresis phase where aggregation effects may be smaller than in later parts. Examining the early parts of the thermophoresis phase may also increase data quality due to less-pronounced convection effects inside the capillaries.
- The MO.Affinity Analysis software offers two options for fitting the dose-response curves. The ‘KD’ model is based on the law of mass-action:

where, T is the labeled target and L is the non-fluorescent ligand.
The fluorescence signal that is dependent on the concentration of the titrated ligand (F(c𝐿)) can be calculated as follows: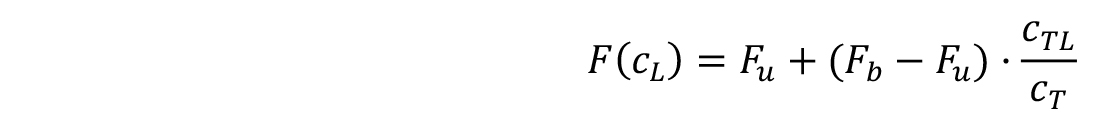
where, Fu and Fb are the fluorescence values in the unbound and bound states, and cTL and cT are the concentrations of formed complex and the fixed concentration of the fluorescently labelled target.
From the fraction bound value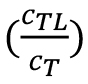 , the KD of an interaction can be derived:
, the KD of an interaction can be derived: 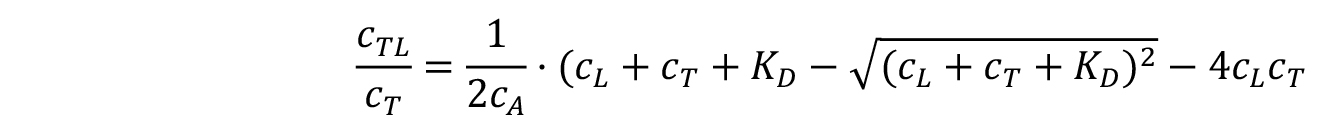
where, KD is the dissociation constant.
Note that this fitting model is only valid for data that describe a 1:1 interaction between the target T and the ligand L with one specific binding site or multiple binding sites with the same affinity. The ‘Hill’ model allows for determining the EC50 value of an interaction:
In this case, the fraction bound is given as: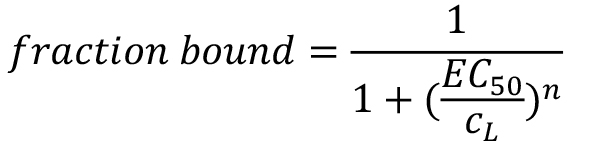
where, cL is the concentration of the titrated, non-labelled ligand.
Note that the ‘Hill’ model is often used for interactions with cooperative binding. It should not be used for fitting data that can be clearly explained with the law of mass action. Also, the ‘Hill’ model only determines the EC50 value of a molecular interaction, which is the concentration of the ligand where 50% of the target molecules are bound. It is important to know that the EC50 value does not directly correspond to the dissociation constant KD. It is more an apparent measure of affinity for one particular experiment that is dependent on the used concentrations and conditions.
- In principle, MST traces can be evaluated with respect to the different underlying physical phenomena. The temperature jump (or T-jump) describes the sudden change if fluorescence emission upon temperature change, which is often characteristic for different fluorophores. The T-jump is highly affected by binding of the ligand in close proximity to the fluorophore. The slower thermophoresis phase of an MST trace is influenced by the movement of the molecules along the induced temperature gradient and is thus highly sensitive to changes in size, charge, and hydration shell of the molecules under study.
- Detection of low fluorophore concentration
Concentrations of labeled targets below 1 nM usually require high excitation light intensities (LED power > 75% (or ‘high’) on the Monolith NT.115pico). Such high intensities can lead to significant photobleaching of the fluorophore and thus introduce additional noise to the data. NanoTemper Technologies offers an anti-photobleaching kit that can help reduce these effects. - About the temperature gradient
The temperature gradient spans 2 °C at an MST power of 20% and 6 °C at an MST power of 80%. The total volume of the heated sample is 2 nl. It is recommended to identify the optimal MST power prior the full binding experiment by performing several measurements with a single capillary and different MST power settings (for example 20%, 40%, and 80%).
Recipes
- NaP buffer
10 mM sodium phosphate pH 7.0
1 mM EDTA
1 mM DTT
0.5 mM PMSF - MST-T buffer
50 mM Tris-HCl pH 7.8
150 mM NaCl
10 mM MgCl2
0.05% Tween-20
Acknowledgments
We thank Corinna Kuttenberger and Clemens Entzian for expert scientific and technical assistance.
References
- Abdiche, Y., Malashock, D., Pinkerton, A. and Pons, J. (2008). Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem 377(2): 209-217.
- Antosz, W., Pfab, A., Ehrnsberger, H. F., Holzinger, P., Kollen, K., Mortensen, S. A., Bruckmann, A., Schubert, T., Langst, G., Griesenbeck, J., Schubert, V., Grasser, M. and Grasser, K. D. (2017). The composition of the Arabidopsis RNA polymerase II transcript elongation complex reveals the interplay between elongation and mRNA processing factors. Plant Cell 29(4): 854-870.
- Arkin, M. R., Tang, Y. and Wells, J. A. (2014). Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol 21(9): 1102-1114.
- Azzarito, V., Long, K., Murphy, N. S. and Wilson, A. J. (2013). Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat Chem 5: 161-173.
- Baaske, P., Wienken, C. J., Reineck, P., Duhr, S. and Braun, D. (2010). Optical thermophoresis for quantifying the buffer dependence of aptamer binding. Angew Chem Int Ed Engl 49(12): 2238-2241.
- Bentley, D. L. (2014). Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15(3): 163-175.
- Boersma, M. D., Haase, H. S., Peterson-Kaufman, K. J., Lee, E. F., Clarke, O. B., Colman, P. M., Smith, B. J., Horne, W. S., Fairlie, W. D. and Gellman, S. H. (2012). Evaluation of diverseα/β-backbone patterns for functional α-helix mimicry: analogues of the Bim BH3 domain. J Am Chem Soc 134(1): 315-323.
- Braun, D. and Libchaber, A. (2002). Trapping of DNA by thermophoretic depletion and convection. Phys Rev Lett 89(18): 188103.
- Buratowski, S. (2009). Progression through the RNA polymerase II CTD cycle. Mol Cell 36(4): 541-546.
- Chen, M., Hu, D., Li, X., Yang, S., Zhang, W., Li, P. and Song, B. (2015). Antiviral activity and interaction mechanisms study of novel glucopyranoside derivatives. Bioorg Med Chem Lett 25(18): 3840-3844.
- Concepcion, J., Witte, K., Wartchow, C., Choo, S., Yao, D., Persson, H., Wei, J., Li, P., Heidecker, B., Ma, W., Varma, R., Zhao, L. S., Perillat, D., Carricato, G., Recknor, M., Du, K., Ho, H., Ellis, T., Gamez, J., Howes, M., Phi-Wilson, J., Lockard, S., Zuk, R. and Tan, H. (2009). Label-free detection of biomolecular interactions using BioLayer interferometry for kinetic characterization. Comb Chem High Throughput Screen 12(8): 791-800.
- Duhr, S., Arduini, S. and Braun, D. (2004). Thermophoresis of DNA determined by microfluidic fluorescence. Eur Phys J E Soft Matter 15(3): 277-286.
- Duhr, S. and Braun, D. (2006). Why molecules move along a temperature gradient. Proc Natl Acad Sci U S A 103(52): 19678-19682.
- Entzian, C. and Schubert, T. (2016). Studying small molecule-aptamer interactions using MicroScale Thermophoresis (MST). Methods 97: 27-34.
- Eskeland, R., Eberharter, A. and Imhof, A. (2007). HP1 binding to chromatin methylated at H3K9 is enhanced by auxiliary factors. Mol Cell Biol 27(2): 453-465.
- Ghai, R., Falconer, R. J. and Collins, B. M. (2012). Applications of isothermal titration calorimetry in pure and applied research--survey of the literature from 2010. J Mol Recognit 25(1): 32-52.
- Gu, X. L., Wang, H., Huang, H. and Cui, X. F. (2012). SPT6L encoding a putative WG/GW-repeat protein regulates apical-basal polarity of embryo in Arabidopsis. Mol Plant 5(1): 249-259.
- Hanlon, A. D., Larkin, M. I. and Reddick, R. M. (2010). Free-solution, label-free protein-protein interactions characterized by dynamic light scattering. Biophys J 98(2): 297-304.
- Harazi, A., Becker-Cohen, M., Zer, H., Moshel, O., Hinderlich, S. and Mitrani-Rosenbaum, S. (2017). The interaction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) and Alpha-actinin 2 is altered in GNE myopathy M743T mutant. Mol Neurobiol 54(4): 2928-2938.
- Heck, A. J. (2008). Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods 5(11): 927-933.
- Ishii, E., Eguchi, Y., and Utsumi, R. (2013). Protein-peptide interaction by surface plasmon resonance. Bio Protoc e321.
- Jacobs, S. A. and Khorasanizadeh, S. (2002). Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 295(5562): 2080-2083.
- Jenuwein, T. and Allis, C. D. (2001). Translating the histone code. Science 293(5532): 1074-1080.
- Jerabek-Willemsen, M., André, T., Wanner, R., Roth, H.M., Duhr, S., Baaske, P., and Breitsprecher, D. (2014). MicroScale thermophoresis: Interaction analysis and beyond. J Mol Struct 1077: 101-113.
- Jerabek-Willemsen, M., Wienken, C. J., Braun, D., Baaske, P. and Duhr, S. (2011). Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol 9(4): 342-353.
- Jeronimo, C., Collin, P. and Robert, F. (2016). The RNA polymerase II CTD: The increasing complexity of a low-complexity protein domain. J Mol Biol 428(12): 2607-2622.
- Josling, G. A., Petter, M., Oehring, S. C., Gupta, A. P., Dietz, O., Wilson, D. W., Schubert, T., Langst, G., Gilson, P. R., Crabb, B. S., Moes, S., Jenoe, P., Lim, S. W., Brown, G. V., Bozdech, Z., Voss, T. S. and Duffy, M. F. (2015). A plasmodium falciparum bromodomain protein regulates invasion gene expression. Cell Host Microbe 17(6): 741-751.
- Kameyama, K. and Minton, A. P. (2006). Rapid quantitative characterization of protein interactions by composition gradient static light scattering. Biophys J 90(6): 2164-2169.
- Kammel, C., Thomaier, M., Sorensen, B. B., Schubert, T., Langst, G., Grasser, M. and Grasser, K. D. (2013). Arabidopsis DEAD-box RNA helicase UAP56 interacts with both RNA and DNA as well as with mRNA export factors. PLoS One 8(3): e60644.
- Kornberg, R. D. (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184(4139): 868-871.
- Kornberg, R. D. (2007). The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A 104(32): 12955-12961.
- Kwon, S. H. and Workman, J. L. (2011). The changing faces of HP1: From heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. Bioessays 33(4): 280-289.
- Lin, W. C., Iversen, L., Tu, H. L., Rhodes, C., Christensen, S. M., Iwig, J. S., Hansen, S. D., Huang, W. Y. and Groves, J. T. (2014). H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc Natl Acad Sci U S A 111(8): 2996-3001.
- Liu, J., Zhang, J., Gong, Q., Xiong, P., Huang, H., Wu, B., Lu, G., Wu, J. and Shi, Y. (2011). Solution structure of tandem SH2 domains from Spt6 protein and their binding to the phosphorylated RNA polymerase II C-terminal domain. J Biol Chem 286(33): 29218-29226.
- Lolas, I. B., Himanen, K., Gronlund, J. T., Lynggaard, C., Houben, A., Melzer, M., Van Lijsebettens, M. and Grasser, K. D. (2010). The transcript elongation factor FACT affects Arabidopsis vegetative and reproductive development and genetically interacts with HUB1/2. Plant J 61(4): 686-697.
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. and Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389(6648): 251-260.
- Lundblad, J. R., Laurance, M. and Goodman, R. H. (1996). Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Mol Endocrinol 10(6): 607-612.
- Milroy, L. G., Grossmann, T. N., Hennig, S., Brunsveld, L. and Ottmann, C. (2014). Modulators of protein-protein interactions. Chem Rev 114(9): 4695-4748.
- Moore, M. J. and Proudfoot, N. J. (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136(4): 688-700.
- Nevola, L. and Giralt, E. (2015). Modulating protein-protein interactions: the potential of peptides. Chem Commun (Camb) 51(16): 3302-3315.
- Pattnaik, P. (2005). Surface plasmon resonance: applications in understanding receptor-ligand interaction. Appl Biochem Biotechnol 126(2): 79-92.
- Pelay-Gimeno, M., Glas, A., Koch, O. and Grossmann, T. N. (2015). Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angew Chem Int Ed Engl 54(31): 8896-8927.
- Pierce, M. M., Raman, C. S. and Nall, B. T. (1999). Isothermal titration calorimetry of protein-protein interactions. Methods 19(2): 213-221.
- Raines, R. T. (2015). Fluorescence polarization assay to quantify protein-protein interactions: an update. Methods Mol Biol 1278: 323-327.
- Ramakrishnan, M., Alves De Melo, F., Kinsey, B. M., Ladbury, J. E., Kosten, T. R. and Orson, F. M. (2012). Probing cocaine-antibody interactions in buffer and human serum. PLoS One 7(7): e40518.
- Schubert, T., Pusch, M. C., Diermeier, S., Benes, V., Kremmer, E., Imhof, A. and Langst, G. (2012). Df31 protein and snoRNAs maintain accessible higher-order structures of chromatin. Mol Cell 48(3): 434-444.
- Seidel, S. A., Dijkman, P. M., Lea, W. A., van den Bogaart, G., Jerabek-Willemsen, M., Lazic, A., Joseph, J. S., Srinivasan, P., Baaske, P., Simeonov, A., Katritch, I., Melo, F. A., Ladbury, J. E., Schreiber, G., Watts, A., Braun, D. and Duhr, S. (2013). Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59(3): 301-315.
- Seidel, S. A., Wienken, C. J., Geissler, S., Jerabek-Willemsen, M., Duhr, S., Reiter, A., Trauner, D., Braun, D. and Baaske, P. (2012). Label-free microscale thermophoresis discriminates sites and affinity of protein-ligand binding. Angew Chem Int Ed Engl 51(42): 10656-10659.
- Shogren-Knaak, M., Ishii, H., Sun, J. M., Pazin, M. J., Davie, J. R. and Peterson, C. L. (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311(5762): 844-847.
- Sperandio, O., Reynes, C. H., Camproux, A. C. and Villoutreix, B. O. (2010). Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discov Today 15(5-6): 220-229.
- Stanfield, R. L. and Wilson, I. A. (1995). Protein-peptide interactions. Curr Opin Struct Biol 5(1): 103-113.
- Stoltenburg, R., Schubert, T. and Strehlitz, B. (2015). In vitro selection and interaction studies of a DNA aptamer targeting protein A. PLoS One 10(7): e0134403.
- Sun, M., Lariviere, L., Dengl, S., Mayer, A. and Cramer, P. (2010). A tandem SH2 domain in transcription elongation factor Spt6 binds the phosphorylated RNA polymerase II C-terminal repeat domain (CTD). J Biol Chem 285(53): 41597-41603.
- Van Lijsebettens, M. and Grasser, K. D. (2014). Transcript elongation factors: shaping transcriptomes after transcript initiation. Trends Plant Sci 19(11): 717-726.
- Wan, C., Wu, B., Song, Z., Zhang, J., Chu, H., Wang, A., Liu, Q., Shi, Y., Li, G., and Wang, J. (2015). Insights into the molecular recognition of the granuphilin C2A domain with PI(4,5)P2. Chem Phys Lipids 186: 61-67.
- Wen, J., Arakawa, T. and Philo, J. S. (1996). Size-exclusion chromatography with on-line light-scattering, absorbance, and refractive index detectors for studying proteins and their interactions. Anal Biochem 240(2): 155-166.
- Wienken, C. J., Baaske, P., Rothbauer, U., Braun, D. and Duhr, S. (2010). Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun 1: 100.
- Yan, J., Zhou, M., Gilbert, J. D., Wolff, J. J., Somogyi, A., Pedder, R. E., Quintyn, R. S., Morrison, L. J., Easterling, M. L., Pasa-Tolic, L. and Wysocki, V. H. (2017). Surface-induced dissociation of protein complexes in a hybrid Fourier transform ion cyclotron resonance mass spectrometer. Anal Chem 89(1): 895-901.
- Zhou, W., Zhu, Y., Dong, A. and Shen, W. H. (2015). Histone H2A/H2B chaperones: from molecules to chromatin-based functions in plant growth and development. Plant J 83(1): 78-95.
- Zillner, K., Jerabek-Willemsen, M., Duhr, S., Braun, D., Langst, G. and Baaske, P. (2012). Microscale thermophoresis as a sensitive method to quantify protein: nucleic acid interactions in solution. Methods Mol Biol 815: 241-252.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Plach, M. G., Grasser, K. D. and Schubert, T. (2017). MicroScale Thermophoresis as a Tool to Study Protein-peptide Interactions in the Context of Large Eukaryotic Protein Complexes. Bio-protocol 7(23): e2632. DOI: 10.21769/BioProtoc.2632.
Category
Molecular Biology > Protein > Protein-protein interaction
Biochemistry > Protein > Interaction > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.