- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection of Internal Matrix Targeting Signal-like Sequences (iMTS-Ls) in Mitochondrial Precursor Proteins Using the TargetP Prediction Tool



(*contributed equally to this work) Published: Vol 8, Iss 17, Sep 5, 2018 DOI: 10.21769/BioProtoc.2474 Views: 9595
Reviewed by: Hassan RasouliAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Mitochondria contain hundreds of proteins which are encoded by the nuclear genome and synthesized in the cytosol from where they are imported into the organelle. Sorting signals encoded in the primary and secondary sequence of these proteins mediate the recognition of newly synthesized precursor proteins and their subsequent translocation through the mitochondrial TOM and TIM translocases. Proteins of the mitochondrial matrix employ aminoterminal matrix targeting signals (MTSs), also called presequences, that are necessary and sufficient for their import into mitochondria. In most cases, these MTSs are proteolytically removed from the mature part of precursor proteins subsequent to their translocation into the matrix. Recently, internal MTS-like sequences (iMTS-Ls) were discovered in the mature region of many precursor proteins. Although these sequences are not sufficient for matrix targeting, they strongly increase the import competence of precursors by supporting their interaction with mitochondrial surface receptors. Due to their similarity to N-terminal MTSs, these iMTS-Ls can be identified using mitochondrial targeting prediction tools such as TargetP which was initially trained to recognize MTSs. In this protocol we describe how TargetP can be used to identify iMTS-Ls in protein sequences.
Keywords: Internal matrix targeting-signal like sequenceBackground
Targeting signals allow the correct intracellular distribution of newly synthesized proteins. Proteins that are transported into their target compartment in a folded conformation often show complex signals that are displayed on the surface of their native, folded structure. These signals are difficult to recognize in the primary sequence since they only can be deciphered when the fold of the native proteins is known.
In contrast, proteins that are threaded through translocases in the membrane of their target compartments in an unfolded conformation typically employ N-terminal signals. This organization allows that protein translocation can commence before translation is completed (Wickner and Schekman, 2005). N-terminal targeting sequences include the signal sequences found on secretory proteins, the transit peptides of chloroplast proteins, the MTSs of mitochondrial proteins and the leader peptides that target bacterial proteins into or across the inner membrane.
Prediction algorithms were developed that recognize and interpret these N-terminal targeting sequences (Juncker et al., 2009). These algorithms can either search for defined characteristic features (such as helicity, hydrophobicity, charge distribution or the presence of specific residues) or they are based on self-learning algorithms that were trained with test sets of proteins of known cellular localizations. One of the latter programs is TargetP which uses the primary sequence of a given protein (in FASTA or text format) to come up with a ‘probability’ score that predicts how likely it is that this sequence starts with a functional matrix targeting sequence (Emanuelsson et al., 2000; Emanuelsson et al., 2007). In the mitochondrial community, TargetP values of larger than 0.6 are typically interpreted as evidence that a given protein is targeted into the mitochondrial matrix.
Recent studies suggested that the important targeting information is not confined to the N-terminal MTS. Obviously, mature parts of proteins are critical for the translocation efficiency in vivo (Yamamoto et al., 2009; Chatzi et al., 2017; Backes et al., 2018). It is known for a long time that strongly folded domains can impede translocation (Eilers and Schatz, 1986; Harner et al., 2011; Schneider, 2018). Hence the unfolding probabilities of cytosolic domains of translocation intermediates strongly influence the velocity and yield of the translocation reaction (Wilcox et al., 2005; Yagawa et al., 2010). On the mitochondrial surface, protein unfolding is supported by cytosolic chaperones and by surface receptors, in particular by Tom70 (Hines et al., 1990; Söllner et al., 1990). The cytosolic domain of Tom70 represents a tetratricopeptide repeat structure which it shares with many co-chaperones of the Hsp90 and Hsp70 chaperone systems (Melin et al., 2015). Indeed, Tom70 cooperates with Hsp90 (in mammals) and Hsp70 (in mammals and fungi) in protein translocation across the outer membrane (Young et al., 2003; Bhangoo et al., 2007; Hoseini et al., 2016; Zanphorlin et al., 2016). It was recently shown that internal sequence stretches that share the typical biochemical properties of mitochondrial presequences support the binding to Tom70 and facilitate protein translocation into mitochondria (Backes et al., 2018, Hansen et al., 2018). Due to their similarity to mitochondrial targeting signals, these internal MTS-like sequences (iMTS-Ls) can be found using the same algorithms that are predicting mitochondrial presequences. The only necessary change one has to make is to relieve the restriction of analyzing only the N-terminus of a given protein, but iterate through the complete amino acid sequence. In this protocol, we explain how TargetP (or a similar tool) can be used to detect and analyze these iMTS-Ls protein sequences of interest. This will be helpful to analyze the features of mature regions of mitochondrial proteins and to estimate their import competence using a simple in silico test. We provide step-by-step instructions for three alternatives that contrast the different settings in data analysis. Depending on linking and scientific background, the detection of iMTS-Ls can be performed using either spreadsheet software (e.g., Microsoft Excel), a language and environment for statistical computing (R) or a general-purpose programming language (F#). While dedicated analysis platforms are the common choice in the field there is an advent for easy and intuitive general-purpose languages to facilitate the integration of the data analysis tasks into production tools and web apps or smooth the linkage of processing with high computational demand and statistical analysis.
Equipment
- Computer or laptop with Internet access
Note: It should work for all common operating systems. However, certain restrictions might apply for specific operating systems. For a standard analysis, any contemporary hardware setup (desktop or notebook) should be suited for a reasonably fast result.
Software
In order to use the scripts provided within this protocol, at least one of the following software packages has to be installed:
- Microsoft Excel or R
A spreadsheet such as Microsoft Excel, F# (https://fsharp.org/) with the libraries FSharp.Plotly and Fsharp.Stats, or R (www.r-project.org) with its packages seqinr, signal and Biostrings (Pagès et al., 2018). - TargetP 1.1 Server or TargetP package
The TargetP algorithm (Emanuelsson et al., 2007) can be used via the interface at the TargetP 1.1 Server (http://www.cbs.dtu.dk/services/TargetP/). Alternatively, the TargetP package can be downloaded and used offline. See the instructions as described here: http://www.cbs.dtu.dk/services/TargetP/instructions.php. For the procedure explained here in this protocol, the online version is sufficient and the installation of the stand-alone software package is not required. However, this stand-alone package offers the possibility to assess large data sets (for example a genome-wide analysis) which is hardly possible with the online service.
Procedure
- Generation of a TargetP profile from a given protein sequence
For each protein sequence you submit, TargetP will give you only one value indicating whether the N-terminus of the sequence looks like an MTS. In order to analyze the complete length of the protein, you have to repeatedly truncate the sequence N-terminally by one residue after the other and submit all these sequences to TargetP. We describe two ways how this can be done efficiently: On the one hand, we give detailed instructions how to use a standard spreadsheet application and online conversion tools to generate a FASTA file containing all truncations of a given sequence. On the other, we provide short scripts written in R and F# which can be used to automatically generate the required FASTA file. The latter is especially useful if you wish to analyze large numbers of sequences, e.g., for genome-wide analyses.
Using spreadsheet software such as Microsoft Excel
Using Microsoft Excel or another common spreadsheet software, this can be simply done by using the following strategy (Figure 1A):- Fill the first column of your spreadsheet with numbers 1, 2, 3, …
- Copy and paste the amino acid sequence into the first line, second column of your spreadsheet (cell B1). In case the stop codon is indicated with an asterisk (*), delete it from the sequence so that the last character is a valid amino acid.

Figure 1. Overview about the different steps in iMTS-L predictions. A. iMTS-L predictions by use of spreadsheet software such as Microsoft Excel. B. Steps used to generate a FASTA file with truncated sequences.- In the second line (cell B2), generate the truncated sequence using the following command: =MID(B1;2;LEN(B1)-1)
This creates a substring from B1 having one letter less than B1 and starting at character number 2. Note that in different versions of Excel (in particular when using a different language setting) and other spreadsheet software, the keywords and syntax of the command might be slightly different. - Click on the lower right corner of cell B2 and drag down the formula to repeatedly apply it to the second, third, … truncation until only the last residue remains in the last cell of the column.
- Highlight both cells A1 and A2 and drag down (or double click) the lower right corner of A2 to fill the first column with numbers 1, 2, 3, … indicating the starting positions of the sequences in the second column.
- Save this file in “tab delimited format” (file extension will typically be .tab or .txt).
- Submit the file you generated to a file conversion tool (e.g., HIV sequence database Format Converter or Bugaco Sequence Conversion) to generate a valid FASTA file. Choose the appropriate options, especially the input and output formats (TAB and FASTA).
- The file you receive should now have the residue numbers 1, 2, 3, … as FASTA headers for the sequentially truncated sequences.
Notes:- This file can now be submitted to TargetP online (http://www.cbs.dtu.dk/services/TargetP/).
- The output will be displayed in the browser. Copy and paste the output into a plain text editor (not Word, OpenOffice or similar!) and save it as a .txt file.
- Import the file into your spreadsheet application. In Excel, open a new spreadsheet and select “Data”, “Import from text”. Choose the appropriate options to import the different values into different columns. The mitochondrial localization score is denoted by “mTP”.
- Note that TargetP limits the size of the input to currently 2,000 sequences with at most 4,000 residues per sequence and 200,000 residues in total. These limits are violated for proteins with a length of more than 631 residues. If you wish to analyze a protein which has more residues, split the FASTA file into different files with fewer sequences and submit them individually.
Using R
The procedure described above is well suited to generate profiles for individual proteins of interest. When larger sets of sequences should be analyzed, it is more convenient to generate the required FASTA files programmatically. In the following paragraphs, we describe how this can be achieved using one of the programming languages R or F#. To use the R function provided, you need the libraries seqinr, signal and Biostrings to read and write FASTA files. To install these, run R with admin privileges and type the following commands into the shell:
source(“https://bioconductor.org/biocLite.R”)
biocLite(“Biostrings”)
biocLite(“seqinr”)
biocLite(“signal”)
To generate a FASTA file with truncated the following steps (Figure 1B):- To make the function available in your own scripts, use:
source(“path/to/the/file/truncateSeq.R”) - Load the amino acid sequence of interest into a character variable. In case you have stored it as a FASTA file, you might use the readAAStringSet function from the Biostrings package, for example:
fastaInput <- readAAStringSet(“input.fasta”)
sequence <- paste(fastaInput) - In case you have multiple sequences in your fasta file, this will create a list of all sequences.
The paste command is used to extract the sequence string from the more complex object created by readAAStringSet. - To generate a table with all truncations and write the corresponding FASTA file, use:
truncateSeq(sequence, write.fasta=TRUE, filename="result.fasta", split.fasta=0)
In case you don’t want to write a FASTA file but only generate the table within your R session, set write.table=FALSE. All other arguments will be ignored in this case.
To automatically split the sequences into several FASTA files with n sequences each, set split.fasta=n. This might be necessary for long sequences (more than 631 residues). - Submit the FASTA file(s) to TargetP and proceed as described above.
We provide two source files. The file truncateSeq.R contains the function to generate the truncated sequences. The file exampleProfile.R can be used as a template to use the procedure described above. It also contains commands to plot and smooth the profile (see Procedure B).
Using F#
F# is a modern strongly typed, cross-platform programming language that due to its functional first approach is very well suited for data analysis tasks. The development environment for every common platform and an introduction on how to get started is available from F# Software Foundation (https://fsharp.org). The following documented F# code (Figure 2) guides through the generation of a TargetP profile from given amino acid sequence(s):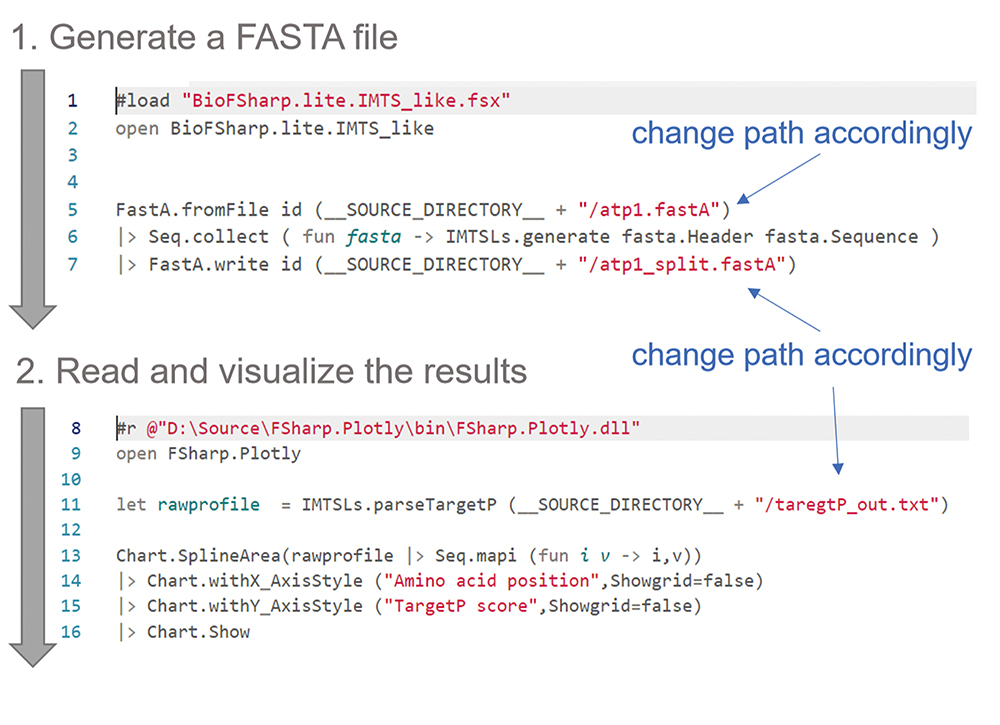
Figure 2. Overview of the workflow using F# for the iMTS-L detection. See text for details.- Load the external function to read and write FASTA files. (Change *path* accordingly)
#load "*path*/BioFSharp.lite.IMTS_like.fsx"
open BioFSharp.lite.IMTS_like - Read the amino acid sequence(s) of interested from a provided file in the fastA format. (*path/input.fastA* needs to be replaced by the filename and path pointing to your target fastA file)
FastA.fromFile id "*path/input.fastA*" - Generate all suffixes sequence of the given amino acid sequence(s).
|> Seq.collect (fun fasta -> IMTSLs.generate fasta.Header fasta.Sequence) - Write all suffixes sequence into the output file in fastA format.(Change *path/output.fastA*)
|> FastA.write id "*path/output.fastA*" - The output file can now be submitted to TargetP online. (http://www.cbs.dtu.dk/services/TargetP/).
- Copy and paste the output into a text file as described above.
- Load the FSharp.Plotly library.(Change *path*)
#r "*path*/FSharp.Plotly.dll*"
open FSharp.Plotly - Read the TargetP scores from the text file.(Change *path/ targetP_out.txt *)
let rawprofile = parseTargetP "*path/targetP_out.txt*" - Generate an area chart showing the sequence position versus the raw TargetP scores.
Chart.SplineArea(rawprofile |> Seq.mapi (fun i v -> i,v))
|> Chart.withX_AxisStyle "Amino acid position"
|> Chart.withY_AxisStyle "TargetP score"
|> Chart.Show
The source file BioFSharp.lite.IMTS_like.fsx summarizes the described procedure and also contains the commands to smooth the profile (see Procedure B). - Smoothing of the TargetP profile to make the iMTS-Ls better visible
Due to the original design of the TargetP algorithm, it calculates the score as a target sequence likelihood under consideration of the N-terminal sequence properties within a fixed windows size. Consequently, removing each first amino acid residue and moving the window along the given sequence leads to fluctuations in the likelihood. We therefore recommend smoothening the raw score to allow a quantitative measure and improve the accuracy to localize the iMTS-Ls (Figure 3B).
While smoothening can be done with many different methods, we can recommend a first order Savitzky-Golay filter (Savitzky and Golay, 1964) with filter length 21 (which is the average length of an MTS).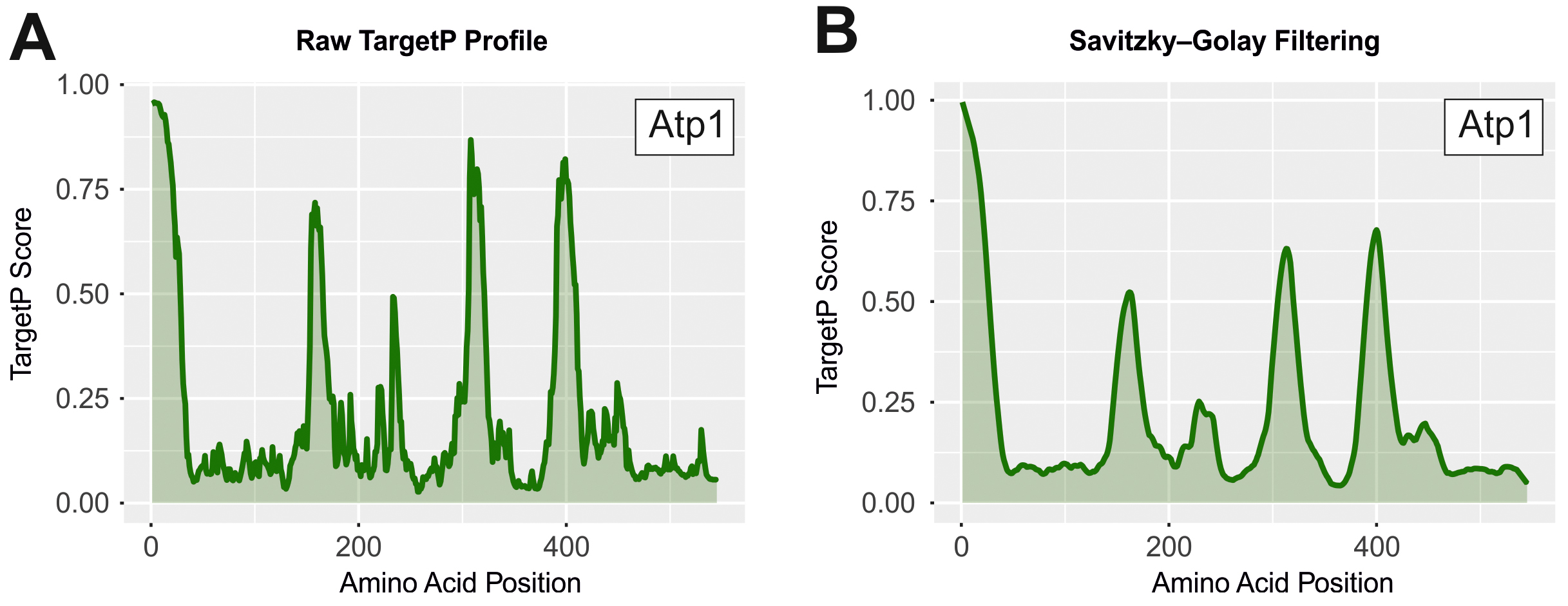
Figure 3. TargetP scores of the model protein Atp1 from S. cerevisiae as examples. Shown are the raw targetP profiles before (A) and after smoothing (B). High TargetP values correlate with iMTS-L sequences which are well visible in both cases.
Using R
The Savitzky-Golay filtering function is included in the R package signal.- Load the signal package library(signal).
- Read the TargetP scores from your output file.
data <- read.csv(“output.fasta”)
You will probably need to specify the options of the read.csv command according to the exact format of your output.fasta file (sep, dec, header and so on). - Apply the Savitzky-Golay filtering. The following command adds a column with the smoothed values to the data frame.
data$smoothed <- sgolayfilt(data$mTP, p=1, n=21, m=0, ts=0)
The Savitzky–Golay filtering function is available in the FSharp.Stats library available from Github or as Nuget Package ("dotnet add package FSharp.Stats").- Load the FSharp.Stats library. (Change *path*)
#r "*path*/FSharp.Stats.dll*"
open FSharp.Stats - Smooth the raw profile using Savitzky–Golay filtering.
let profile = rawprofile |> Signal.Filtering.savitzky_golay 21 1 0 0 - Generate an area chart showing the sequence position versus the TargetP profile scores.
Chart.SplineArea(profile |> Seq.mapi (fun i v -> i,v))
|> Chart.withX_AxisStyle "Amino acid position"
|> Chart.withY_AxisStyle "TargetP profile score"
|> Chart.Show
Data analysis
The profiles generated with one of the procedures described above can readily be used to draw conclusions about the presence and position of putative iMTS-L sequences. It should be noted that these are predictions that typically should be addressed experimentally, for example by mutating the identified stretches (Backes et al., 2018).
To compare larger sets of proteins, the iMTS-L profiles can be used to calculate statistical measures. For example, a measure of quantification of iMTS-L content in a protein, the iMTS-L propensity, was introduced in Backes et al. (2018). It can be used to summarize iMTS-L information from a complete profile in a single number.
Notes
- It is important to always select appropriate options when submitting the sequences to TargetP. Especially the organism (plant/non-plant) can affect the profiles considerably.
- The approach described in this protocol is not limited to the use of TargetP. It is straightforward to adapt the method for other prediction algorithms, such as MitoFates (Fukasawa et al., 2015) or PSort (Horton et al., 2007). We did not test these, but they might serve as complementary predictions to identify “high confidence iMTS-Ls”. Of course, also prediction programs for features other than mitochondrial presequences can be used. TargetP also offers scores for signal sequences or plastidal transit peptides.
- While iMTS-Ls are reported to serve as Tom70 binding sites in mitochondrial precursors (Backes et al., 2018), they also occur in non-mitochondrial proteins. Given that mitochondrial presequences typically constitute good binding sites for chaperones such as Hsp70 (Okamoto et al., 2002), the detection of iMTS-Ls might also be interesting for the study of protein-protein interactions, especially protein-chaperone interactions.
Acknowledgments
The potential of TargetP to identify internal targeting sequences was initially recognized by Michael W. Wöllhaf (Woellhaf et al., 2016). By experimental lab work, Sandra Backes, Steffen Hess and Sabrina Gödel verified that the internal TargetP profiles are reliable indicators to predict the import competence of mitochondrial precursor proteins (Backes et al., 2018). This work was supported by grants from the Deutsche Forschungsgemeinschaft (He2803/9-1, DIP Mito-Balance and IRTG 1830 to JMH) and the Landesschwerpunkt BioComp.
Competing interests
The authors declare that they have no competing interests.
References
- Backes, S., Hess, S., Boos, F., Woellhaf, M. W., Godel, S., Jung, M., Muhlhaus, T. and Herrmann, J. M. (2018). Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J Cell Biol 217(4): 1369-1382.
- Bhangoo, M. K., Tzankov, S., Fan, A. C., Dejgaard, K., Thomas, D. Y. and Young, J. C. (2007). Multiple 40-kDa heat-shock protein chaperones function in Tom70-dependent mitochondrial import. Mol Biol Cell 18(9): 3414-3428.
- Chatzi, K. E., Sardis, M. F., Tsirigotaki, A., Koukaki, M., Sostaric, N., Konijnenberg, A., Sobott, F., Kalodimos, C. G., Karamanou, S. and Economou, A. (2017). Preprotein mature domains contain translocase targeting signals that are essential for secretion. J Cell Biol 216(5): 1357-1369.
- Eilers, M. and Schatz, G. (1986). Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature 322(6076): 228-232.
- Emanuelsson, O., Brunak, S., von Heijne, G. and Nielsen, H. (2007). Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2(4): 953-971.
- Emanuelsson, O., Nielsen, H., Brunak, S. and von Heijne, G. (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300(4): 1005-1016.
- Fukasawa, Y., Tsuji, J., Fu, S. C., Tomii, K., Horton, P. and Imai, K. (2015). MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics 14(4): 1113-1126.
- Hansen, K. G., Boos, F. and Herrmann, J. M. (2018). Accessory signals in protein translocation. Aging (Albany NY) 10(4): 530-531.
- Harner, M., Neupert, W. and Deponte, M. (2011). Lateral release of proteins from the TOM complex into the outer membrane of mitochondria. EMBO J 30(16): 3232-3241.
- Hines, V., Brandt, A., Griffiths, G., Horstmann, H., Brutsch, H. and Schatz, G. (1990). Protein import into yeast mitochondria is accelerated by the outer membrane protein MAS70. EMBO J 9(10): 3191-3200.
- Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J. and Nakai, K. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res 35(Web Server issue): W585-587.
- Hoseini, H., Pandey, S., Jores, T., Schmitt, A., Franz-Wachtel, M., Macek, B., Buchner, J., Dimmer, K. S. and Rapaport, D. (2016). The cytosolic cochaperone Sti1 is relevant for mitochondrial biogenesis and morphology. FEBS J 283(18): 3338-3352.
- Juncker, A. S., Jensen, L. J., Pierleoni, A., Bernsel, A., Tress, M. L., Bork, P., von Heijne, G., Valencia, A., Ouzounis, C. A., Casadio, R. and Brunak, S. (2009). Sequence-based feature prediction and annotation of proteins. Genome Biol 10(2): 206.
- Melin, J., Kilisch, M., Neumann, P., Lytovchenko, O., Gomkale, R., Schendzielorz, A., Schmidt, B., Liepold, T., Ficner, R., Jahn, O., Rehling, P. and Schulz, C. (2015). A presequence-binding groove in Tom70 supports import of Mdl1 into mitochondria. Biochim Biophys Acta 1853(8): 1850-1859.
- Okamoto, K., Brinker, A., Paschen, S. A., Moarefi, I., Hayer-Hartl, M., Neupert, W. and Brunner, M. (2002). The protein import motor of mitochondria: a targeted molecular ratchet driving unfolding and translocation. EMBO J 21(14): 3659-3671.
- Pagès, H., Aboyoun, P., Gentleman, R. and DebRoy, S. (2018). Biostrings: Efficient manipulation of biological strings. R package version 2.48.0.
- Savitzky, A., and Golay, M. J. E. (1964). Smoothing and differentiation of data by simplified least squares procedures. Anal Chem 36(8): 1627-1639.
- Schneider, A. (2018). Dihydrofolate reductase and membrane translocation: evolution of a classic experiment: Classic landmark papers, irrespective of their age, can teach students how best science is practiced and inspire new experiments. EMBO Rep 19(3).
- Söllner, T., Pfaller, R., Griffiths, G., Pfanner, N. and Neupert, W. (1990). A mitochondrial import receptor for the ADP/ATP carrier. Cell 62(1): 107-115.
- Wickner, W. and Schekman, R. (2005). Protein translocation across biological membranes. Science 310(5753): 1452-1456.
- Wilcox, A. J., Choy, J., Bustamante, C. and Matouschek, A. (2005). Effect of protein structure on mitochondrial import. Proc Natl Acad Sci U S A 102(43): 15435-15440.
- Woellhaf, M. W., Sommer, F., Schroda, M. and Herrmann, J. M. (2016). Proteomic profiling of the mitochondrial ribosome identifies Atp25 as a composite mitochondrial precursor protein. Mol Biol Cell 27(20): 3031-3039.
- Yagawa, K., Yamano, K., Oguro, T., Maeda, M., Sato, T., Momose, T., Kawano, S. and Endo, T. (2010). Structural basis for unfolding pathway-dependent stability of proteins: vectorial unfolding versus global unfolding. Protein Sci 19(4): 693-702.
- Yamamoto, H., Fukui, K., Takahashi, H., Kitamura, S., Shiota, T., Terao, K., Uchida, M., Esaki, M., Nishikawa, S., Yoshihisa, T., Yamano, K. and Endo, T. (2009). Roles of Tom70 in import of presequence-containing mitochondrial proteins. J Biol Chem 284(46): 31635-31646.
- Young, J. C., Hoogenraad, N. J. and Hartl, F. U. (2003). Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112(1): 41-50.
- Zanphorlin, L. M., Lima, T. B., Wong, M. J., Balbuena, T. S., Minetti, C. A., Remeta, D. P., Young, J. C., Barbosa, L. R., Gozzo, F. C. and Ramos, C. H. (2016). Heat shock protein 90 kDa (Hsp90) has a second functional interaction site with the mitochondrial import receptor Tom70. J Biol Chem 291(36): 18620-18631.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Boos, F., Mühlhaus, T. and Herrmann, J. M. (2018). Detection of Internal Matrix Targeting Signal-like Sequences (iMTS-Ls) in Mitochondrial Precursor Proteins Using the TargetP Prediction Tool. Bio-protocol 8(17): e2474. DOI: 10.21769/BioProtoc.2474.
- Backes, S., Hess, S., Boos, F., Woellhaf, M. W., Godel, S., Jung, M., Muhlhaus, T. and Herrmann, J. M. (2018). Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J Cell Biol 217(4): 1369-1382.
Category
Biochemistry > Protein > Structure
Cell Biology > Organelle isolation > Mitochondria
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.