- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Organotypic Brain Cultures: A Framework for Studying CNS Infection by Neurotropic Viruses and Screening Antiviral Drugs

(§Deceased) Published: Vol 7, Iss 22, Nov 20, 2017 DOI: 10.21769/BioProtoc.2605 Views: 14015
Reviewed by: Kae-Jiun ChangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
According to the World Health Organization (WHO), at least 50% of emerging viruses endowed with pathogenicity in humans can infect the Central Nervous System (CNS) with induction of encephalitis and other neurologic diseases (Taylor et al., 2001; Olival and Daszak, 2005). While neurological diseases are progressively documented, the underlying cellular and molecular mechanisms involved in virus infection and dissemination within the CNS are still poorly understood (Swanson and McGavern, 2015; Ludlow et al., 2016). For example, measles virus (MeV) can infect neural cells, and cause a persistent brain infections leading to lethal encephalitis from several months to years after primary infection with no available treatment (Reuter and Schneider-Schaulies, 2010; Laksono et al., 2016). The Organotypic Brain Culture (OBC) is a suitable model for the virology field to better understand the CNS infections. Indeed, it allows not only studying the infection and the dissemination of neurotropic viruses within the CNS but it could also serve as screening model of innovative antiviral strategies or molecules, such as our recently published studies about fusion inhibitory peptides and the HSP90 chaperone activity inhibitor, 17-DMAG (Welsch et al., 2013; Bloyet et al., 2016). Based on our previous work, we propose here an optimized method to prepare OBC of hippocampi and cerebellums which are suitable for small rodent models based virus studies, including mice, rats as well as hamsters at a post-natal stage, between P6 to P10. We notably took into account the stress of the slice procedure on the tissue and the subsequent cellular reactions, which is essential to fully characterize the model prior to any use in infectious conditions. With this knowledge, we propose a protocol highlighting the requirements, including potential trouble shootings of the slicing parameters, to consider the variations we observed according to the structure and animal studied. This framework should facilitate the use of OBC for better conclusive studies of neurotropic viruses.
Keywords: Organotypic brain cultureBackground
Since 1958 neurobiologists have continuously developed organotypic brain cultures (OBC) with a tremendous increase in their usage over the last two decades in the fields of neurodevelopment, neurodegenerative diseases or neuropharmacology (Bornstein and Murray, 1958; Kim et al., 2013; Humpel, 2015). In contrast, despite the advantages of this model, very few studies of virus infection, tropism or dissemination have been published (Mayer et al., 2005; Braun et al., 2006; Stubblefield Park et al., 2011). Indeed, experiments using OBC are inherently more complex to set up than classical cellular primary cultures (i.e., purified neurons or dissociated brain cultures). However, the elegance of this approach resides in the possibility to maintain major cell types in a preserved three-dimensional tissue architecture that allows studying in real time viral invasion throughout brain structures and cell subsets, in more physiological environment and without the impact of the peripheral immune system. Furthermore, since the cellular composition of the tissue is maintained, including neurons, oligodendrocytes, microglial cells and astrocytes, it becomes possible to assess and decipher the involvement and the response of each cell population during the viral infection (Lossi et al., 2009). This model also presents the advantage to reduce the animal payload compared to in vivo experiment which fits perfectly with the recommendations and regulation of animal usage in life science by the Institutional Animal Care and Use Committee (IACUC). Indeed, it is possible to generate at least 10 to 15 slices per structure and thus it allows expanding the number of tested conditions per animal. Furthermore, most of the equipment required for its implementation is easy to acquire or already available in laboratories using tissue culture approaches with interest in neuro-virology. This protocol details the preparation of cultured rodent brain slices obtained from either hippocampus or cerebellum, assessment of its viability, analysis of brain cell types, morphological rearrangements and kinetic during one-week culture. Finally, this protocol offers an example of utilization of OBC to study viral brain infection with measles virus (MeV) in rodent explants.
Materials and Reagents
- Sterile pipette tips, 1,000 μl (Corning, catalog number: 9032 )
- Sterile filtered pipette tips, 10 μl (Corning, catalog number: 4807 )
- Sterile Falcon 6-well flat bottom plate (Corning, Falcon®, catalog number: 353046 )
- Feather 81-S razor blades (Dominique Dutscher, catalog number: 711164B)
Manufacturer: Feather Safety Razor, model: 81-S . - Scalpel blades N°10 (Dominique Dutcher, catalog number: 132510 )
- Sterile 50 ml sterile Falcon tubes (Corning, catalog number: 430290 )
- Sterile Petri dishes, 35 mm (Corning, Falcon®, catalog number: 351008 )
- Sterile pipettes for cell culture 5 ml Falcon (Corning, Falcon®, catalog number: 356543 )
- Sterile Whatman paper (for the hippocampal slicing process) (GE Healthcare, catalog number: 10347510 )
- Sterile PTFE plate 60 x 60 x 5 mm for cerebellum slicing (ePlastics, 0.250” PTFE Sheet 12” x 12”)
- Sterile syringe filter with a pore size of 0.22 µm (EMD Millipore, catalog number: SLGV033RS )
- Sterile Millicell Cell Culture insert, 30 mm, hydrophilic PTFE, 0.4 µm (EMD Millipore, catalog number: PICM0RG50 )
- 96-well, white plate flat clear bottom with lid (Corning, catalog number: 3610 )
- Falcon 12-well flat bottom plate (Dominique Dutscher, catalog number: 064023 )
- Slide and coverslip
- Filtration unit Stericup GP Millipore, pores 0.2 µm (EMD Millipore, catalog number: SCGPU05RE )
- Needle (Hamilton Bell, catalog number: 6980 )
- Neonate rodent (mouse, rat, hamster) between postnatal day P6 to P10 (males and/or females)
Note: Based on our experience, the sex of the animals did not affect our results, but this parameter should be considered carefully when working with other viruses than MeV. - Example of virus: recombinant measles virus (IC323 strain) coding for enhanced green fluorescent protein (MeV-EGFP–1.107 pfu/ml)
- 70% ethanol
- Ketamine hydrochloride (MWI Animal Health, NDC 13985-584-10)
- Propidium iodide solution (Sigma-Aldrich, catalog number: P4864 )
- Dulbecco’s phosphate buffer saline (DPBS) 1x, w/o calcium/magnesium (Thermo Fisher Scientific, catalog number: 14190094 )
- AlarmarBlue® Cell Viability Reagent–Stock solution 10x (Thermo Fisher Scientific, InvitrogenTM, catalog number: DAL1025 )
- Anti-Glial Fibrillary Acidic Protein (GFAP) rabbit polyclonal (Agilent Technologies, Dako, catalog number: Z0334 ) used at 1/700 in BPS
- Anti-NeuN rabbit polyclonal (EMD Millipore, catalog number: ABN78 ) used at 1/500 in BPS
- Anti-calbindin D-28 K rabbit polyclonal (Swant, catalog number: CB38 ) used at 1/700 in BPS
- Anti-Iba1 (Wako Pure Chemical Industries, catalog number: 019-19741 ) used at 1/250 in BPS
- Anti-olig2 (Oligodendrocyte Lineage Transcription Factor 2) (R&D Systems, catalog number: AF2418 ) used at 1/200 in BPS
- Anti-rabbit IgG Fab2 Alexa Fluor® 488 (Cell Signaling Technology, catalog number: 4412S ) used at 1/750 in BPS
- Anti-goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Thermo Fisher Scientific, catalog number: A-11055 ) used at 1/750 in BPS
- Anti-rabbit IgG Fab2 Alexa Fluor® 555 (Cell Signaling Technology, catalog number: 4413S ) used at 1/750 in BPS
- Fluoprep (BioMérieux, catalog number: 75521 )
- Opti-MEM reduced serum medium (Thermo Fisher Scientific, GibcoTM, catalog number: 31985062 )
- RNA extraction kit NucleoSpin® RNA (MACHEREY-NAGEL, catalog number: 740955.250 )
- RNase Away®, 475 ml (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 7002 )
- iScriptTM cDNA Synthesis Kit (Bio-Rad Laboratories, catalog number: 170-8891 )
- Platinum® SYBR® Green qPCR SuperMix-UDG w/ROX (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11744500 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266-1KG )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: S8045-1KG )
- Hydrochloric acid solution (HCl), 1.0 N, BioReagent, suitable for cell culture (Sigma-Aldrich, catalog number: H9892-100ML )
- Recombinant human insulin (Sigma-Aldrich, catalog number: 91077C-100MG )
- Minimum Essential Media (MEM), HEPES, GlutaMAXTM Supplement, 500 ml (Thermo Fisher Scientific, GibcoTM, catalog number: 42360081 )
- Heat-inactivated horse serum, 100 ml (Thermo Fisher Scientific, GibcoTM, catalog number: 26050070 )
- D-glucose cell culture grade 5 g/L (Sigma-Aldrich, catalog number: G7528 )
- Kynurenic acid (Sigma-Aldrich, catalog number: K3375-5G )
- HEPES 1 M, 100 ml (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080 )
- Hibernate®-A medium (Thermo Fisher Scientific, GibcoTM, catalog number: A1247501 )
- Crystalline PFA (Sigma-Aldrich, catalog number: P6148 )
- Fetal bovine serum (FBS), 500 ml (Eurobio, catalog number: CVFSVF0001 )
- TritonTM X-100 (Sigma-Aldrich, catalog number: T8787 )
- 2-Mercaptoethanol (Sigma-Aldrich, catalog number: M6250 )
- 1 M MgCl2 solution (see Recipes)
- 0.1 N NaOH solution (see Recipes)
- Human insulin 50 mg/ml (see Recipes)
- Organotypic brain culture medium (see Recipes)
- 10x kynurenic acid solution (see Recipes)
- Dissection medium (see Recipes)
- 8% paraformaldehyde (PFA) (see Recipes)
- 4% paraformaldehyde (PFA) (see Recipes)
- Blocking and permeabilization solution (BPS) (see Recipes)
Equipment
- Straight tweezers, type N°5 length 11 cm for removing the skin and the skull (Dominique Dutscher, catalog number: 005092 )
- Ice container
- 5% CO2 incubator maintained at 37 °C with humidified atmosphere (Thermo Fisher Scientific, Thermo ScientificTM, model: Series 8000 Water-Jacketed , catalog number: 3423)
- Biosafety cabinets
Note: Work in a horizontal flow hood is recommended for the slices preparation and BSL2 vertical flow hood is recommended for the viral infection step with BSL2 pathogens and infection follow-up. However, if the protection glass on the BSL2 cabinet can be maintained up, the slices can be prepared the same way. Based on our experience and even if the sterility is not well preserved under these conditions, the contamination rate remains very low. In any case, the biosafety level has to be adapted depending on the virus considered (BSL2, BSL3 or BSL4) for the viral infection and follow up steps. - Stainless steel dissecting scissors length 11 cm (Dominique Dutscher, catalog number: 005064 )
- Beaker, 250 ml
- McIlwain tissue chopper (Campden Instruments, model: TC752 )
- Pipette bulb (Fisher Scientific, catalog number: 03-448-29 )
- Stainless steel dissecting scissors ultra-fine length 12 cm for cutting and removing the skull (Dominique Dutscher, catalog number: 005068 )
- Dumont tweezers #5 for the dissection of the brain and the meninges removal, 0.1 x 0.06, Dumoxel (World Precision Instruments, catalog number: 14098 )
- Stainless steel forceps rounded ends length 130 mm for holding the brain during the dissection procedure and the slices separation (Dominique Dutscher, catalog number: 442256 )
- Lanceolate tip spatula for the midbrain removal (imLab, catalog number: NE010 )
- Curved tweezers type N°7 length 11 cm for the midbrain removal and harvesting the slices from the culture insert (Dominique Dutcher, catalog number: 005093 )
- P1000 Pipetman (Gilson, catalog number: F123602 )
- P20 Pipetman (Gilson, catalog number: F123600 )
- KOLLE needle holder for the slices separation (Hamilton Bell, catalog number: 6780 )
- Water bath
- Widefield fluorescence microscope (ZEISS, model: Axioplan 2 ) with a cooled monochrome camera (Photometrics, model: CoolSNAP HQ2 ) and a fluorescence filter set for propidium iodide (for example: excitation 550-580 nm, emission 600-660 nm)
- Tecan Infinite® 200 PRO series plate reader (Tecan, model: M Plex )
- Confocal spectral microscope (Leica, model: Leica TCS SP5 )
- TPersonal 48 Thermal Cycler (Analytik Jena, Biometra, model: T-Personal 48 , catalog number: 846-050-551)
- StepOnePlusTM Real-Time PCR System (Thermo Fisher Scientific, Applied BiosystemsTM, model: StepOnePlusTM, catalog number: 4376600 )
- Stereomicroscope for dissection (Leica, model: Leica LED2000 )
- Fume hood
- -20 °C freezer
- -80 °C freezer
Software
- ImageJ (https://imagej.nih.gov/ij/ [Schneider et al., 2012]) with the plugin ‘Auto Local Threshold’ from Gabriel Landini (http://fiji.sc/Auto_Local_Threshold#Installation)
Note: Fiji (http://fiji.sc/ [Schindelin et al., 2012]) could be used instead of ImageJ because it bundles the required plugin. Download the Macro file ‘OBC_IP_mortality.ijm’ for the analysis of the propidium iodide staining. Save the file in the ImageJ/plugins folder. ‘OBC IP mortality’ should appear in the Plugins menu. - GraphPad Prism (GraphPad software–https://www.graphpad.com/scientific-software/prism/) and/or R software (https://www.r-project.org/)
- StepOnePlus software (https://www.thermofisher.com/us/en/home/technical-resources/software-downloads/StepOne-and-StepOnePlus-Real-Time-PCR-System.html)
Procedure
- Culture plate preparation
- The day before slicing the brain explants, prepare 6-well culture plates by adding 1 ml of organotypic culture medium (see Recipes) into each well and place the culture inserts on the top using straight tweezers in order to reverse the hydrophobicity of PTFE and to allow a good feeding of the slices (Table 1).
Table 1. Time duration recommended for each step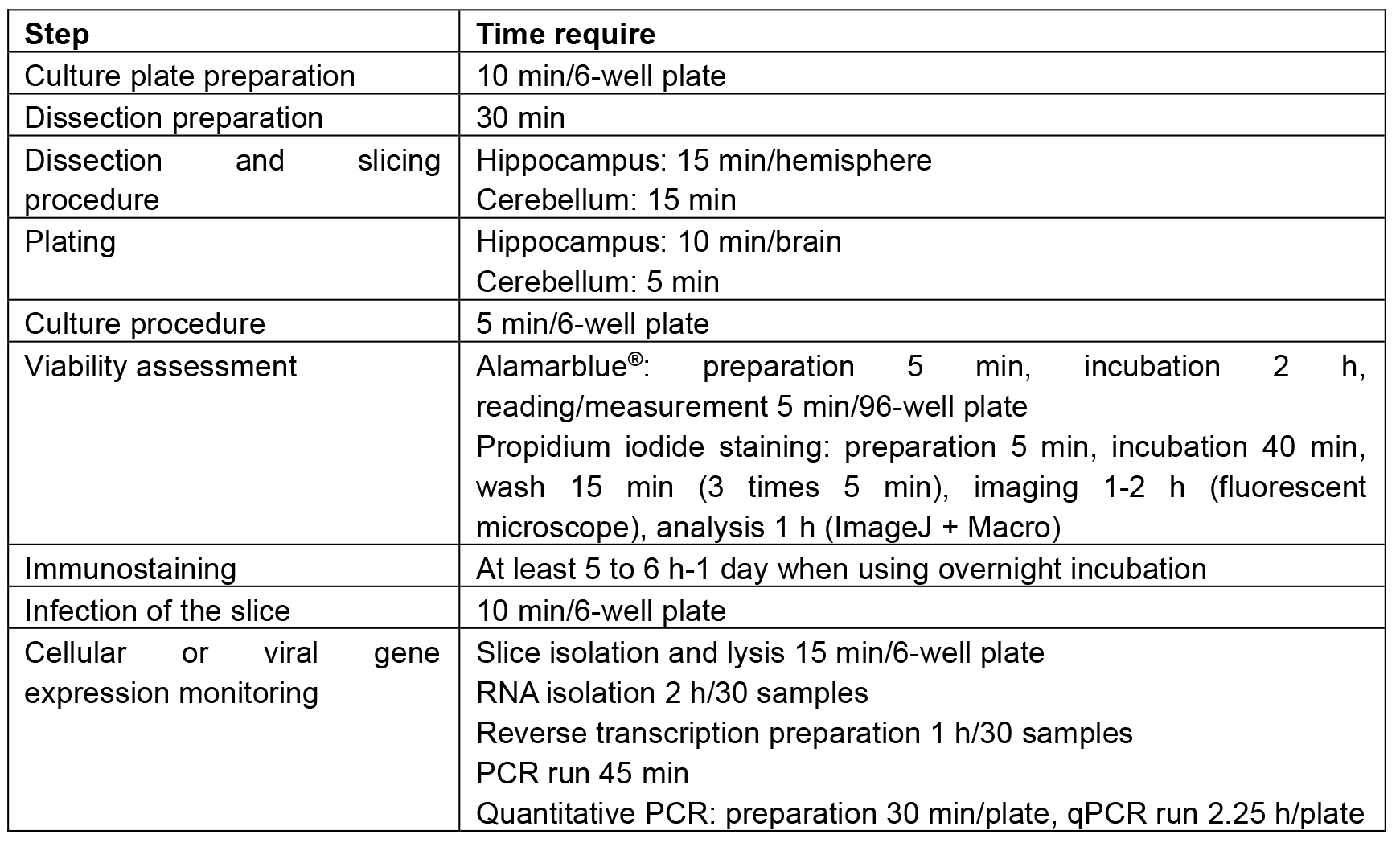
- Incubate the plate overnight at 37 °C in a humidified 5% CO2 atmosphere.
- The day before slicing the brain explants, prepare 6-well culture plates by adding 1 ml of organotypic culture medium (see Recipes) into each well and place the culture inserts on the top using straight tweezers in order to reverse the hydrophobicity of PTFE and to allow a good feeding of the slices (Table 1).
- Dissection preparation
- Decontaminate the walls and floor of the biosafety cabinet using 70% ethanol in water and UV irradiate for 15 min if available.
- Before the dissection procedure, sterilize all equipment, i.e., scissors, tweezers, PTFE plate by autoclaving and put them under the biosafety cabinet. Prepare a beaker half-filled with 70% ethanol in water in order to decontaminate the dissection material during the procedure (Table 1).
- Clean the tissue chopper, the razor blade and the stereomicroscope with 70% ethanol, ensure that the slicing platform is correctly decontaminated and then let everything dry. Mount the razor blade on the tissue chopper using the screw and nut and place the instruments under the biosafety cabinet and spray again with ethanol (Table 1).
- Place a sterilized container filled with ice under the biosafety cabinet.
- Prepare the dissection medium (see Recipes) as described in the ‘Recipes’ section using a sterile 50 ml conical tube. Put 6 ml of medium in the bottom of the 35 mm Petri dishes (1 Petri dish for the hippocampus, 1 Petri dish for the cerebellum) and keep the cover for the dissection. The dissection medium must be kept on ice and the entire dissection procedure must be performed at 4 °C (cold medium) to limit tissue damage and cell degeneration.
- Fill a Petri dish cover with 5 ml of dissection medium in order to have cold fresh medium throughout the slicing procedure.
- Remove the conical extremity of a 5 ml plastic pipette with scalpel blade and insert the top part with the carded cotton into the pipette bulb (defined as 5 ml truncated pipette).
- Decontaminate the walls and floor of the biosafety cabinet using 70% ethanol in water and UV irradiate for 15 min if available.
- Dissection and slicing procedure
Notes:- Refer to the Table 1 for the duration of each step.
- Table 2 summarizes the troubleshooting and the proposed solution.
- Gently spray the neck and the head of the animal with 70% ethanol and proceed rapidly to the following step.
- We recommend using 6 to 10 days-old animals. Since cervical dislocation is not suitable for suckling animals, proceed with decapitation using scissors. This action must be performed by skilled accredited personnel. At this stage, it is necessary to work as quickly as possible.
Notes:- When animals give rise to a small number of babies, the suckling animals could be much bigger and the decapitation more difficult. In order to prevent potential pain and allow a more comfortable procedure for the experimenter, an anesthetic can be used. We recommend the use of ketamine at a dose of 150 mg/kg, which is an efficient activation-dependent channel blocker of N-methyl-d-aspartate (NMDA) receptors leading to a reduction of the excitotoxicity and prevent neuronal death. In combination with the kynurenic acid contained in the dissection medium, ketamine provides good neuroprotection against excitotoxicity.
- Brains from younger animals (between P3 to P6) are more difficult to cut into regular parallel slices because they are softer. In addition, numerous non-fully differentiated cells endowed with migration properties may alter the reproducibility of infection experiments. Brains from older animals (beyond P10 to P12) are poorly suitable because of increased cell death that can add bias to the analyses.
- This procedure has to follow the local animal ethics protocols and has to mention the species and strains used. Any change in the species has to be amended and validated on the ethics protocol prior to any experimentation.
- When animals give rise to a small number of babies, the suckling animals could be much bigger and the decapitation more difficult. In order to prevent potential pain and allow a more comfortable procedure for the experimenter, an anesthetic can be used. We recommend the use of ketamine at a dose of 150 mg/kg, which is an efficient activation-dependent channel blocker of N-methyl-d-aspartate (NMDA) receptors leading to a reduction of the excitotoxicity and prevent neuronal death. In combination with the kynurenic acid contained in the dissection medium, ketamine provides good neuroprotection against excitotoxicity.
- Hold the head between the thumb and index, and remove as much skin and flesh as possible using ultra-fine scissors.
- Cut the skull in a rostral way from the cavity where the top part of the spinal cord is visible to the cavity containing the olfactory bulbs using the tip of ultra-fine scissors.
- Remove the brain from the head by inserting the tip of the Dumont tweezers under the olfactory bulbs and lift gently until detachment from the cranium.
- Put the brain directly in cold dissecting medium in the 35-mm dish. At this stage, it is necessary to isolate and dissect the brain structure in dissection medium to maintain cell viability.
- Separate the cerebellum (slicing process–from steps C16 to C20) from the brain using a scalpel blade (Figure 1A) and conserve it in cold dissection medium during the preparation of the hippocampal slices as follows (dissecting and slicing process–from steps C8 to C15).
- Separate the two hemispheres along the midline (Figure 1B) and remove the midbrains (Figure 1C), holding the half brain with forceps and using the lanceolate tip spatula or alternatively a curved tweezers (with an appropriate training approximately 1 min is needed per brain). At this time, the hippocampus is visible (arrow head–Figure 1D).
- With a scalpel blade cut and remove the rostral part containing the olfactory bulbs (Figure 1D–about 10 sec per hemisphere).
- Remove the meninges using the Dumont tweezers #5. The hippocampus and cortex are ready for slicing process.
- Put the hippocampus face down in rostral orientation on Whatman paper (Figure 1E–about 1 min needed).
- Place it onto the slicing platform of the McIlwain tissue chopper (see Figure 1–arrow 2–about 1 min needed).
- With the 5 ml truncated pipette add one drop of dissection medium to prevent the tissue from sticking to the razor blade.
- Proceed rapidly to the slicing by turning on the apparatus (Figure 1E–30 sec required to slice two hippocampi).
Note: All slicing parameters, i.e., thickness (typically between 200 and 500 µm), blade force (usually maximum force) and blade speed (typically one slice per second) should be established in preliminary tests. - Remove the Whatman paper from the slicing platform and abundantly add cold dissection media on the tissue.
- With the lanceolate tip spatula, remove the tissue from the Whatman paper by gently sliding the tip of the spatula between the tissue and the paper.
- Place the tissue back in Petri dish containing cold dissection medium during the dissection of the cerebellum.
- In cold dissection medium and under the stereomicroscope, hold the cerebellum using forceps and gently remove meninges using Dumont tweezers #5, the inferior colliculus and brain stem (Figure 1B’).
- Place the cerebellum on the PTFE plate following a sagittal plan compared to the razor blade (Figure 1C’).
Note: The rostro-caudal plane for the slicing process is not critical and the cerebellum can be sliced with the rostral or the caudal part facing the experimenter when placed on the slicing platform. However, in our experiment the caudal part of the cerebellum, as shown in Figure 1A and 1B’, was always facing the experimenter during the slicing procedure. - With a P1000 pipette remove the entire medium in order to improve the cerebellum adhesion to the PTFE plate.
- Proceed to the slicing as described for the hippocampus (Figure 1D’).
Table 2. Troubleshooting table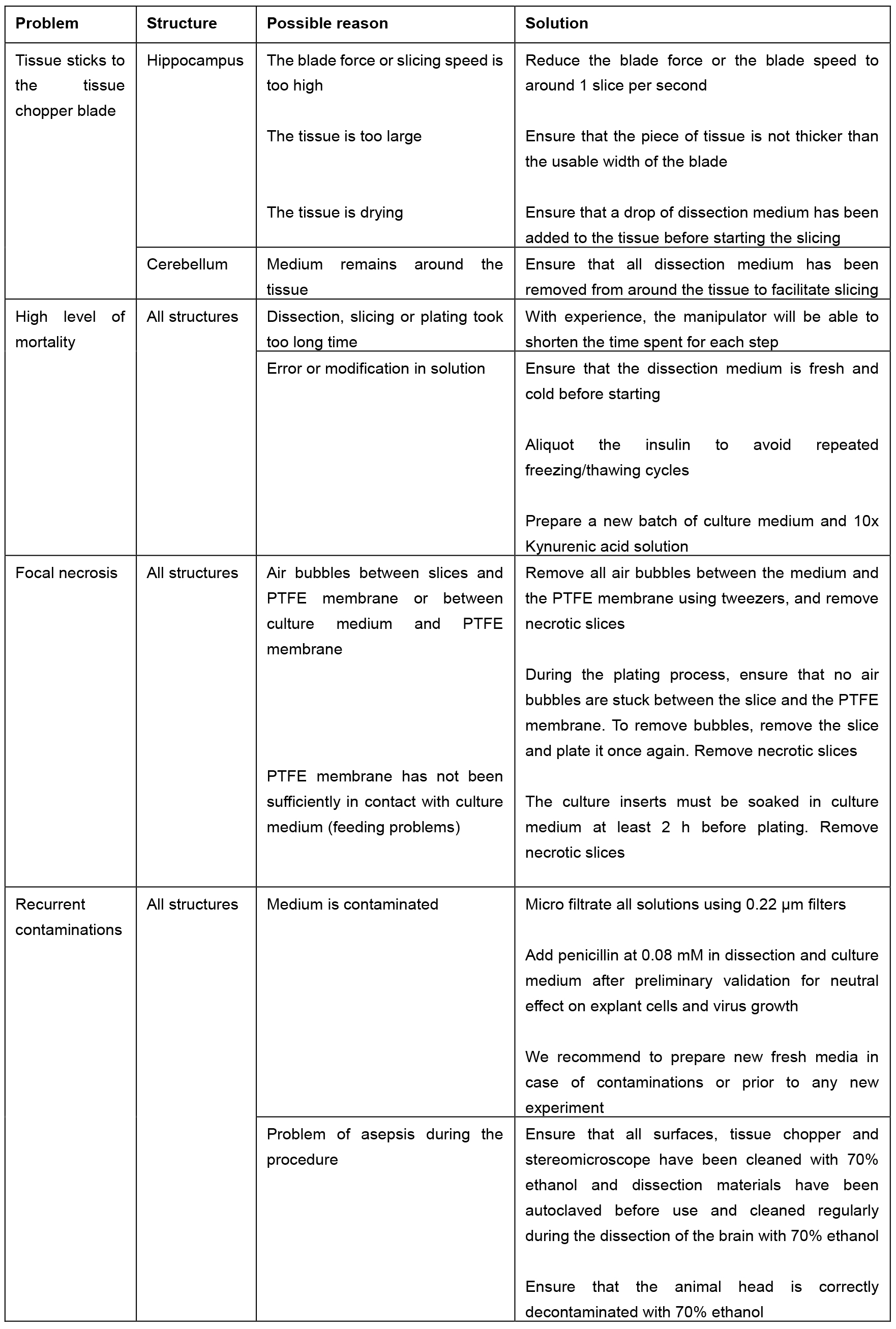
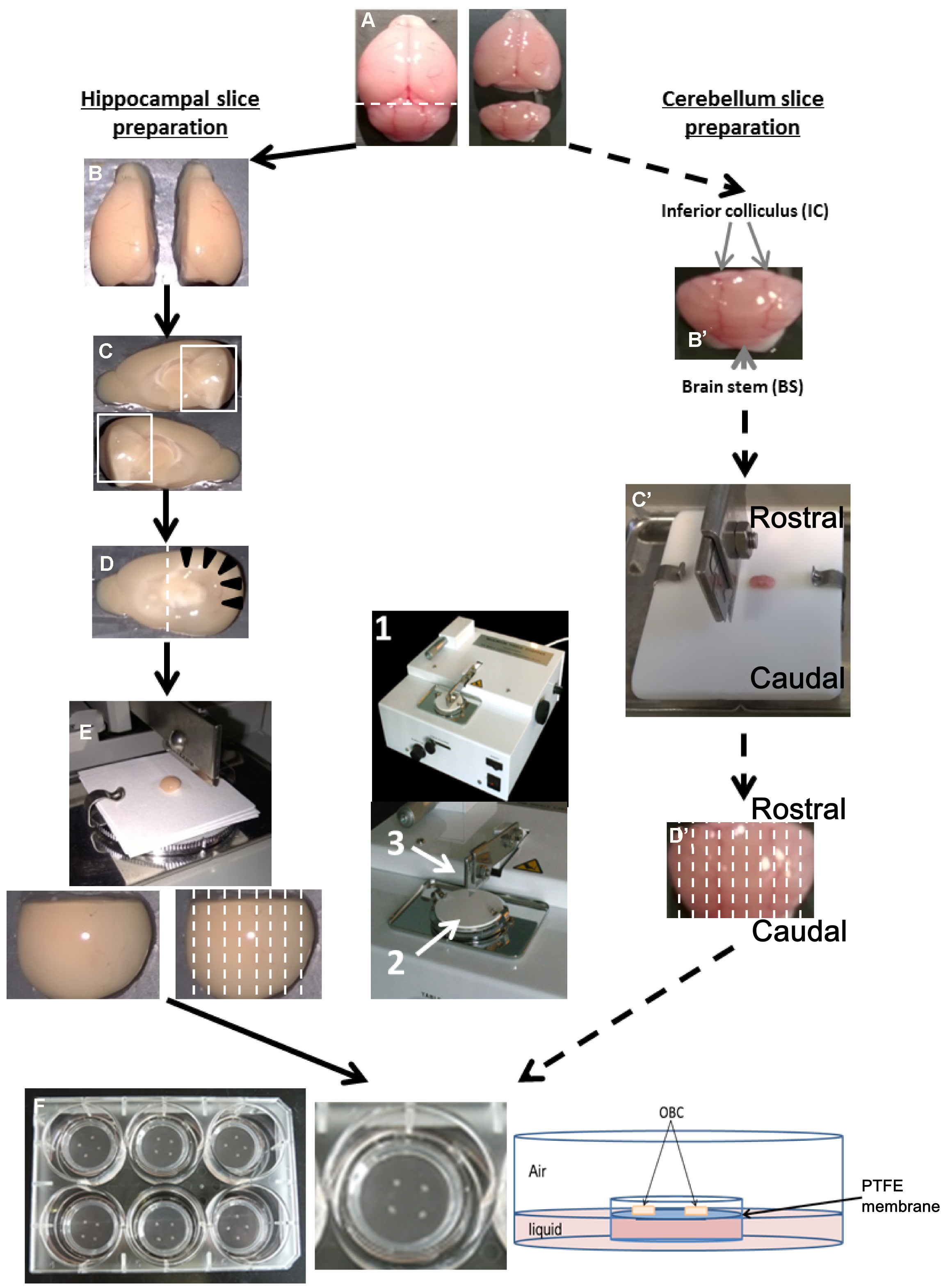
Figure 1. Preparation of OBC from suckling rodent brain between P6 to P10. Hippocampal slice preparation (A-F, left panel). A. Collect the brain after decapitation and separate the cerebellum with a scalpel blade following the dotted line. B. Separate the two hemispheres using a scalpel blade. C. Remove the midbrain to expose the hippocampus area. D. Remove the rostral part of the brain (containing the olfactory bulbs), in order to visualize the hippocampus (black arrow head). E. Put and orientate the dissected brain in the antero-posterior axis onto the slicing platform (2) of a tissue chopper (1) with perpendicular cut plan to the razor blade (3) and cut following dotted line. F. Dissociate and transfer slices to millicell insert culture systems relying on the OBC medium. Cerebellum slice preparation (A, B’-D’, F, right panel). A. Separate the cerebellum from the rest of the brain. B’. Remove the inferior colliculi (IC) and the brain stem (BS) from the cerebellum. C’. Put cerebellum on a Teflon plate according to the antero-posterior axis, and transfer to the slicing platform of the tissue chopper. D’. Cut sagittally following the dotted line. F. Transfer in a Petri dish containing dissection medium, dissociate carefully the slices from each other and plate them on the insert culture systems. - Carefully transfer the tissue into a Petri dish containing cold dissection medium.
- Under the stereomicroscope, separate carefully the hippocampal and/or cerebellum slices using forceps to hold the tissue and a needle mounted on KOLLE needle holder in cold dissection medium. With a good focus, mainly on the top of the tissue, the spaces between slices are easy to visualize which helps to precisely separate them (the slices separation takes 10 to 15 min for the hippocampus and only 5 min for the cerebellum).
Note: Ensure the light source of the stereomicroscope is cold (i.e., optic fiber) to avoid cell-stress induced by overwarming of the medium during the slice dissociation. - Select slices for the culture under the stereomicroscope.
Note: The hippocampus should contain the CA (Cornu Ammonis) region and the dentate gyrus. The cerebellum should contain the deep nucleus and the different cellular layer such as the Purkinje Cell (PC) layer or the molecular layer (ML) should be visible. Refer to a brain Atlas for more details such as the Allen Brain Atlas. - Plate a maximum of 4 hippocampal or 5 cerebellum slices per Millipore cell culture insert membrane using the 5 ml truncated pipette mounted on a pipette bulb. To favor the appropriate feeding and oxygenation of slices, remove as much dissection medium as possible around each brain explant after plating on the insert and incubate the slices overnight at 37 °C, 5% CO2 in a humidified atmosphere.
- Refer to the Table 1 for the duration of each step.
- Culture procedure
Notes:- Change the medium the day after the slicing procedure, and then every 2-3 days.
- Refer to the Table 1 for the duration of each step.
- Hold the edge of the insert from the 6-well plate with tweezers and remove old medium with a P1000 Pipetman.
- Put the insert back in place in the 6-well plate and add 1 ml of fresh culture medium pre-warmed at 37 °C in the water bath to avoid any thermal shock.
- Make sure that no air bubble is trapped between the PTFE membrane and the medium. If so, bend the 6-well plate at an angle of ~45° and gently shake it to allow the air bubbles to reach the edge of the culture insert.
- Change the medium the day after the slicing procedure, and then every 2-3 days.
- Viability of cultured brain explants: Cellular metabolism/mortality assessment–Alamarblue® and propidium iodide staining
Notes:- Prior to infection studies, it is extremely important to ensure that the brain explant is healthy ex vivo and determine the best time window during which the slices can be used.
- Based on our experience, the viability evolution is reproducible if the OBC preparation is kept identical as described in this protocol and can be assessed once for all using three independent batches of slices, for a total of 15 slices analysis per time point. However, we recommend periodically assessing the viability, at least with one of the two proposed approach, in order to make sure that the procedure is still correctly set up.
- Detach slices from PTFE membrane by flushing culture medium gently on the edge of the explant.
- Gently transfer the tissue into a 96-well plate using a truncated 5 ml pipette.
Note: After lengthy culture time (4 to 7 days), slices might be too adhesive to be detached without damages. The PTFE membrane of the inserts is suitable for fluorescence microscopy and does not affect the observation of the IP staining, the reading of the Alamarblue assay and the confocal analysis (no autofluoresence), hence we recommend using this type of insert to support the OBC slices.
Propidium iodide staining
- In a 96-well plate, immerse slices in 200 µl of propidium iodide solution at 5 µg/ml in culture medium and incubate the plate for 45 min at 37 °C, 5% CO2 in a humidified atmosphere.
Note: Propidium iodide enters into dying cells leading to a red-fluorescent nuclear staining. The surface density of propidium iodide labeled nuclei over the total tissue surface is used to monitor cell death and provides necessary quantification for statistical analysis (see Data analysis section). Note that there is no need to wash the slices prior to the staining. - Remove the propidium iodide solution and wash slices 3 times with 200 µl of 1x DPBS (37 °C).
- Transfer slices into a 12-well plate and fix them with 1 ml of 4% paraformaldehyde solution (see Recipes) for 30 min at room temperature.
- Mount gently slices between a slide and coverslip. Make sure to not crush the tissues, especially on thick slice (300 and 500 µm).
- Acquire images with a widefield fluorescence microscope, at least 6 randomized fields per slice and 5 slices per time point. Use a 5x objective, to acquire large fields of view.
- Analyze the images in ImageJ with the provided Macro (i.e., script for ImageJ). Run the Macro (click on Plugins>‘OBC IP mortality’) and follow the instructions: first, choose the image to analyze. It will be opened in ImageJ with an enhanced display (see Figure 2). Click on the areas outside the slice (like the region outlined in yellow in Figure 2). If the region is too small or too large, the tolerance of the wand tool can be adjusted: double-click on the wand tool in the ImageJ menu (in the red frame in Figure 2), and adjust the value of the tolerance (which is 200 by default). When the region is correctly outlined, press the T key to save it in the ROI manager. Repeat for all the regions in the image. At the end, the windows should look like Figure 3.
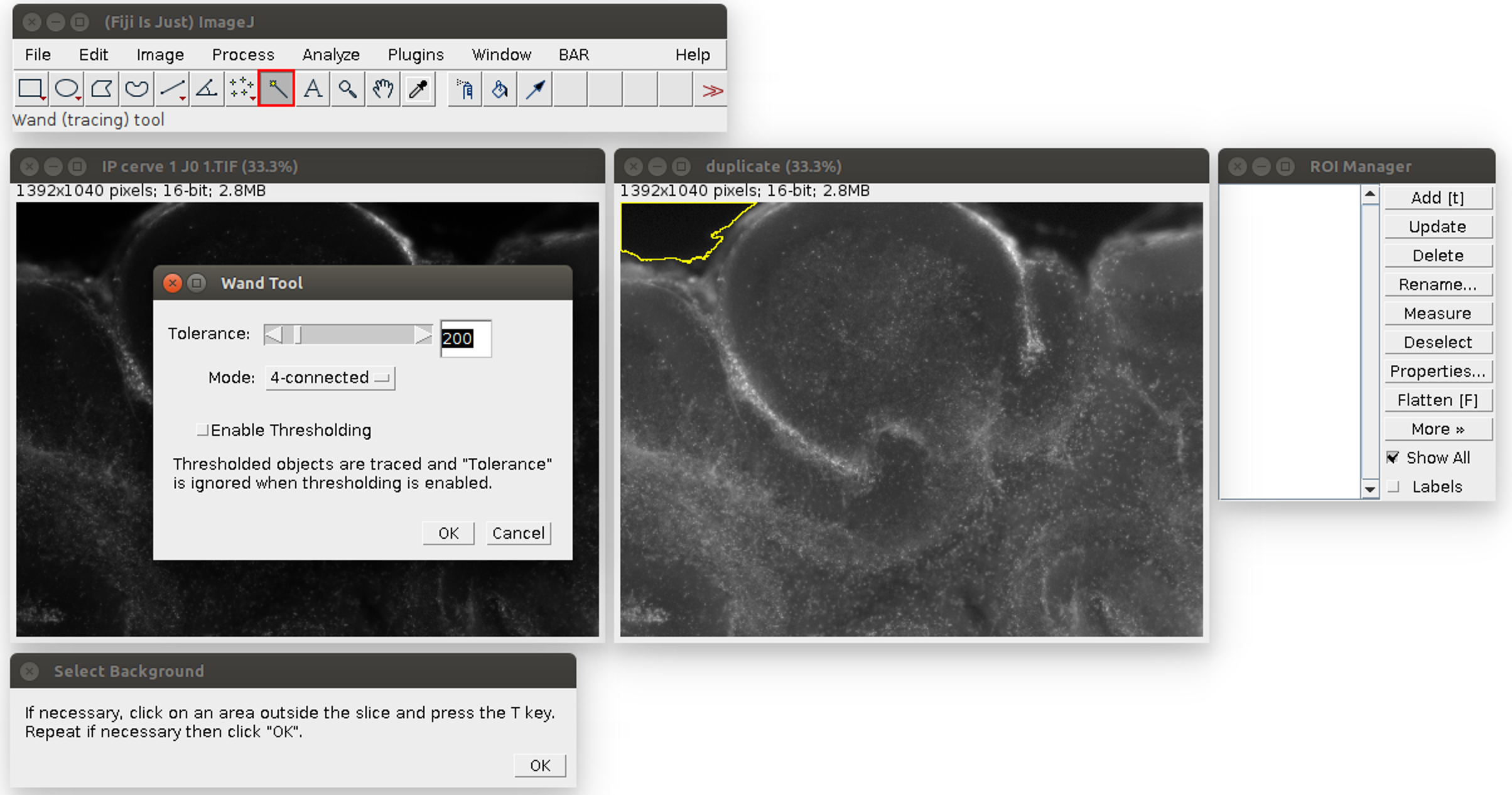
Figure 2. Analysis of the propidium iodide staining in ImageJ with the Macro. The windows can be arranged at the user’s convenience. The original image is on the left, its duplicate with an enhanced display on the right. The instructions are at the bottom. The tolerance of the wand tool can be adjusted by double-clicking in the red square.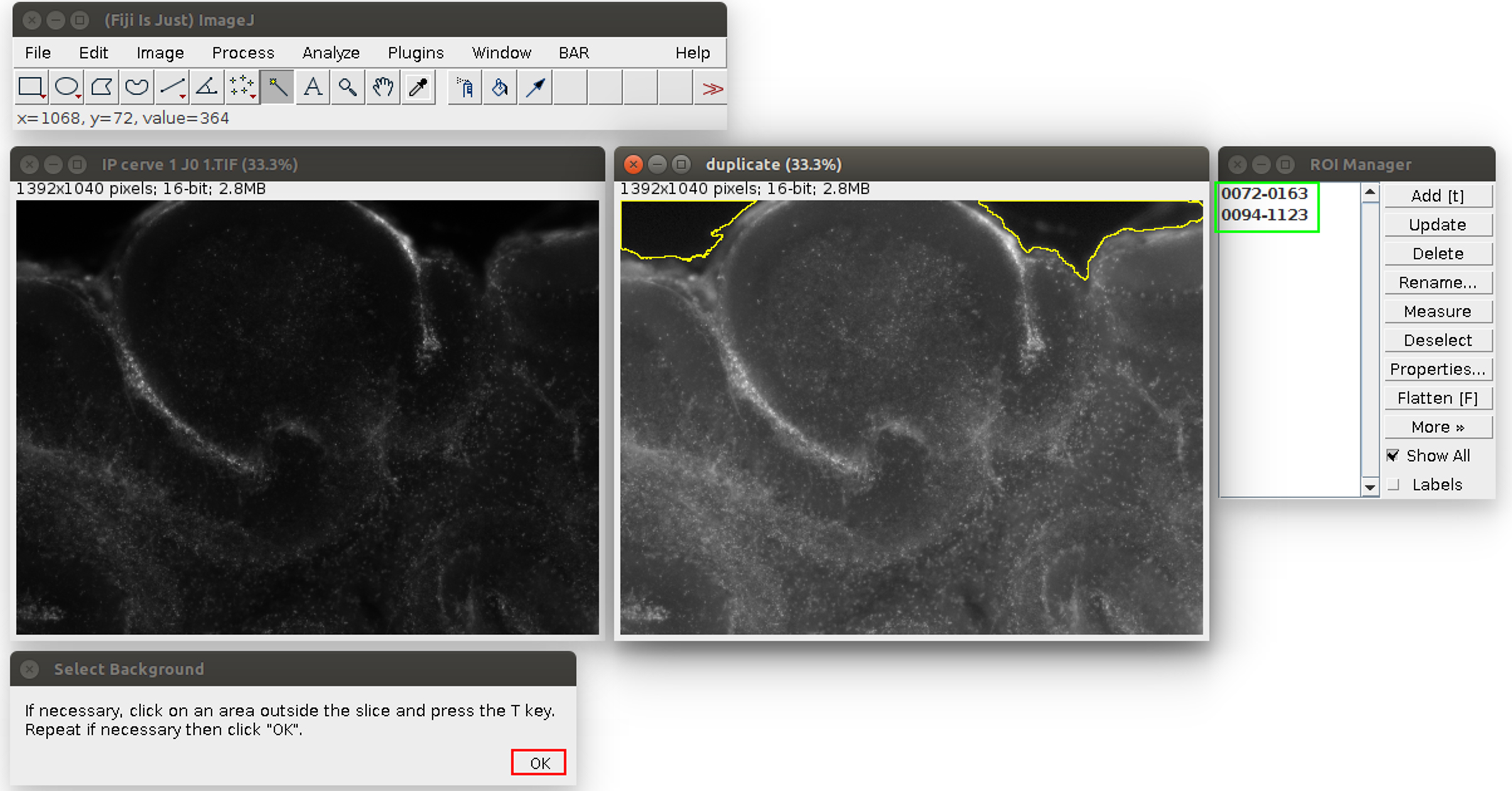
Figure 3. Second step of the propidium iodide staining analysis. The regions are saved in the ROI manager on the right, in the green frame. The user can click on ‘OK’ in the red frame. - Click on ‘OK’ (see the red square in Figure 3). The Macro will do an automatic segmentation of the propidium iodide staining (with the ‘Auto Local Threshold’ plugin https://imagej.net/ Auto_Local_Threshold#Bernsen), and measure the area of this staining, and the area of the slice.
- When the process is finished, the propidium iodide area is displayed as a red overlay on the image and the slice area is surrounded in yellow to visually check the results of the segmentation (see Figure 4). The red overlay can be activated/deactivated by checking/unchecking the Channel 1 box in the ‘Channels’ window (see Figure 4). The numerical results (the propidium iodide area ‘Area’ and the whole slice area ‘Slice area’ in pixels, and their ratio in %) are displayed in a spread sheet which can be saved (for more details see Data analysis section point 3). Several images can be analyzed in batch and the results saved in the same file: close only the previous image and run again the Macro in the same way from step E8. The results will be added at the bottom of the spread sheet.
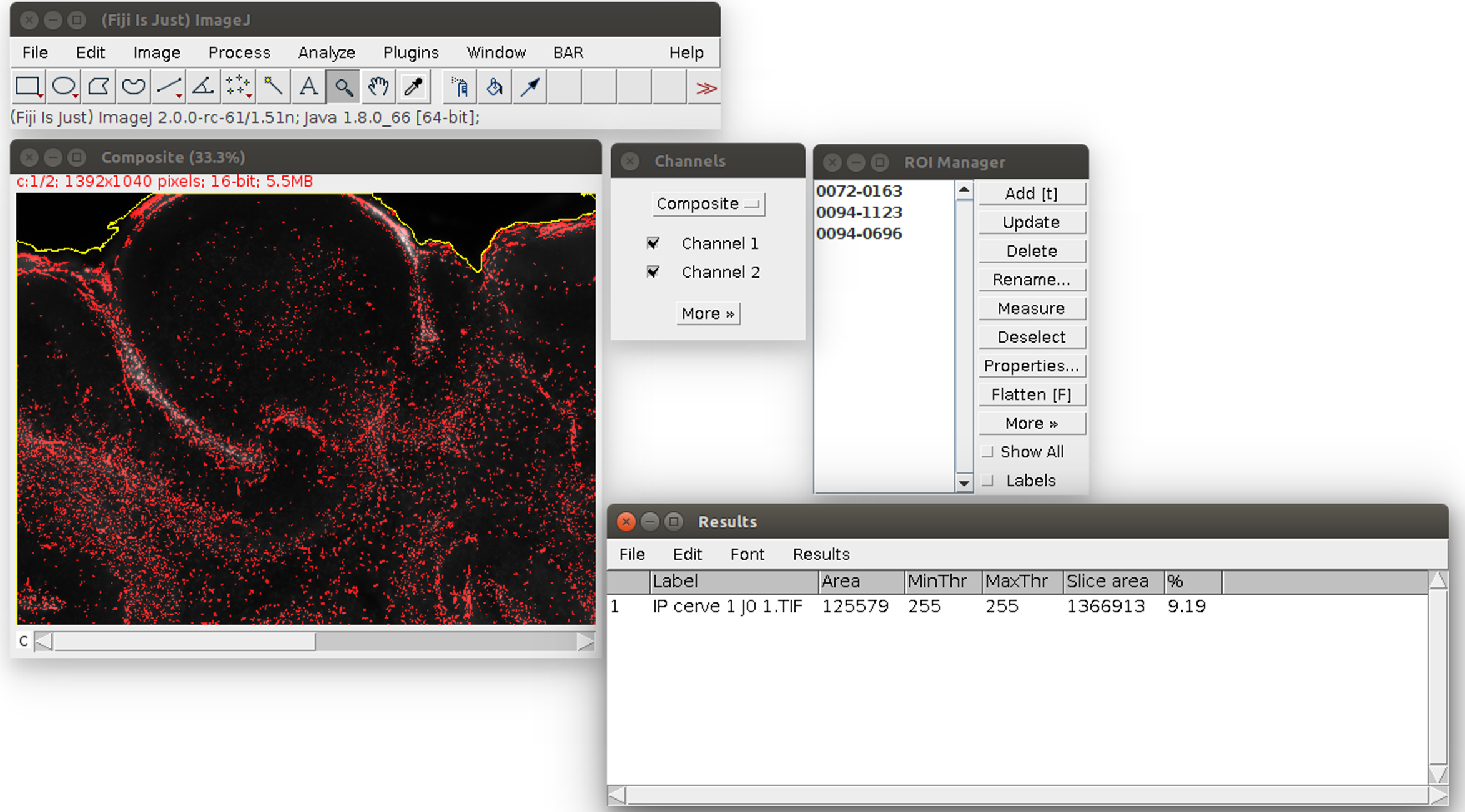
Figure 4. Result of the propidium iodide staining analysis. The area of the staining is displayed in red. The area of the slice is outlined in yellow. On the bottom right, the results table can be saved with its menu File>Save As.
Alamarblue®
- In a 96-well white plate, immerse slices in 200 µl of 1x Alamarblue® solution in culture medium.
- Incubate the plate for 2 h at 37 °C, 5% CO2 in a humidified atmosphere.
Note: The Alamarblue® reagent becomes red proportionally to the cellular metabolism of the slices reflecting the proportion of active cells and thus the global OBC health state. - Read the fluorescence emission at 580-610 nm according to the manufacturer’s protocol using a Tecan Infinite® 200 PRO series microplate reader.
- To analyze metabolic activity, only the animal and slice (random) factors are evaluated. We applied the Kruskal-Wallis tests since the data showed violation of the assumptions of ANOVA.
- Variation of mortality (Figures 5A and 5C) and metabolic activity (Figures 5B and 5D) across times (D0, D1, and D7) were analyzed using the Kruskal-Wallis test. For each day, the coefficients of variation (the ratio of standard deviation and mean) were compared between mice and hamsters using the Wilcoxon test (for more details see Data analysis section point 3) (Figure 6).
Note: Based on our experience, we would not recommend working with OBC when one of these two parameters exceeds or varies more than 20% during the first two days since it may greatly impact the maintenance of the cell population in the slices and their chemokine response and thus the reproducibility of the results.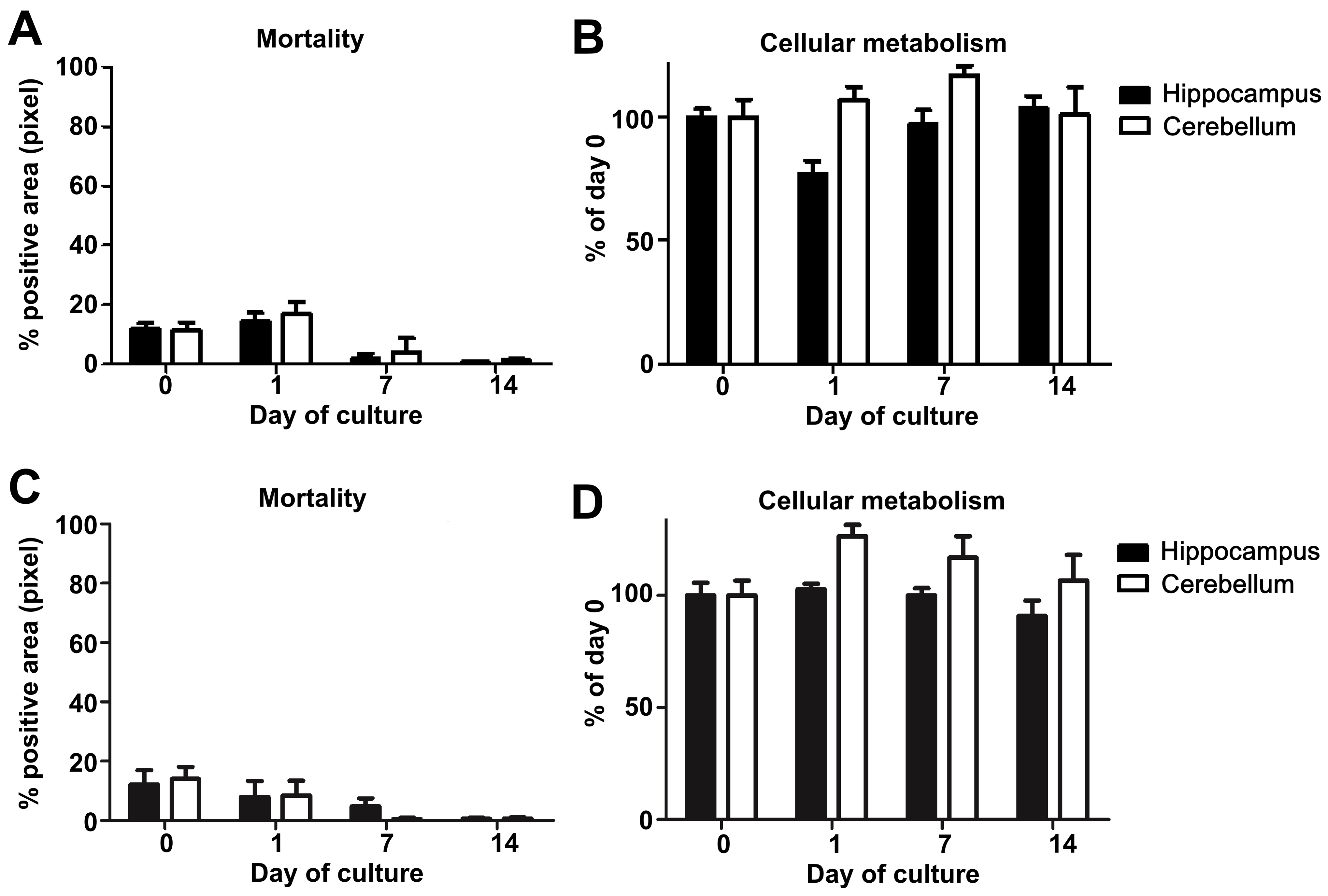
Figure 5. Study of mouse and hamster OBC viability over 14 days of culture. A. Mouse or C. hamster (3 animals/group, at least 4 slices of each substructure/animal) hippocampus and cerebellum slice mortality were evaluated by propidium iodide staining and analyzed with an Axioplan Imager fluorescence microscope. B. Mouse or D. hamster (n = 3), at least 3 slices of each substructure/animal) hippocampus and cerebellum slice metabolism activity were assessed by incubation with Alamarblue reagent kit and analyzed using an Infinite 200 PRO Tecan microplate reader. The graphics show the evolution of cellular metabolism (reflecting health) over the time of the culture.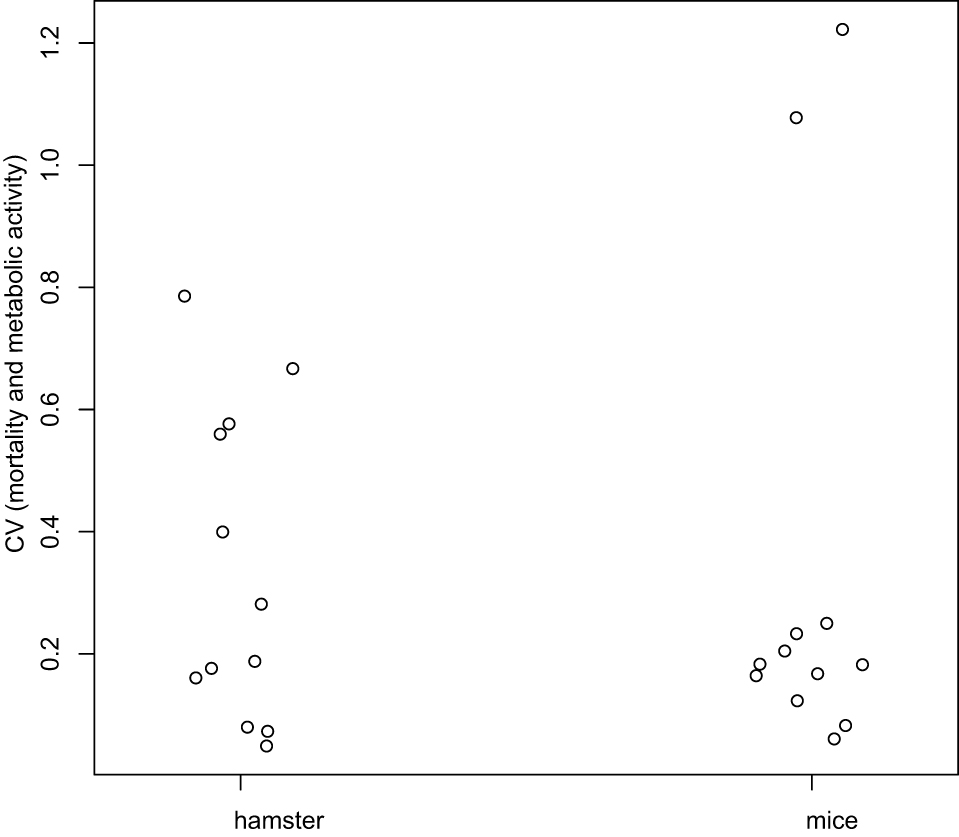
Figure 6. Coefficients of variation analysis. Coefficients of Variation (CV) for mortality and metabolic activity were computed at each day, and globally compared between hamster and mice using the Wilcoxon test. There is no difference statistically significant between mice and hamster (P-value = 0.7987).
- Prior to infection studies, it is extremely important to ensure that the brain explant is healthy ex vivo and determine the best time window during which the slices can be used.
- Immunostaining of brain explants
Note: As preliminary test, we recommend to assess the maintenance of the major cell population (Figure 7) as well as the structure preservation during the culture (Figure 8) by immunostaining before starting infection studies to exclude any variation related to the experimenter.- Detach, place the slices in 12-well plates using the 5 ml truncated pipette and fix them with PFA at room temperature as described above.
Note: In the case of infected slices, the PFA fixation can be performed directly on the culture insert. The culture medium has to be removed, and 1 ml of PFA is added below and above the culture insert in order to completely cover the slices. - Proceed to the blocking and permeabilization of tissue with 1 ml of the blocking and permeabilization solution (BPS) (see Recipes) for at least 45 min at room temperature under gentle agitation.
- Remove the BPS and add the primary antibody (Ab) (i.e., astrocytic marker GFAP, neuronal marker NeuN or CB-28K–microglia marker Iba1 or oligodendrocyte marker Olig-2–see Figure 7) diluted in BPS and incubate for 2 h at room temperature (also possible ON at 4 °C).
Notes:- Ensure that the Ab can bind the epitope in the presence of Triton X-100 present in the BPS. If not, remove the Triton X-100 at this step and wash slices 3 times in a solution of 1x DPBS/4% FBS.
- By using a 48-well plate, the volume of the staining solution can be as minimal as 150 µl.
- For example, to highlight the hippocampal and the cerebellum structures, use respectively the neuronal marker NeuN (Figure 8A) or the Purkinje cells marker CB-28K (Figure 8B). The CB-28K decreased staining is mostly due to the Purkinje cells loss and the reorganization of the neurons into the slice. However, the apparent lower staining of the hamster CB-28K at day 7, is due to the acquisition of this image. Indeed, all the left panels of this figure are a tile reconstitution coupled with 100 µm z. Due to the irregular shape of the tissue and the mounting process, it can be difficult to keep a homogenous staining even if the exposure time is identical, since the density of the cells throughout the slice can be different.
- Ensure that the Ab can bind the epitope in the presence of Triton X-100 present in the BPS. If not, remove the Triton X-100 at this step and wash slices 3 times in a solution of 1x DPBS/4% FBS.
- Remove the Ab and wash 3 times each for 10 min with 1x DPBS.
- Dilute the secondary Ab in BPS and incubate slices for at least 2 h at room temperature.
- Repeat the wash step.
- Mount the slices between slide and coverslip using fluoprep mounting medium.
Note: The slices are still quite thick at this step; thus the immunostainings imaging have to be performed using confocal microscopy.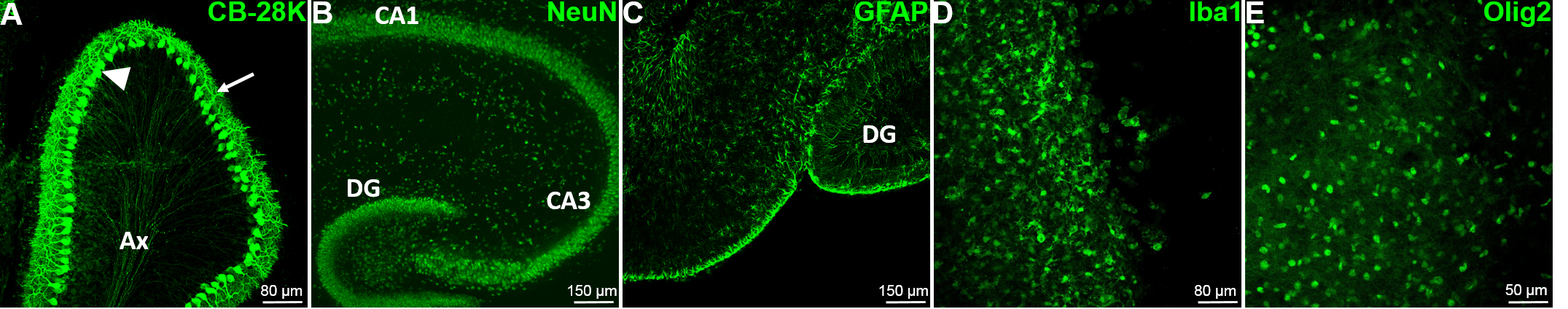
Figure 7. The major cell types of the brain are present and easily detectable by immunofluorescence. A. The characteristic Purkinje cells (PC) of the cerebellum (GABAergic neurons), have been detected in mouse cerebellum slice with the Calbindin-28k marker at DIV0. The arrow head shows the soma of the PC; the arrow shows the dendritic tree of the PC and Ax designate the regrouping PC axons. B. The mouse hippocampal neurons have been stained at DIV0 using the specific neuron nuclear marker NeuN to reveal the internal organization of the hippocampus. C. The mouse hippocampal astrocytes have been stained using the specific Glial Fibrillary Astrocyte Protein (GFAP) marker at DIV0. D. The microglia have been stained at DIV7 using the specific marker Iba1 in mouse hippocampus. E. The oligodendrocytes have been stained at DIV5 using the Olig2 specific nuclear marker in mouse hippocampus. CA1: Cornu Ammonis area 1, CA3: Cornu Ammonis area 3, DG: Dentate Gyrus.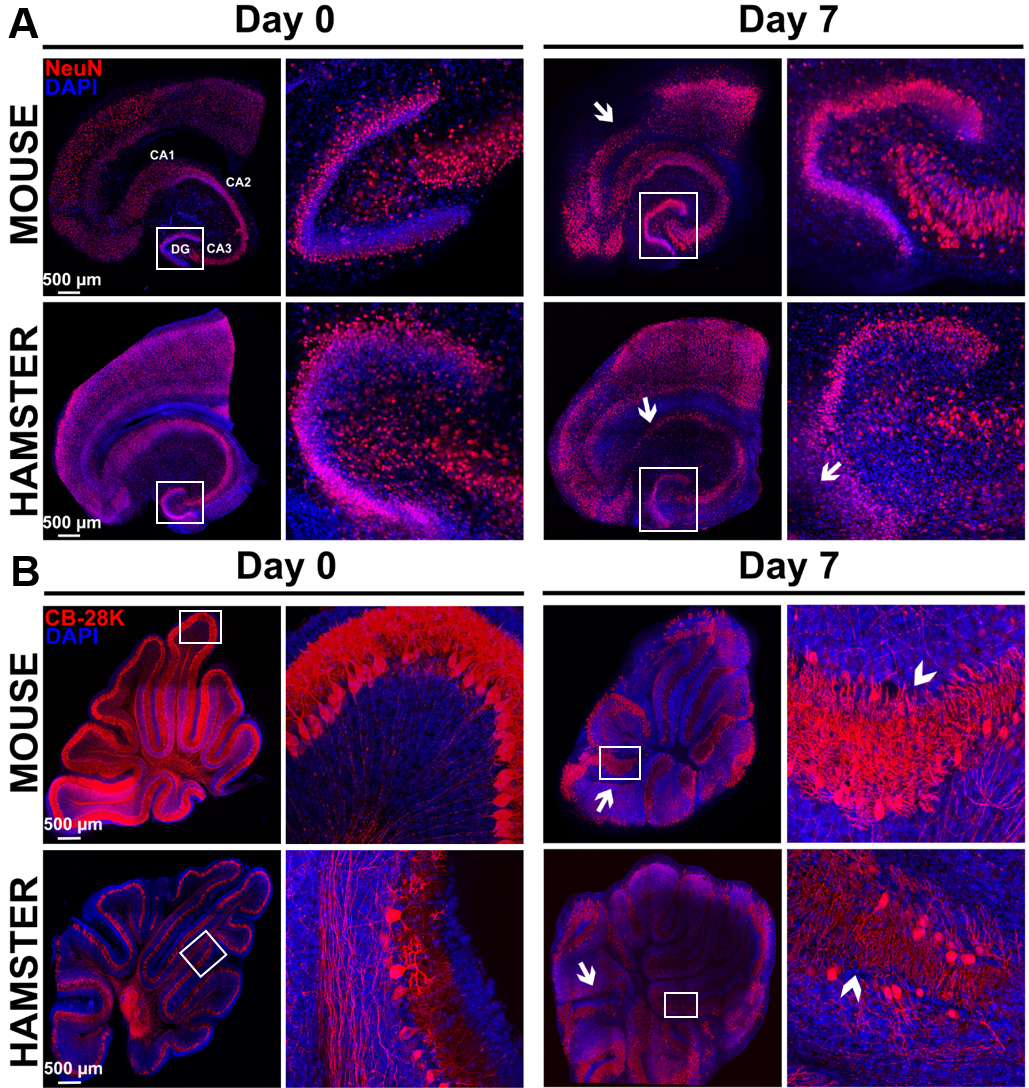
Figure 8. Study of morphological rearrangements and kinetic of mouse or hamster OBC viability over 7 days of culture. A. Hippocampus slices from mice or hamsters were stained with anti-NeuN antibody (neurons marker) at day 0 and day 7 of culture. Pictures show a conserved general organization of the hippocampus over at least 7 days, including the Ammon’s horn (Cornu Ammonis CA) regions and dentate gyrus (DG), which showed only limited loss of cell density (white arrows) possibly related to neuron death. B. Cerebellum slices from mice or hamsters were stained with Calbindin 28K (CB 28K–Purkinje Cells marker) at day 0 and day 7 of culture. Arrows in the pictures show neuronal loss and arrow heads show an interruption of the Purkinje cells layer. Nuclei were counterstained with DAPI. Images taken at the indicated times of culture with a Leica SP5 confocal microscope show morphological evolution within slices at low (reconstructed tile–on left column for each day) or high magnification on right column for each day (20x objective in left panel of each days and 40x objective in right panel of each days corresponding to the white square) over the culture duration.
- Detach, place the slices in 12-well plates using the 5 ml truncated pipette and fix them with PFA at room temperature as described above.
- Infection procedure of brain explants
- Dilute the virus stock in Opti-MEM medium in order to infect cultured slices (in 6-well plates) with a defined number of plaque forming units (pfu). For example, with measles virus (MeV-EGFP) 104 to 4 x 104 pfu are used. For the mock control slices, apply the same volume of Opti-MEM as for the infection.
Note: The dose necessary for the infection should be determined experimentally for each virus/brain structure/animal model combination. - Using a P20 pipette, deposit gently 2 to 10 µl of diluted viral stock on the top of the slices, and make sure that the inoculum is distributed homogeneously.
Note: A micro-injector (with adequate equipment) can be used at this step if the study requires a single site of entry for virus dissemination studies (Ehrengruber et al., 2002). - Incubate plates at 37 °C, 5% CO2 in humidified atmosphere.
- In case of EGFP expressing virus, the fluorescence resulting of the viral replication can be monitored using a fluorescence microscope (Figure 9).
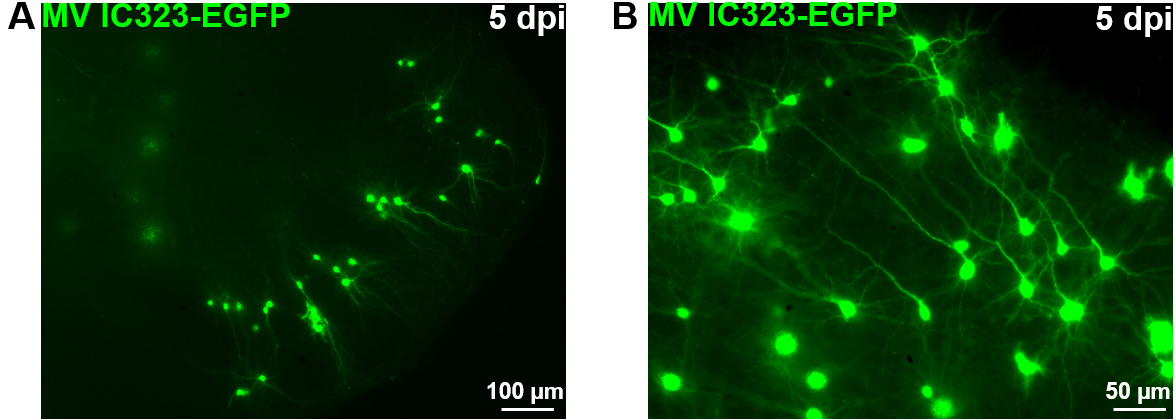
Figure 9. OBC infection with a recombinant measles virus expressing the EGFP. Rat cerebellum slices have been infected with 2 x 104 pfu of MeV IC323-EGFP the day of the slice preparation (DIV 0). MeV-EGFP fluorescence was observed after 5 days using an inverted fluorescence microscope (ZEISS) and camera (AxioCam, ZEISS) at low (A) or high magnification (B).
- Dilute the virus stock in Opti-MEM medium in order to infect cultured slices (in 6-well plates) with a defined number of plaque forming units (pfu). For example, with measles virus (MeV-EGFP) 104 to 4 x 104 pfu are used. For the mock control slices, apply the same volume of Opti-MEM as for the infection.
- Monitoring of cellular or viral gene expression by RT-qPCR
- Using curved tweezers, take the slice to be analyzed out of the culture insert and place it in lysis buffer RA1-10% 2-mercaptoethanol according to the manufacturer’s recommendations (RNA extraction kit–Macherey Nagel).
- Between treating each slice, remove RNase with RNase Away solution.
- Decontaminate tweezers with absolute ethanol.
- Rinse with RNase free water.
- Freeze the lysates at -80 °C and then proceed to the RNA isolation using the RNA extraction kit according to Macherey-Nagel recommendations.
- Prepare cDNA using iScriptTM cDNA Synthesis Kit using a TPersonal 48 Thermal Cycler.
- Perform the quantitative PCR on targeted cDNA, i.e., viral or cellular, using Platinum® SYBR® Green qPCR SuperMix-UDG w/ROX Kit according to manufacturer’s recommendations and StepOnePlusTM Real-Time PCR System.
- All results should be normalized to mRNA from a housekeeping gene such as GAPDH and analyzed as detailed elsewhere (Welsch et al., 2013).
- Using curved tweezers, take the slice to be analyzed out of the culture insert and place it in lysis buffer RA1-10% 2-mercaptoethanol according to the manufacturer’s recommendations (RNA extraction kit–Macherey Nagel).
Data analysis
- Analysis of immunostaining requires the use of a confocal microscope, since the OBC are relatively thick usually between 150 to 500 µm (Figures 7 and 8).
- For experimental design, analysis of cellular or viral gene expression requires at least 12 slices per condition and/or time point coming from separated animals, i.e., 2 slices from 3 different donors (n = 6) repeated at least 2 times (n = 12). For immunofluorescence experiment, we recommend using 1 slices from 3 different donors repeated at least 3 times (n = 9).
Note: Regarding the viability and the structures’ evolution analyses, our results showed that the OBC from mice or hamsters (also true for rats–data not shown) lead to a similar evolution of these parameters, indicating that our protocol can be used with the main laboratory rodent species and confirming the versatility of our method. - For the viability statistical analysis, the mortality and metabolic activity variables should be analyzed separately (Table 3). Concerning mortality, several factors are evaluated at different days of culture (D0, D1 and D7): animal, slices from the same animal and repeats for the same slice (i.e., several images from the same slice). Hence, the experiment has a hierarchical structure, and the model is written as:
Mij = µ + Ai + Sj(i) + eij
where, Mij is the mortality value of the jth slice from the ith animal, µ the general mean, Ai the effect of the ith animal, Sj(i) the effect of the jth slice from the ith animal, and eij the random effect.
These various effects are statistically tested using the nested analysis of variance (nested ANOVA) implemented in R software as follows:
anova(lm(formula = Mortality ~ Animal + slice %in% Animal + image %in% slice, data = file.name)).
This analysis is performed for each day of culture (D0, D1 and D7).
Caution: Assumptions for ANOVA should be checked (normality of the dependent variable, homoscedasticity, graphical diagnostic procedures like residuals vs. predicted values plot and normal QQ-plot of residuals). If not, the non-parametric Friedman test should be used for comparison of image differences within a slice, and slice differences within an animal, and the non-parametric Kruskal-Wallis test for animal differences. - For the statistical analysis, we used GraphPad Prism and R software. Alternatively, a website called ‘Handbook of Biological Statistics’ (John H. McDonald) detailing the method and providing spreadsheets and advices for 2 to 4-level nested ANOVA is accessible at this address: http://www.biostathandbook.com/nestedanova.html. For example, if you want to analyze the time effect, use the spreadsheet dedicated to 4-level nested ANOVA and fill the column as follow: Day/animal/slice/image/fluorescence value respectively representing group/subgroup/ subsubgroup/subsubsubgroup/observation. For the analysis of the animal effect you can perform the 3-level nested ANOVA and fill the column as follow: animal/slice/image/fluorescence value respectively representing group/ subgroup/ subsubgroup/ observation (Note again that you need to respect the ANOVA assumption, and if not simply apply a Kruskall-Wallis test).
Table 3. Statistical analysis of OBC viability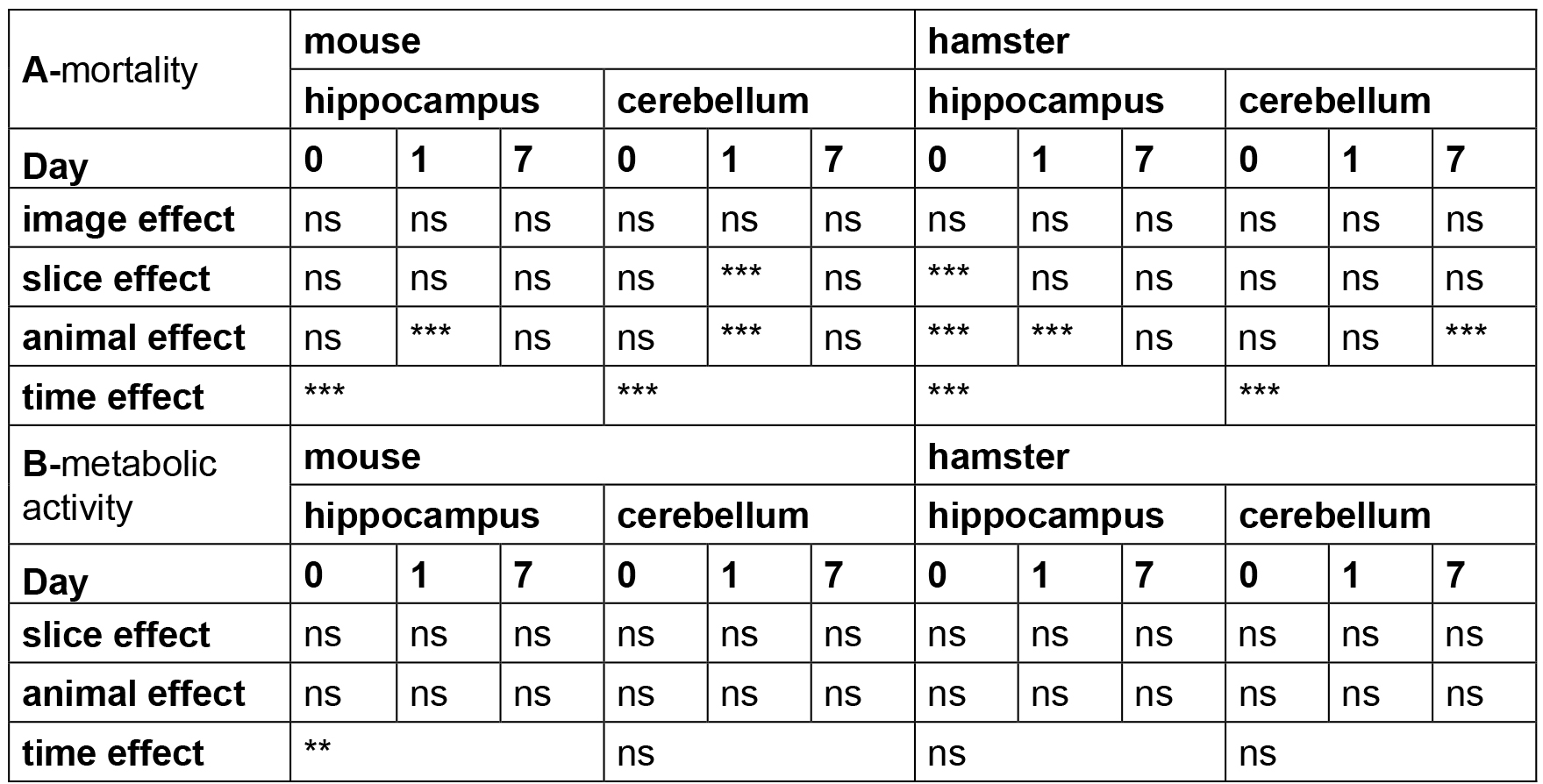
Note: The time, animal, slice and image effects were statistically estimated according to the statistical analyses described above. P-values are reported for each test performed with ns (non-significant P > 0.05), ** P < 0.01, *** P < 0.001.
Recipes
Note: All solutions must be sterilized using a Stericup filtration unit for large volumes (organotypic culture medium, dissection media and 10x kynurenic acid) or with a syringe filter with 0.22 µm pore size.
- 1 M MgCl2 (1 L)
- Weight 95.21 g of MgCl2
- Bring to 1 L with distilled water
- Keep at room temperature up to 1 year
- Weight 95.21 g of MgCl2
- 0.1 N NaOH (500 ml)
- Weight 1.99 g of NaOH
- Bring to 500 ml with distilled water
- Keep at room temperature up to 1 year
- Weight 1.99 g of NaOH
- Insulin solution at 50 mg/ml (2 ml)
- Reconstitute the totality of the vial in 2 ml of 0.005 N HCl solution
- Aliquot (1 ml) and store at -20 °C
- Reconstitute the totality of the vial in 2 ml of 0.005 N HCl solution
- Organotypic brain culture medium (500 ml)
Add all the components:
375 ml of MEM GlutaMAX
125 ml of heat-inactivated horse serum (store the serum at -20 °C)
2.5 g of D-glucose
1 ml of human insulin at 50 mg/ml (final concentration 0.1 mg/L)
Filter to sterilize (0.22 μm) and store at 4 °C for up to 2 weeks
Note: Addition of antibiotics is not necessary when the procedure is kept aseptic, but may be helpful to prevent contamination. When using penicillin, the concentration should be kept below 0.08 mM final concentration to avoid potential interference with synaptic activity (see Table 2). - 10x kynurenic acid solution (concentrated excitotoxicity inhibitor solution) (200 ml)
- Dissolve 378 mg of kynurenic in 170 ml of H2O
- Add 20 ml of 1 M MgCl2
- Adjust pH at 7.4 using 0.1 N NaOH
- Add 1 ml of HEPES
- Adjust volume to 200 ml
- Filter to sterilize, protect from light and keep up to 2 weeks at room temperature
- Dissolve 378 mg of kynurenic in 170 ml of H2O
- Dissection medium (Hibernate®-A/5 g/L D-Glucose/1x Kynurenic acid) (50 ml)
- Prepare 500 ml of Hibernate®-A supplemented with glucose by adding 2.5 g of D-glucose
- Filter sterilize using a Stericup filtration unit and store at 4 °C up to 6 months
- The day of dissection, mix 45 ml of Hibernate®-A/5 g/L D-Glucose with 5 ml of 10x kynurenic acid
- Keep on ice during the dissection
- Prepare 500 ml of Hibernate®-A supplemented with glucose by adding 2.5 g of D-glucose
- 8% paraformaldehyde (PFA) (100 ml)
In a fume hood:- Weigh 8 g of crystalline PFA in a beaker and add 100 ml of 1x DPBS
- Heat the solution until the powder is fully dissolved to 65 °C (not higher as PFA will degrade)
- Aliquot and store at -20 °C
- Dilute to 4% with 1x DPBS (make fresh prior to use)
- Weigh 8 g of crystalline PFA in a beaker and add 100 ml of 1x DPBS
- Blocking and permeabilization solution (BPS) (1x DPBS/0.3% Triton X-100/4% FBS) (50 ml)
Add all the components:
47.85 ml of 1x DPBS
2 ml of FBS
150 µl of Triton X-100
Vortex and incubate at 37 °C in a water bath in order to help dissolving Triton X-100 for 30 min
Store at 4 °C up to 2 weeks
Note: Ensure that no bacteria or fungus proliferated in solution prior to use, and if so prepare a new batch.
Acknowledgments
This work was supported by French ANR NITRODEP grant (project ANR-13-PDOC-0010-01) (http://www.agence-nationale-recherche.fr) and LABEX ECOFECT (ANR-11-LABX-0048) of Lyon University, within the program “Investissements d’Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR). We are grateful to Dr. Isabelle Dussart (Université Pierre et Marie Curie, Université Paris 06, CNRS - UMR 7102, 75005 Paris, France) and Dr. Helene Clot-Faybesse (INMED, INSERM U29, Université de la Méditerranée, Marseille, France) for precious advice on OBC cultures. This protocol was adapted from Stoppini et al. (1991) with minor changes. The authors declare no conflicts or competing of interests.
References
- Bloyet, L. M., Welsch, J., Enchery, F., Mathieu, C., de Breyne, S., Horvat, B., Grigorov, B. and Gerlier, D. (2016). Requirement of HSP90 chaperoning in addition to phosphoprotein for folding but not for supporting enzymatic activities of measles and Nipah virus L polymerases. J Virol 90:JVI.00602-16.
- Bornstein, M. B. and Murray, M. R. (1958). Serial observations on patterns of growth, myelin formation, maintenance and degeneration in cultures of new-born rat and kitten cerebellum. J Biophys Biochem Cytol 4(5): 499-504.
- Braun, E., Zimmerman, T., Hur, T. B., Reinhartz, E., Fellig, Y., Panet, A. and Steiner, I. (2006). Neurotropism of herpes simplex virus type 1 in brain organ cultures. J Gen Virol 87(Pt 10): 2827-2837.
- Ehrengruber, M. U., Ehler, E., Billeter, M. A. and Naim, H. Y. (2002). Measles virus spreads in rat hippocampal neurons by cell-to-cell contact and in a polarized fashion. J Virol 76(11): 5720-5728.
- Humpel, C. (2015). Organotypic brain slice cultures: A review. Neuroscience 305: 86-98.
- Kim, H., Kim, E., Park, M., Lee, E. and Namkoong, K. (2013). Organotypic hippocampal slice culture from the adult mouse brain: a versatile tool for translational neuropsychopharmacology. Prog Neuropsychopharmacol Biol Psychiatry 41: 36-43.
- Laksono, B. M., de Vries, R. D., McQuaid, S., Duprex, W. P. and de Swart, R. L. (2016). Measles virus host invasion and pathogenesis. Viruses 8(8): E210.
- Lossi, L., Alasia, S., Salio, C. and Merighi, A. (2009). Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog Neurobiol 88(4): 221-245.
- Ludlow, M., Kortekaas, J., Herden, C., Hoffmann, B., Tappe, D., Trebst, C., Griffin, D. E., Brindle, H. E., Solomon, T., Brown, A. S., van Riel, D., Wolthers, K. C., Pajkrt, D., Wohlsein, P., Martina, B. E. E., Baumgartner, W., Verjans, G. M. and Osterhaus, A. (2016). Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol 131(2): 159-184.
- Mayer, D., Fischer, H., Schneider, U., Heimrich, B. and Schwemmle, M. (2005). Borna disease virus replication in organotypic hippocampal slice cultures from rats results in selective damage of dentate granule cells. J Virol 79(18): 11716-11723.
- Olival, K. J. and Daszak, P. (2005). The ecology of emerging neurotropic viruses. J Neurovirol 11(5): 441-446.
- Reuter, D. and Schneider-Schaulies, J. (2010). Measles virus infection of the CNS: human disease, animal models, and approaches to therapy. Med Microbiol Immunol 199(3): 261-271.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Stoppini, L., Buchs, P. A. and Muller, D. (1991). A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182.
- Stubblefield Park, S. R., Widness, M., Levine, A. D. and Patterson, C. E. (2011). T cell-, interleukin-12-, and gamma interferon-driven viral clearance in measles virus-infected brain tissue. J Virol 85:3664-76.
- Swanson, P. A., 2nd and McGavern, D. B. (2015). Viral diseases of the central nervous system. Curr Opin Virol 11: 44-54.
- Taylor, L. H., Latham, S. M. and Woolhouse, M. E. (2001). Risk factors for human disease emergence. Philos Trans R Soc Lond B Biol Sci 356(1411): 983-989.
- Welsch, J. C., Talekar, A., Mathieu, C., Pessi, A., Moscona, A., Horvat, B. and Porotto, M. (2013). Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J Virol 87(24): 13785-13794.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Welsch, J. C., Lionnet, C., Terzian, C., Horvat, B., Gerlier, D. and Mathieu, C. (2017). Organotypic Brain Cultures: A Framework for Studying CNS Infection by Neurotropic Viruses and Screening Antiviral Drugs. Bio-protocol 7(22): e2605. DOI: 10.21769/BioProtoc.2605.
Category
Microbiology > Microbe-host interactions > Virus
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > 3D cell culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.