- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Staining of Membrane Receptors with Fluorescently-labeled DNA Aptamers for Super-resolution Imaging

Published: Vol 7, Iss 17, Sep 5, 2017 DOI: 10.21769/BioProtoc.2541 Views: 10404
Reviewed by: Gal HaimovichShalini Low-NamAnca Flavia Savulescu
Original research article
The authors used this protocol in:

Feb 2017
Advertisement






Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
One of the most prominent applications of fluorescent super-resolution microscopy is the study of nanodomain arrangements of receptors and the endocytic pathway. Staining methods are becoming crucial for answering questions on the nanoscale, therefore, the use of small and monovalent affinity probes is of great interest in super-resolution microscopy with biological samples. One kind of affinity probe is the aptamer. Aptamers are single DNA or RNA sequences that bind with high affinity to their targets and due to their small size they are able to (i) place the fluorophore in close proximity to the protein of interest and (ii) bind to most of the protein of interest overcoming the steric hindrance effect, resulting in better staining density. Here we describe a detailed protocol with which to stain live cells using aptamers and to image them with Stimulated Emission Depletion (STED) microscopy. In this protocol, the stainings were performed with commercially available aptamers that target the epidermal growth factor receptor (EGFR), the human epidermal growth factor receptor 2 (HER2 or ErbB2) and the ephrin type-A receptor 2 (Epha2). Since aptamers can be coupled to most of the popular fluorophores, we believe that the procedure presented here can be extended to the large majority of the current super-resolution microscopy techniques.
Keywords: MicroscopyBackground
Recent advances in super-resolution imaging techniques have led to the search for more accurate methodologies to tag cellular elements. Diffraction unlimited imaging instruments provide excellent resolutions, however standard sample staining methodologies, such as immunostaining, lack the necessary precision for the detection of cellular elements. Due to their large dimension (~15 nm in length) and high molecular weight (~150 kDa), antibodies can poorly penetrate into biological samples. Additionally, the primary/secondary antibody complex places the fluorophores at approximately 25 nm away from the target, compromising the detection accuracy. Moreover, due to the large size of the primary/secondary antibody complex, a smaller fraction of the targets can be labelled due to the steric hindrance (Fornasiero and Opazo, 2015). This leads to lower labelling density, a crucial parameter for super-resolution microscopy, especially in recognizing and describing nanostructures. To circumvent these problems, small affinity probes that bind to single targets (monovalently) such as aptamers, affibodies or nanobodies have been tested in recent years (Rothbauer et al., 2006; Opazo et al., 2012; Ries et al., 2012) and are becoming valuable tools for super-resolution microscopy. Nanobodies, affibodies and aptamers have a relatively small linear size (~3 nm, ~2 nm and ~3 nm, respectively). This property allows them to place the fluorescent dye in close proximity to the target, penetrate samples in a more efficient manner and bind to a higher fraction of target proteins, bypassing the effect of steric hindrance observed by antibodies (Ries et al., 2012; Mikhaylova et al., 2015). In a previous study we made a systematic comparison between three commercially available aptamers and antibodies that target the epidermal growth factor receptor (EGFR), the human epidermal growth factor receptor 2 (HER2 or ErbB2) and the ephrin type-A receptor 2 (Epha2). Our results showed that aptamers were able to find more epitopes (resulting in higher labeling density). As a consequence, several structural features of the subcellular components that were imaged became more apparent. Among these, the inner lumen and the complex morphology of endocytic organelles were visible. For these reasons, smaller imaging tools are becoming the preferred choice over antibodies, in order to improve the quality of an immunolabeling super-resolution approach and allow a more precise description the localization and the distribution of membrane receptors (Gomes de Castro et al., 2017).
Materials and Reagents
- Biospin 6 column (Micro Bio-SpinTM P-6 Gel Columns, Tris Buffer) (Bio-Rad Laboratories, catalog number: 7326222 )
- Cell culture 12-well plates (Thermo Fisher Scientific, catalog number: 150628 )
- 18 mm diameter round glass coverslips (Gerhard Menzel, catalog number: CB00180RA1 )
- 15 ml tubes
- PCR tubes
- Parafilm M
- Soft paper tissue
- Gloves
- Microscopy glass slides
- 2 ml tubes
- Vacuum filtration (e.g., VWR® Vacuum Filtration Systems, Standard Line) (VWR, catalog number: 10040-436 )
- 0.2 µm syringe filter
- Transfer pipette
- The images shown in Figure 1 are from aptamers against human:
EGFR (5’-SH-EGFR aptamer-3’, seq # 2369-27-02, 50 mer)
ErbB2 (5’-SH-ErbB2 aptamer-3’, seq # 1194 ± 35, 40 mer)
Epha2 (5’-SH-EphA2 aptamer-3’, seq # 2176-01-01, 76 mer)
Note: They were produced by Aptamer Sciences, Inc., South Korea and supplied by AMS Biotechnology, Europe. All three aptamers contain the chemical modification 5-(N-benzylcarboxyamide)-2’-deoxyuridine (5-BzdU) in unrevealed locations. - Triethylammonium acetate buffer pH 7.0, 1 M (TEAA) (AppliChem, catalog number: A3846 )
- Maleimide Atto647N dye (Atto-TEC, catalog number: AD 647N-41 )
- Dimethyl sulfoxide, anhydrous (DMSO) (Sigma-Aldrich, catalog number: 276855 )
- Sodium chloride (NaCl)
- Ethanol
- 1x Dulbecco’s phosphate buffered saline (1x DPBS) (Sigma-Aldrich, catalog number: D8662 )
- Acetonitrile
- Trypsin-EDTA solution (Lonza, catalog number: 17-161E )
- Ultrapure DNase- and RNase-free distilled water (Carl Roth, catalog number: T143.2 )
- Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (Sigma-Aldrich, catalog number: C4706 )
- Sodium hydroxide (NaOH)
- DMEM, high glucose medium, no glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 11960044 )
- Fetal bovine serum (FBS) (Biochrom, catalog number: S 0615 )
- L-Glutamine 200 mM (Lonza, catalog number: BE17-605E )
- Penicillin/streptomycin 10,000 U/ml, each (Lonza, catalog number: 17-602E )
- RPMI 1640 medium, no glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 21870076 )
- Poly-L-lysine (PLL) (Sigma-Aldrich, catalog number: P5899-5MG )
- Sodium phosphate dibasic (Na2HPO4)
- Potassium phosphate monobasic (KH2PO4)
- Potassium chloride (KCl)
- Magnesium chloride hexahydrate (MgCl2·6H2O)
- Salmon sperm DNA, sheared, 10 mg/ml (Thermo Fisher Scientific, catalog number: AM9680 )
- Dextran sulfate sodium salt (Sigma-Aldrich, catalog number: 31404 )
- Paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: P6148 )
- Glycine (Sigma-Aldrich, catalog number: G8898 )
- Mowiol® (Sigma-Aldrich, catalog number: 81381 )
- 1 M Tris (2-carboxyethyl) phosphine hydrochloride stock solution (TCEP) (see Recipes)
- Complete DMEM medium (see Recipes)
- Complete RPMI medium (see Recipes)
- Poly-L-lysine (PLL) stock solution (see Recipes)
- 5x phosphate buffer saline (5x PBS) (see Recipes)
- 25 mM MgCl2 solution (5x MgCl2) (see Recipes)
- Blocking solution (see Recipes)
- 4% paraformaldehyde (PFA) (see Recipes)
- Quenching solution (see Recipes)
- Mowiol® (see Recipes)
- (Optional) Buffer A (see Recipes)
- (Optional) Buffer B (see Recipes)
Equipment
- Microcentrifuge (e.g., Eppendorf, model: 5415 or similar)
- Nucleosil 100-5 C18 column
- (Optional) Dionex DNAPac PA200 4 x 250 mm column
- Hemocytometer
- Cell culture hood
- Thermal cycler
- Aluminum metal plate (length x width x thickness [cm]: e.g., 20 x 12 x 2)
- Cell culture incubator
- Half-curved-forceps
- Oven
- STED microscope, Leica pulsed STED setup composed by a True Confocal System (TCS) STED SP5 (Leica Microsystems, model: Leica TCS SP5 ) fluorescence microscope equipped with a 100x 1.4 NA HCX PL APO oil objective (Leica Microsystems, Germany)
- Pulsed laser (PicoQuant, Germany)
- Sapphire tunable laser (Mai Tai Broadband, Spectra-Physics, USA)
- Glass beaker
- Magnetic stirrer
- Lab coat, eye protection
Software
- ImageJ (http://imagej.nih.gov/ij/docs/index.html)
- MATLAB (MathWorks, Massachusetts, USA)
Procedure
- Coupling of dye to aptamers
During this step a thiol-maleimide cross-linking reaction is performed to conjugate the fluorophore to aptamers. Alternatively, aptamers with a 3’-amino group can be used for further conjugation with NHS esters (e.g., Atto647N NHS-ester). Some fluorescently labeled aptamers are also commercially available.- Add 10 nmoles of the thiolated aptamer with 10 mM Tris (2-carboxyethyl) phosphine solution (TCEP; see Recipe 1) in 100 μl of 0.1 M TEAA. Heat the tube at 70 °C for 3 min and incubate the mixture at room temperature for 1 h.
- Desalt this reaction by size exclusion chromatography on a Biospin 6 column according to the manufacturer’s instructions.
- Add to the reduced aptamer (from step A2) 4 µl of 10 µg/µl maleimide-functionalized Atto647N (previously dissolved in DMSO) and mix the contents well by pipetting or vortexing.
- Incubate overnight at 4 °C.
- Recover the fluorophore-labelled aptamer by ethanol precipitation: add 100 µl of 300 mM NaCl and 450 µl cold ethanol, mix carefully by inverting the closed sample several times and freeze the sample in dry ice for 20 min (or at -20 °C for several hours), centrifuge at 15,000 x g (rcf) at 4 °C for 30 min, discard the supernatant and wash the pellet in 50 µl of 70% ethanol.
- Resuspend in 50 µl 1x DPBS and desalt on a Biospin 6 column into 1x DPBS.
- Measure the absorbance ratio at 260 nm and 650 nm.
- Confirm the labeling efficiency and absence of free dye by reversed phase HLPC (e.g., on a Nucleosil 100-5 C18 4 x 250 mm column using a gradient of 0-40% acetonitrile in TEAA buffer in 30 min with a flow rate of 1 ml/min at 30 °C) or by anion exchange HPLC (e.g., Dionex DNAPac PA200 4 x 250 mm column using a gradient of 0-75% buffer B (see Recipe 12) in buffer A (see Recipe 11) in 40 min with a flow rate of 1 ml/min at 60 °C.
- Store the labeled aptamer sample at -20 °C until use.
- Add 10 nmoles of the thiolated aptamer with 10 mM Tris (2-carboxyethyl) phosphine solution (TCEP; see Recipe 1) in 100 μl of 0.1 M TEAA. Heat the tube at 70 °C for 3 min and incubate the mixture at room temperature for 1 h.
- Cell culture preparation prior to staining
Cell lines containing the receptor of interest (and cell lines lacking it, used as negative controls) should be seeded into 12-well plates containing coated coverslips with poly-L-lysine (PLL) one day before the aptamer staining. Depending on the cell line, choose adequate cell numbers (use the hemocytometer to count the cells) to reach approximately 70-80% confluence after 12-16 h incubation. The entire procedure described below must be performed under sterile conditions under a cell culture hood.- HeLa (Epha2a positive) cells are grown in plates with complete DMEM medium (see Recipe 2) and A-431 (EGFR positive) and SKBR3 (ErbB2R positive) cells are cultured in complete RPMI medium (see Recipe 3). All cell lines are maintained at 37 °C and 5% CO2.
- To split cells, first wash cells with sterile 1x DPBS, add 2-3 ml of trypsin-EDTA to cover the surface of the plate and incubate the plate for 1-5 min at 37 °C or until cells are completely detached. Add 10 ml of complete DMEM medium or RPMI medium to inactivate trypsin. Gently pipette up and down the medium containing the inactivated trypsin to detach all cells from the plate and transfer them into a sterile 15 ml tube.
- Centrifuge the cells at 1,000 rpm (~250 x g) for 4 min at RT.
- Aspirate the supernatant and resuspend the cell pellet in 10 ml of complete DMEM medium/RPMI medium. Dilute the resuspended cells to the desired concentration in fresh complete DMEM or RPMI medium and add 1 ml to every well of the 12-well plates containing PLL treated coverslips (for PLL stock solution preparation and dilution, see Recipe 4). Incubate the plates at 37 °C and 5% CO2 until staining.
- HeLa (Epha2a positive) cells are grown in plates with complete DMEM medium (see Recipe 2) and A-431 (EGFR positive) and SKBR3 (ErbB2R positive) cells are cultured in complete RPMI medium (see Recipe 3). All cell lines are maintained at 37 °C and 5% CO2.
- Preparation of functional aptamer: folding reaction
During this step, aptamers are exposed to a high temperature at an appropriate magnesium concentration which allows them to attain a proper folding prior to the staining procedure. For better results, the folding reaction should be freshly performed each time before staining. Before starting the folding reaction, prepare a stock solution diluting aptamers in 1x DPBS to a final concentration of 30 µM and store it protected from light at 4 °C.- Prepare 10 µl of 10 µM functional aptamer by mixing 3.3 µl of fluorescently labelled aptamer (30 µM aptamer stock solution), 2 µl of 5x PBS (see Recipe 5), 2 µl of 5x MgCl2 (see Recipe 6) and 2.7 µl of ultrapure DNase- and RNase-free distilled water. Please note that this reaction should be carried out in PCR tubes that fit the thermal cycler.
- Heat up the aptamer solution to 75 °C for 3 min and then cool down to 20 °C at a rate of 1 °C/min using a thermal cycler.
- Prepare 10 µl of 10 µM functional aptamer by mixing 3.3 µl of fluorescently labelled aptamer (30 µM aptamer stock solution), 2 µl of 5x PBS (see Recipe 5), 2 µl of 5x MgCl2 (see Recipe 6) and 2.7 µl of ultrapure DNase- and RNase-free distilled water. Please note that this reaction should be carried out in PCR tubes that fit the thermal cycler.
- Aptamer live cells staining
- Aspirate the medium from the cells prepared during Procedure B, rinse briefly once with complete DMEM or RPMI depending on the cell line and incubate for 10 min at 37 °C and 5% CO2 (in the cell culture incubator) with 500 µl/well of freshly prepared blocking solution containing sheared salmon sperm DNA and dextran sulfate (see Recipe 7).
- In the meantime, prepare the metal plate used for staining/incubation at 37 °C. Fix with tape a piece of Parafilm® M (large enough to fit all coverslips to be stained) on the surface (see Video 1) and pre-heat the metal plate containing the Parafilm® M in the cell culture incubator.
 Video 1. Aptamers live staining part I. This video shows the metal plate preparation for aptamer live staining. Please note that this video is for demonstration only and that this step should be performed inside a cell culture hood.
Video 1. Aptamers live staining part I. This video shows the metal plate preparation for aptamer live staining. Please note that this video is for demonstration only and that this step should be performed inside a cell culture hood. - After blocking (step D1), carefully remove the coverslips from the 12-well plate using half-curved-forceps, remove the excess blocking solution by gently tapping the edges of the coverslips with a soft paper tissue and place the coverslips upside down on 60 µl of staining solution (complete DMEM supplemented with 100 μg/ml sheared salmon sperm DNA and 250 nM folded aptamer) spotted onto the Parafilm® M fixed to the metal plate (made in step D2) (see Video 2). Incubate the cells for 60 min at 37 °C and 5% CO2. Video 2. Aptamers live staining part II. This video shows removal of coverslips containing cells from the 12-well plates using half-curved-forceps and incubation of cells (on coverslips) with staining solution. Please note that this video is for demonstration only and that this step should be performed inside the cell culture hood. To avoid contamination, gloves should be sprayed with 70% ethanol.
- After incubation, carefully take the coverslips off the metal plate using half-curved-forceps, remove the excess staining solution by gently tapping the edges of the coverslips with a soft paper tissue, and then submerge it several times in large volumes (e.g., 20-40 ml in a small beaker) of ice-cold 1x DPBS. Briefly remove the excess 1x DPBS from the coverslips by tapping them on a tissue paper and place each one (cell-side-up) into a well of a new 12-well plate filled with 1 ml/well ice-cold 4% PFA (see Recipe 8) for fixation (see Video 3). Video 3. Aptamers live staining part III. This video shows the washing step after staining procedure. Please note that video is in open air for demonstration only and that 4% PFA should only be handled in the fume hood.
- Fix the cells for 20 min on ice and subsequently at room temperature for another 25 min. During fixation, keep the plate protected from light to avoid fluorophore bleaching.
- Aspirate the PFA solution and add 1 ml of quenching solution (see Recipe 9) to each well. Incubate for 15 min at room temperature protected from light.
- Wash twice for 5 min with 1x DPBS (1 ml/well) and mount the coverslips with Mowiol® (see Recipe 10; e.g., 8-9 µl for an 18 mm coverslip) on microscopy glass slides.
- Dry the mounted coverslips in an oven at 37 °C for 20-30 min or overnight at RT and store at 4 °C protected from light until imaging.
- Aspirate the medium from the cells prepared during Procedure B, rinse briefly once with complete DMEM or RPMI depending on the cell line and incubate for 10 min at 37 °C and 5% CO2 (in the cell culture incubator) with 500 µl/well of freshly prepared blocking solution containing sheared salmon sperm DNA and dextran sulfate (see Recipe 7).
Data analysis
Imaging: The images shown in this protocol (Figure 1) were acquired using a 100x 1.4 NA HCX PL APO oil objective. Excitation of Atto647N fluorophore was achieved with a 635 nm pulsed laser (PicoQuant, Germany), and the 750 nm depletion STED beam was obtained with a pulsed infrared titanium: sapphire tunable laser (Mai Tai Broadband, Spectra-Physics, USA). The pixel size was set to 20.2 nm, scanning speed to 1 kHz, line average to 96 times, pinhole to one Airy unit and signal was detected with an avalanche photodiode detector (APD).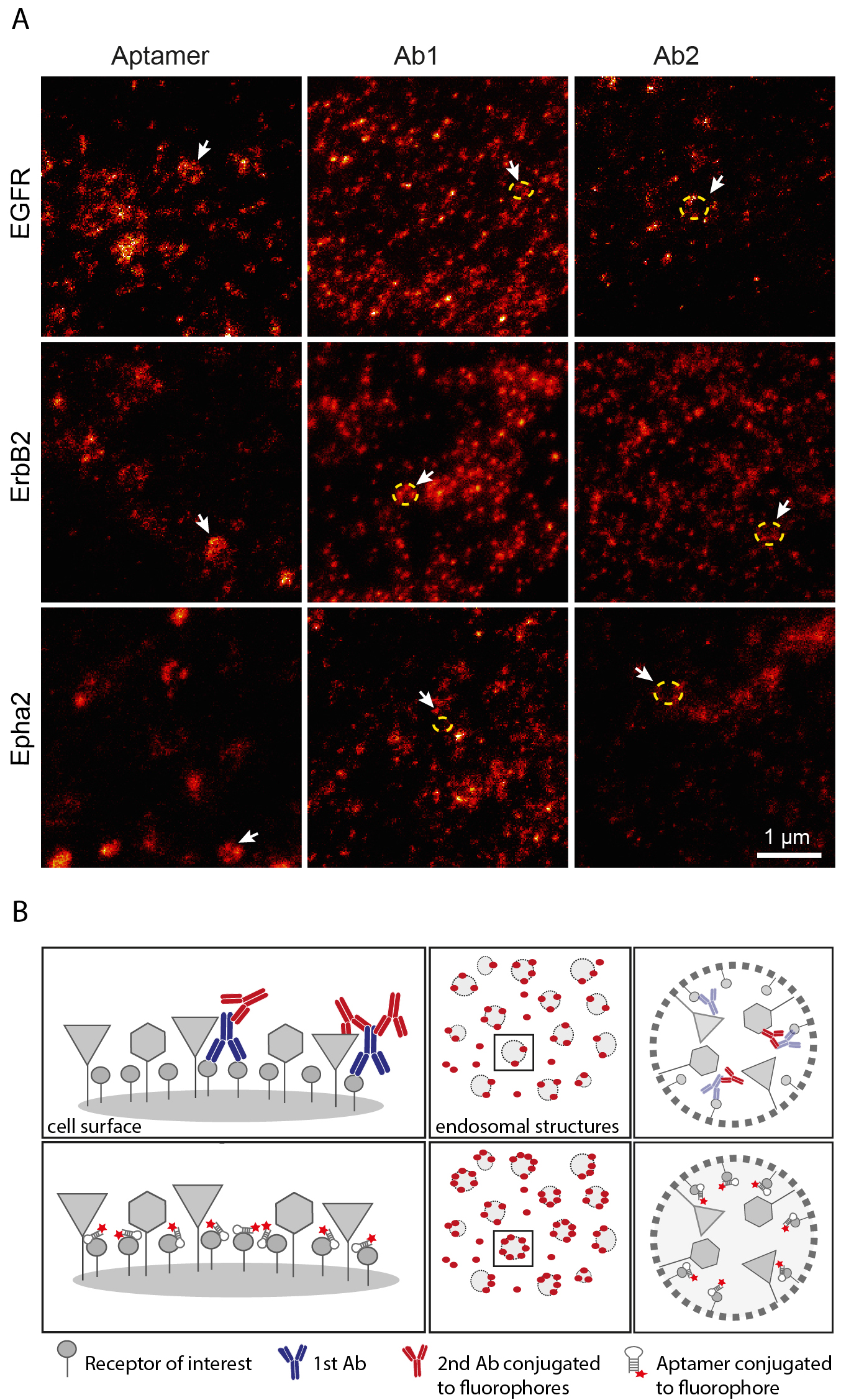
Figure 1. Recognition of endosome-like structures in aptamer or antibody stained cells. A. STED images comparing the cells stained with aptamers and antibodies against the same receptors (Ab1 and Ab2, under saturating conditions as described in (Gomes de Castro et al., 2017). Arrowheads point to some examples of endosome-like structures. Staining using antibodies resulted in discontinuous labelling of the organelle contours (indicated by yellow doted circles in the STED image). B. The scheme represents our current hypothesis, indicating that large affinity probes like antibodies (upper panel) might not detect all available epitopes and that small molecules like aptamers (lower panel) decorate the target structures better.
Notes
- To determine the saturating concentration or optimal staining concentration (best signal-to-noise ratio) for the aptamer of interest fixing the incubation time of staining (e.g., 60 min) and testing different aptamers concentrations ranging from 10-500 nM (or more) in positive cell lines is recommended. After staining with different concentrations of the aptamers, fixation and mounting, cells can be imaged in any epifluorescence microscope and fluorescent intensities can be calculated using as an example ImageJ or MATLAB.
- After defining the saturating conditions for the staining procedure, the next step is to test the binding specificity. This is achieved by evaluating the staining in the cells expressing the receptor and the cells that do not express it. Importantly, the staining conditions, the aptamer concentrations and the incubation times must be the same for both positive and negative cells. Once stained and fixed, cells can be imaged with an epifluorescence or confocal microscope. Negative cell lines should show virtually no specific fluorescent signal. For a proper control of the unspecific staining background, it is useful to test the immunolabeling with a random aptamer conjugated to the same fluorophore as the aptamer binding the specific target, in parallel. This control should not have any fluorescence signal. An additional control is the evaluation of the binding specificity by fluorescence flow cytometry analysis using the same controls.
- Every receptor has its own internalization kinetics. In this work and protocol we intend to maximize the labeling by internalization and therefore choose longer incubation times.
- If the aptamers contain BzdU (5-(N-benzylcarboxyamide)-2’-deoxyuridine) or any other hydrophobic groups, we recommend the addition of dextran sulfate to the blocking solution. An optimization of the dextran sulfate concentration in the pre-blocking solution strongly reduces the background caused by nonspecific binding due to electrostatic interaction of aptamers to the PLL-treated coverslip.
- If unspecific binding of the aptamer persists, increase the concentration of sheared salmon sperm DNA and/or dextran sulphate in the staining solution. The polyanionic competitor/blocking agent dextran sulfate also substantially reduces the nonspecific binding caused by electrostatic attraction of polyanionic aptamers to positively charged sites in nuclei, such as histones.
- For identification and quantification of endosome-like structures, colocalization studies are strongly recommended (Gomes de Castro et al., 2017).
Recipes
- 1 M Tris (2-carboxyethyl) phosphine hydrochloride stock solution (TCEP)
For 100 ml:- Add 11.47 g TCEP to 35 ml cold ultrapure water
- Bring the pH to 7.0 with 10 N NaOH and adjust the volume to 100 ml
- Aliquot into 2.0 ml tubes and store at -20 °C
- Add 11.47 g TCEP to 35 ml cold ultrapure water
- Complete DMEM medium
- DMEM supplemented with 10% FBS, 4 mM L-glutamine and 100 U/ml each of penicillin and streptomycin
- Sterilize by vacuum filtration
- Store at 4 °C
- DMEM supplemented with 10% FBS, 4 mM L-glutamine and 100 U/ml each of penicillin and streptomycin
- Complete RPMI medium
- RPMI supplemented with 10% FBS, 4 mM L-glutamine and 100 U/ml each of penicillin and streptomycin
- Sterilize by vacuum filtration
- Store at 4 °C
- RPMI supplemented with 10% FBS, 4 mM L-glutamine and 100 U/ml each of penicillin and streptomycin
- Poly-L-lysine (PLL) stock solution
- Prepare 2 mg/ml stock solution in ultrapure water and sterilize through a syringe filter of 0.2 µm
- Make aliquots and store at -20 °C
- Prepare the plates containing PLL-treated coverslips under a sterile hood
- Add 1 ml of 0.1 mg/ml PLL into every well containing a coverslip
- Incubate for 1 h at RT
- After incubation, wash twice with ultrapure water and leave the plates to air-dry inside the cell culture hood
- Store the plates at 4 °C until use
- Prepare 2 mg/ml stock solution in ultrapure water and sterilize through a syringe filter of 0.2 µm
- 5x phosphate buffer saline (5x PBS)
- Dilute the 10x concentrated PBS stock solution in ultrapure water. Filter the solutions inside a cell culture hood with a 0.2 µm syringe filter or autoclave. Store at RT
- For 1 L 10x concentrated PBS stock solution:
14.4 g sodium phosphate
2.4 g potassium phosphate
2 g KCl
80 g NaCl
Dissolved in DNase-and RNase-free water
Adjust the pH to 7.4
Sterilize by filter sterilization or autoclaving
Store at RT
- Dilute the 10x concentrated PBS stock solution in ultrapure water. Filter the solutions inside a cell culture hood with a 0.2 µm syringe filter or autoclave. Store at RT
- 25 mM MgCl2 solution (5x MgCl2)
- Dilute the 1 M MgCl2 stock solution to 25 mM in DNase- and RNase-free distilled water. To avoid particles and contamination, filter the solutions inside a cell culture hood with a 0.2 µm syringe filter. Store at room temperature
- For 100 ml of 1 M MgCl2 stock solution:
Dissolve 20.3 g MgCl2·6H2O in 70 ml DNase-and RNase-free water and adjust the volume to 100 ml
Store at RT
- Dilute the 1 M MgCl2 stock solution to 25 mM in DNase- and RNase-free distilled water. To avoid particles and contamination, filter the solutions inside a cell culture hood with a 0.2 µm syringe filter. Store at room temperature
- Blocking solution
DMEM or RPMI complete medium supplemented with 100 μg/ml sheared salmon sperm DNA and 1 mM dextran sulfate - 4% paraformaldehyde (PFA)
For 1 L PFA 4% preparation:- Add approximately 600 ml of 1x PBS to 40 g of PFA in a glass beaker and stir using a magnetic stirrer at ~50 °C
- Adjust the pH to between 7 and 8
- Adjust the volume to 1 L with 1x PBS, make aliquots and store at -20 °C
Note: PFA is highly toxic: use gloves, lab coat, respiratory and eye protection while handling PFA. - Add approximately 600 ml of 1x PBS to 40 g of PFA in a glass beaker and stir using a magnetic stirrer at ~50 °C
- Quenching solution
- 0.1 M glycine in 1x DPBS
Store at RT until use - To prepare 100 ml of glycine 1 M stock solution:
Dissolve 7.5 g glycine in ultrapure water
Filter it through a 0.2 µm syringe filter and store at RT
- 0.1 M glycine in 1x DPBS
- Mowiol®
- Mix 24 g glycerol, 9.6 g Mowiol® 4-88 reagent, 62.4 ml distilled water and 9.6 ml 1 M Tris buffer in a conical cylinder with a magnetic stirrer for 5-7 days
- Optionally heat the mixture at 40-50 °C to dissolve Mowiol®
- After precipitation divide the supernatant in aliquots (e.g., in 2.0 ml tubes) and store at 4 °C
- Mix 24 g glycerol, 9.6 g Mowiol® 4-88 reagent, 62.4 ml distilled water and 9.6 ml 1 M Tris buffer in a conical cylinder with a magnetic stirrer for 5-7 days
- (Optional) Buffer A
25 mM Tris-HCl, pH 8
6 M urea - (Optional) Buffer B
0.5 M NaClO4 in 25 mM Tris-HCl pH 8, 6 M urea
Acknowledgments
This protocol described here in more detail has been published in (Gomes de Castro et al., 2017). This work was supported by the Cluster of Excellence and DFG Research Center Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB).
References
- Fornasiero, E. F. and Opazo, F. (2015). Super-resolution imaging for cell biologists: concepts, applications, current challenges and developments. Bioessays 37(4): 436-451.
- Gomes de Castro, M. A., Hobartner, C. and Opazo, F. (2017). Aptamers provide superior stainings of cellular receptors studied under super-resolution microscopy. PLoS One 12(2): e0173050.
- Mikhaylova, M., Cloin, B. M., Finan, K., van den Berg, R., Teeuw, J., Kijanka, M. M., Sokolowski, M., Katrukha, E. A., Maidorn, M., Opazo, F., Moutel, S., Vantard, M., Perez, F., van Bergen en Henegouwen, P. M., Hoogenraad, C. C., Ewers, H. and Kapitein, L. C. (2015). Resolving bundled microtubules using anti-tubulin nanobodies. Nat Commun 6: 7933.
- Opazo, F., Levy, M., Byrom, M., Schafer, C., Geisler, C., Groemer, T. W., Ellington, A. D. and Rizzoli, S. O. (2012). Aptamers as potential tools for super-resolution microscopy. Nat Methods 9(10): 938-939.
- Ries, J., Kaplan, C., Platonova, E., Eghlidi, H. and Ewers, H. (2012). A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat Methods 9(6): 582-584.
- Rothbauer, U., Zolghadr, K., Tillib, S., Nowak, D., Schermelleh, L., Gahl, A., Backmann, N., Conrath, K., Muyldermans, S., Cardoso, M. C. and Leonhardt, H. (2006). Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods 3(11): 887-889.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gomes de Castro, M. A., Höbartner, C. and Opazo, F. (2017). Staining of Membrane Receptors with Fluorescently-labeled DNA Aptamers for Super-resolution Imaging. Bio-protocol 7(17): e2541. DOI: 10.21769/BioProtoc.2541.
Category
Cell Biology > Cell imaging > Fixed-cell imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.