- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Image-Based Analysis of Mitochondrial Area and Counting from Adult Mouse Dopaminergic Neurites
Published: Vol 8, Iss 16, Aug 20, 2018 DOI: 10.21769/BioProtoc.2471 Views: 9590
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2018
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Mitochondria form dynamic cytoplasmic networks which undergo morphological changes in order to adapt to cellular stresses and signals. These changes can include alterations in size and number within a given cell. Analysis of the whole network can be a useful metric to assess overall mitochondrial health, particularly in neurons, which are highly sensitive to mitochondrial dysfunction. Here we describe a method which combines immunofluorescence and computerized image analysis to measure mitochondrial morphology (quantification of number, density, and area) in dopaminergic neurites of mice expressing mitochondrially-targeted eYFP.
Keywords: Neurite mitochondriaBackground
Mitochondria are double-membraned organelles present in essentially all the cells of every complex organism. Their main function is to supply the majority of cellular energy as ATP, but they also play roles in apoptosis, buffering intracellular Ca2+, reactive oxygen species production, and regulation of membrane potential (Neupert and Herrmann, 2007; Hamanaka and Chandel, 2010; Shutt and McBride, 2013).
These organelles, often depicted as single “bean-like” structures, are in reality components of a dynamic cytoplasmic network. They can undergo major morphological changes regulated by dynamic processes of membrane fusion and fission, a process believed to be involved in the elimination of dysfunctional organelles through a process called mitophagy. The mitochondrial network can also increase as a response to high cellular energy demand (Sheng, 2017; Devine and Kittler, 2018). The morphology of the mitochondrial network can be altered in response to different stressors, and there is a wide range of possible morphologies which are cell-type, or even cell-compartment dependent (Picard et al., 2013). The localization of mitochondria in the dendrites and axons of neurons, in particular, play a crucial role as they provide, in these specific cells, the energy necessary for synaptic transmission (Chang and Reynolds, 2006; Misgeld and Schwarz, 2017).
Mitochondria localization and dynamics in one particular group of cells, the dopaminergic neurons in the substantia nigra, have been extensively studied during the last decades. These neurons’ projections reach the striatum and their loss causes a depletion of striatal dopamine which is the cause of the classical motor symptoms of Parkinson’s disease (PD) (Dauer and Przedborski, 2003; Braak et al., 2004).
The involvement of mitochondrial dysfunctions in these neurons has been investigated since the 80’s (Kopin and Markey, 1988), but the study of mitochondrial dynamics attracted particular interest especially since the discovery that genes mutated in monogenic forms of Parkinson’s disease (in particular Parkin and PINK1) have an essential role in mitochondrial fission/fusion, mitochondrial transport and mitophagy (Koh and Chung, 2010; Narendra and Youle, 2011).
We recently investigated the consequences of the loss of Parkin in a mouse model of PD in which degeneration of dopaminergic neurons was caused by mtDNA depletion and mitochondrial dysfunction (Pinto et al., 2018). In this context, we also found that lack of Parkin affected mitochondrial morphology in dopaminergic axons.
A challenge in studying the mitochondria morphology in vivo is the clear visualization of the organelles and in discerning the ones present in the axons. To study mitochondrial morphology in dopaminergic neurons, we used immunofluorescence microscopy on mice specifically expressing eYFP in the mitochondria of dopaminergic neurons, as well as computerized image analysis software to study mitochondrial number, density and size, as a measure of mitochondrial health (Pinto et al., 2018).
Materials and Reagents
- 500 ml Vacuum Filter (Corning, catalog number: 431097 )
- Single edge razor blades (VWR, catalog number: 55411-050 )
- 24-well Tissue Culture Plate (VWR, catalog number: 10062-896 )
- Microscope Cover Glasses (VWR, catalog number: 16004-098 )
- Micro Slides, Superfrost Plus (VWR, catalog number: 48311-703 )
- Syringes (Insulin Syringes) (BD, catalog number: 328468 )
- Mouse strains: males mito-eYFP DAT-tTA were analyzed at 4 months of age
- Super glue (Henkel, Loctite, catalog number: 1399967 )
- Ketamine (Ketamine HCl, Hospira, catalog number: 02051-05 )
- Xylazine (AnaSed, NDC 59599-110-20)
- Reagent Alcohol (Sigma-Aldrich, catalog number: 793213 )
- Paraformaldehyde (Sigma-Aldrich, catalog number: 441244 )
- 10x PBS (Merck, Calbiochem, catalog number: 6505-4L )
- Hard Set Fluorescent Mounting Medium (Vector Laboratories, catalog number: H-1400 )
- Sodium azide (Sigma-Aldrich, catalog number: S8032 )
- 1x PBS (see Recipes)
- 4% Paraformaldehyde (see Recipes)
- 0.02% (w/v) Sodium azide (see Recipes)
Equipment
- Perfusion machine (Gilson, model: MINIPULS® 3 )
- 23 G Surshield Safety Winged Infusion Set (Terumo Medical, catalog number: SV*S23BL )
- Blunt scissors (Thermo Fisher Scientific, catalog number: 78702 )
- Sharp scissors (Fisher Scientific, catalog number: 13-808-2 )
- Forceps (Fisher Scientific, catalog number: 13-812-39 )
- Metal Spatula (Fisher Scientific, catalog number: 14-374 )
- Acrylic Brain Matrix for Adult Mouse, Coronal Slices, 1 mm spacing (World Precision Instruments, catalog number: RBMA-200C )
- Vibratome (Leica Biosystems, model: Leica VT1000 S )
- Confocal Microscope (ZEISS, Laser Scanning Microscope LSM 710 Observer Z1)
Software
- Zen Software Package (ZEN 2010B SP1, Version 6,0,0,485, Configuration 5.00.01)
- FIJI (FIJI Is Just ImageJ) (Version 2.0.0–rc–67/1.52d)
Notes:- It is a distribution package of ImageJ with plugins for image analysis
- It is available for free download here (https://imagej.net/Fiji/Downloads)
- Microsoft® Excel
Procedure
- Generation of mito-eYFP DAT-tTA mice
The mito-eYFP DAT-tTA mice are generated by crossing mice harboring one allele of the dopamine transporter promoter-driven tetracycline-transactivator with mice harboring one allele of the CMV-tetracycline response element driven mitochondrial-targeted eYFP. We genotyped the offspring following Jackson laboratory protocols and primer sequences. In this study, we used 4 months old males but we do not foresee limitations in using different ages and both males and females. Mice were continuously fed with normal food (no DOX). Because DAT is a protein specifically expressed by dopaminergic neurons, mice expressed eYFP exclusively in the mitochondria of dopaminergic neurons (Figure 1). Mice were sacrificed at 4 months.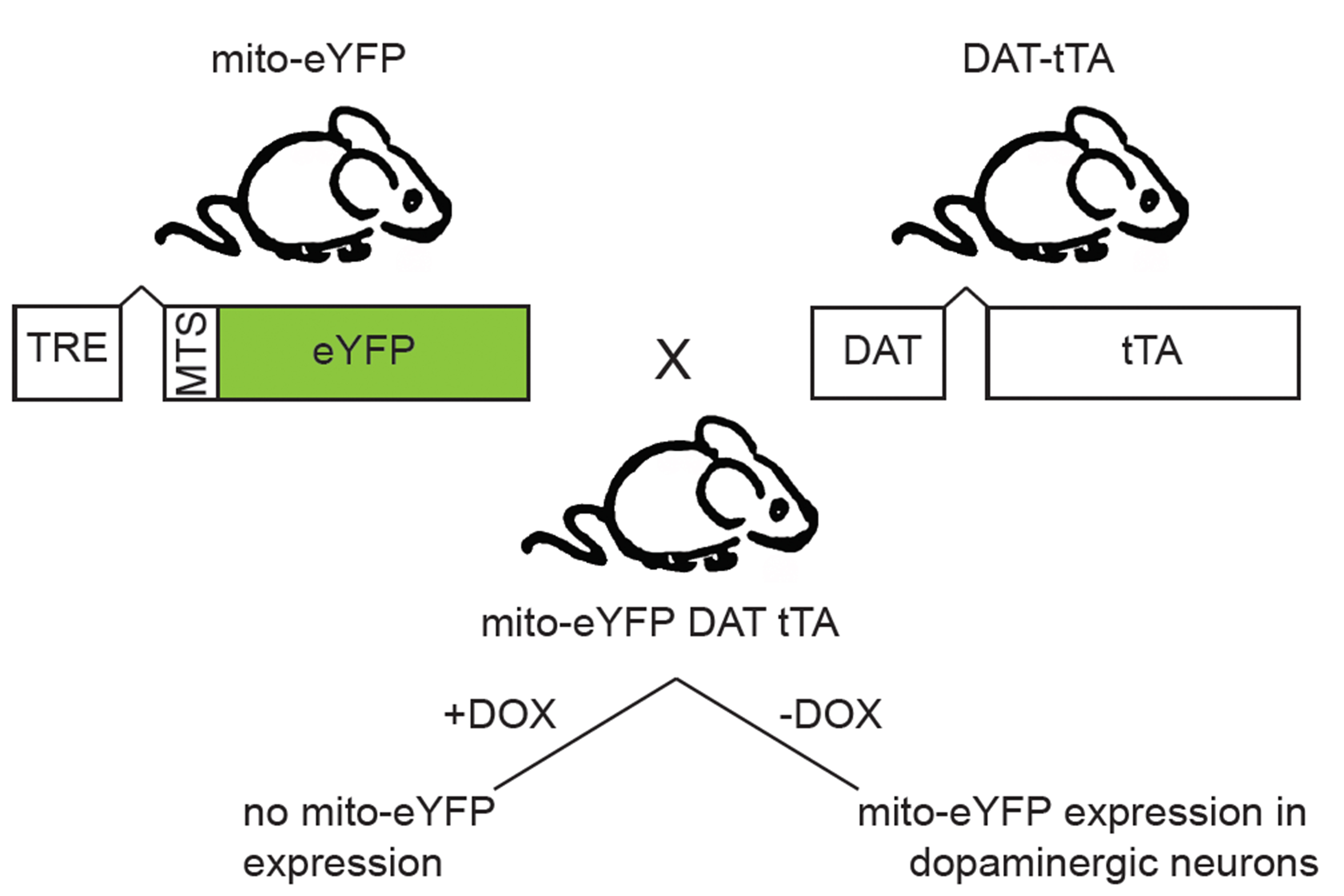
Figure 1. Breeding strategy to express mito-eYFP specifically in dopaminergic neurons. This system involves the induction of mito-eYFP expression when tetracycline/doxycycline are removed from the diet of the double-transgenic mice. - Transcardial perfusion and whole brain fixation (Gage et al., 2012)
- Anesthetize Mito-eYFP DAT-tTA mice with an intraperitoneal injection of Ketamine (90-120 mg/kg) and Xylazine (5-10 mg/kg) mixed together starting from lower dose (90 mg/kg of Ketamine and 5 mg/kg of Xylazine). If the mouse is still responsive after 5 min, increase the dose (usually until 120 mg/kg of Ketamine and 10 mg/kg of Xylazine). Check that the mice no longer exhibit reflex movement (pinch the paws and tail with a tweezer until it does not react to the stimulus) and place them on their back. Spray 70% ethanol on the fur to prevent it from sticking to the exposed tissues or the tools.
- Using blunt scissors, cut open the abdomen, identify the diaphragm, pinch it until the lungs collapse. Cut the diaphragm and the rib cage exposing the heart.
- Holding the heart with forceps, insert a 23 G needle perfusing ice-cold PBS into the left ventricle and snip the right atrium using small, sharp scissors. The PBS perfusion continues at 7 ml/minute for at least 5 min, until the liver appears clear from blood.
- Switch the perfusion solution to ice-cold 4% PFA in PBS (Twenty milliliter per mouse or until the mouse is properly perfused and the body is mostly rigid).
- Open the skull using scissors and forceps, and carefully remove the brain using a metal spatula.
- Substantia nigra is located in the midbrain. To isolate the midbrain section containing the substantia nigra, position the mouse brain on the matrix and with two single edge razor blades, cut the brain at Bregma 0 mm and Bregma -0.5 mm (coronal direction). Incubate the midbrain section in 4% PFA in PBS overnight at 4 °C.
- Brain slices preparation
- Glue the PFA-fixed brain (Bregma 0 mm) to the plate of the vibratome (Leica VT1000 S) and submerge the brain in ice-cold PBS.
- Cut 50 μm thick coronal brain sections. The slices will float in the PBS, so with a small brush they can be fished and collected. Cut slices approximately from Bregma -2.46 mm to Bregma -4.04 mm (total of about 30 slices).
The settings for Leica VT1000 S vibratome are as follows:- Speed set 3
Note: Max speed set on Leica VT1000 S vibratome is 10. - Frequency set 7
Note: Max frequency set on Leica VT1000 S vibratome is 10.
- Speed set 3
- Collect three slices per well into a 24-well plate containing PBS + Sodium azide (0.02% w/v). For the counting, analyze one slice per well for a total of 10 slices per mouse.
- If you choose to perform immunohistochemistry on slices, it can be done at this stage. Otherwise, using a small brush, carefully transfer slices on a glass slide, put 20 μl of a hard-set fluorescent mounting media on the slice, and cover the slice with a glass coverslip.
Notes:
- Counterstaining slices with an anti-TH antibody can be done to confirm the specificity of the mito-eYFP DAT-tTA expression.
- Slices can be stored at 4 °C in PBS protected from light, although we suggest not to exceed 14 days before the analysis as the EYFP fluorescence intensity might decrease over time.
- Imaging of brain slices
- From the 10 mounted slices, choose 5 with the clearer staining (enough and distinguishable neurites). Using a Zeiss laser scanning confocal microscope (Zeiss Laser Scanning Microscope, LSM 710, Observer Z1), and associated ZEN software package (ZEN 2010B SP1, Version 6,0,0,485, Configuration 5.00.01) take Z-Stack images from the midbrain region at 20x magnification.
- Take each Z-Stack using a minimum of 10 steps. Save files with a “.lsm” extension for image analysis. Use the same settings (pinhole, intensity, contrast) for all the Z-stacks.
- In our study, the settings used for image acquisition on the Zeiss LSM 710 were:
Laser: Type Argon, Excitation 480-515 nm, Emission 500-545 nm, Power 5.00, Gain 700, Digital Offset 0.0, Digital Gain 1.0
Image Size: 1024 x 1024 pixels
Pinhole: 89.9 μm
Exposure Time: Pixel Dwell 50.42 μsec, Scan Time 1 min 2 sec
- Quantification of mitochondrial size and neurite length
Quantification is done using FIJI Software Package, and Figures 2-9 show step by step image processing in the immunofluorescent images leading to the quantification of mitochondrial size and neurite length.- Open .lsm files with the FIJI Software package.
- OPTIONAL (if immunohistochemistry has been performed with additional channels). To separate the channels and only keep the Mito-eYFP information, click on the “Image” drop-down menu: Color → Split channels → Close all channels except for the mito-eYFP channel (Figure 2).
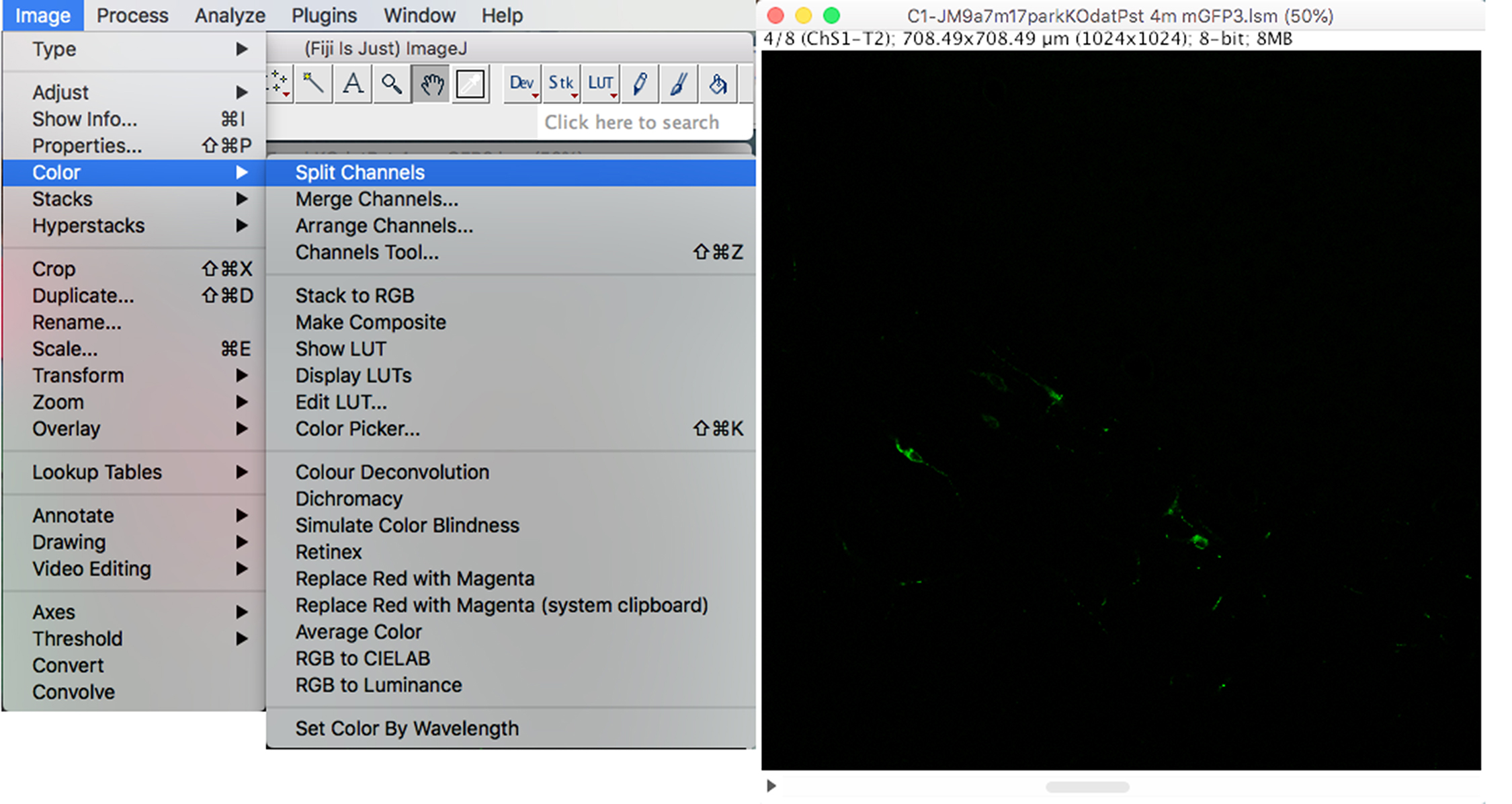
Figure 2. Retention of mito-eYFP channel - To flatten the Z-Stack and create a maximum intensity image, click on the “Image” drop-down menu: Stacks → Z Project → Max Intensity (Figure 3).
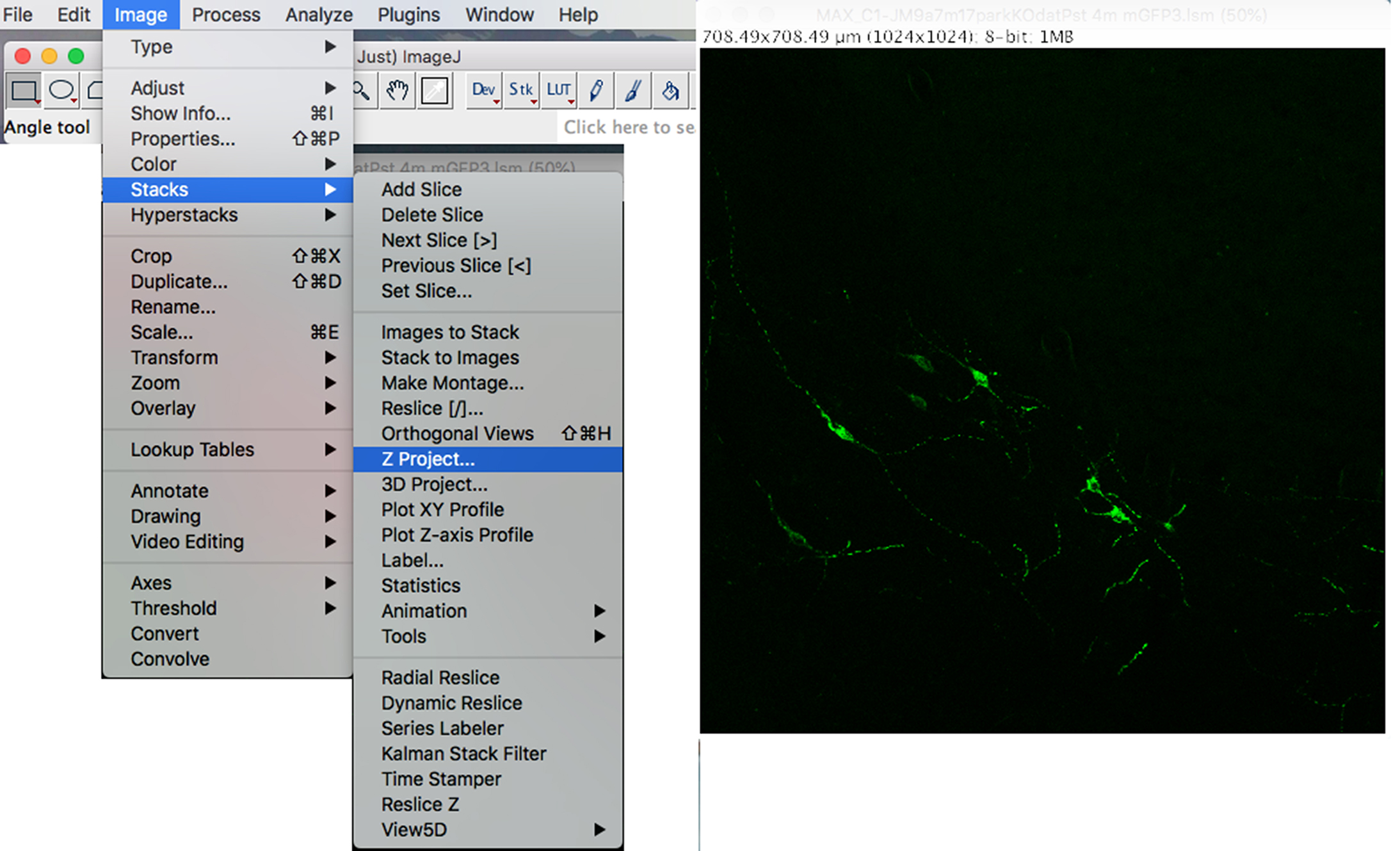
Figure 3. Maximum projection of Z-stack
- To convert the image to an 8-bit image for analysis, click on the “Image” drop-down menu: Type → 8 bit (Figure 4).
Note: Converting to the image to binary instead of 8-bit would create too much background.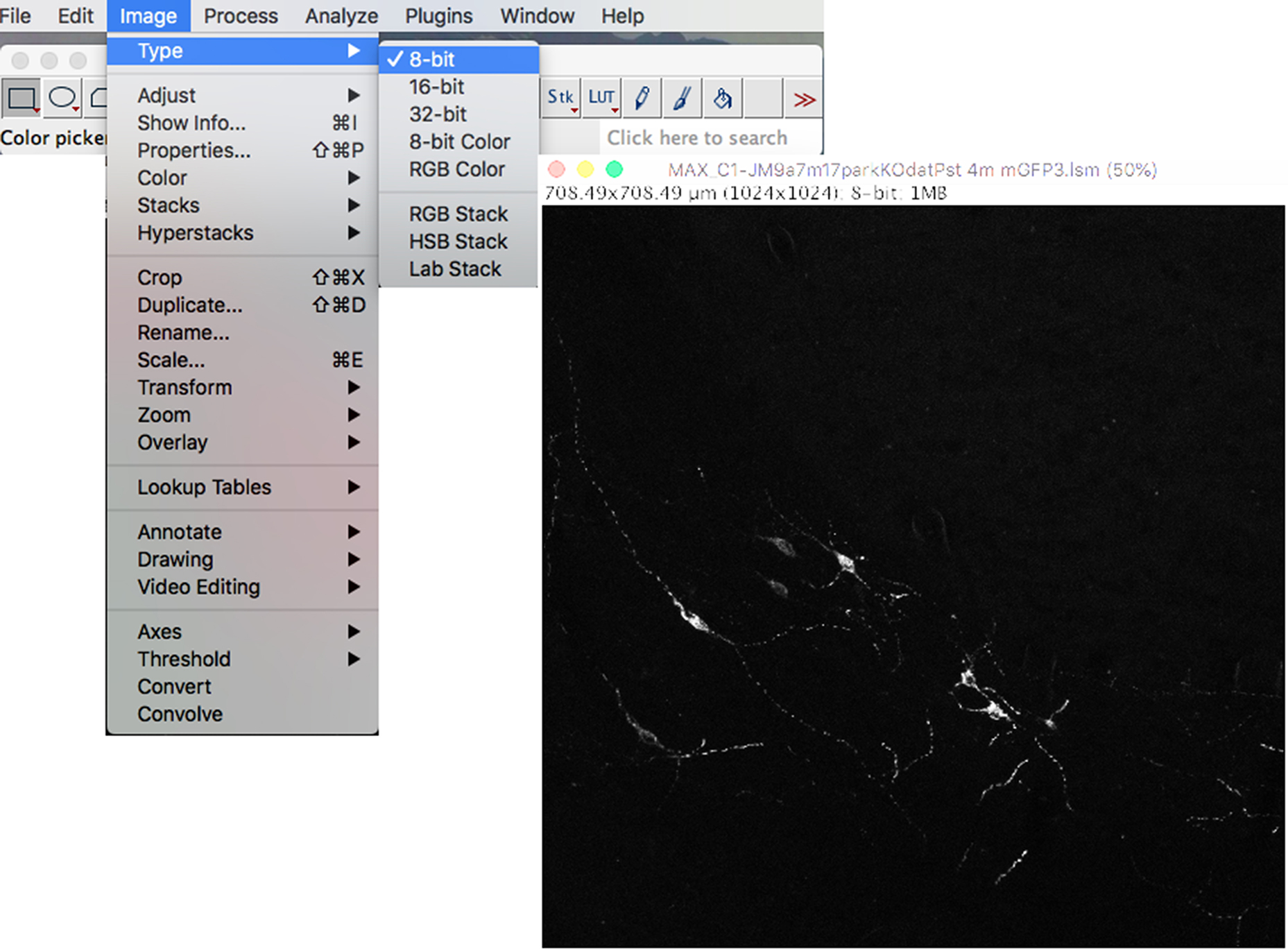
Figure 4. Conversion to 8-bit type
- To invert the colors of the image, click on the “Edit” drop-down menu: Invert (Figure 5).
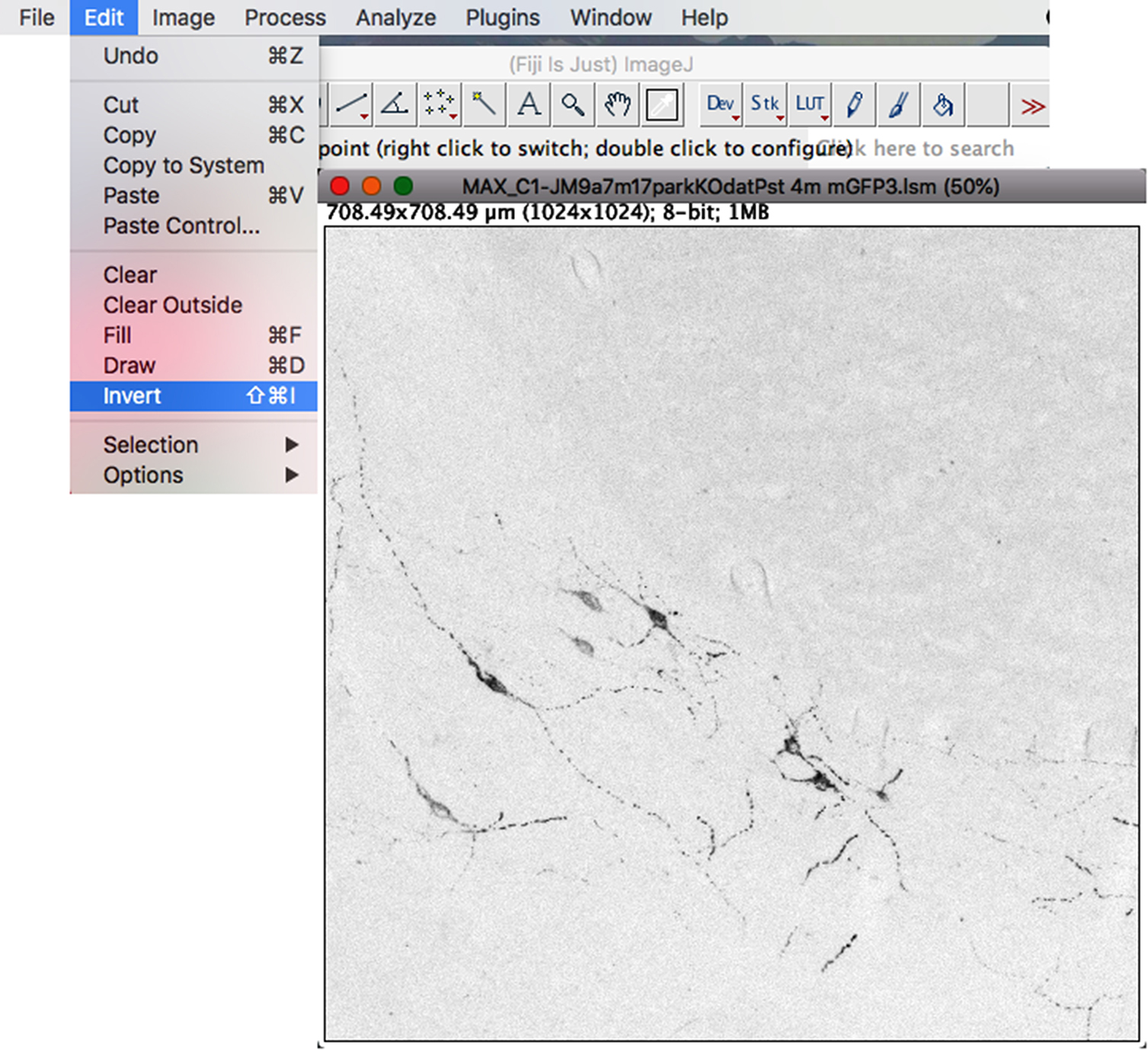
Figure 5. Inverting channel colors
- To better separate the background from the actual image in order to see the full neurite but not oversaturate the pixel intensity, the image threshold must be adjusted. In 8 bit greyscale images, there are 256 (28) intensity graduations which can be assigned to a pixel. A pixel with an intensity of 0 is black, a pixel with a value of 255 is white, everything in between is a shade of grey.
To set the threshold for image analysis, click on the “Image” drop-down menu: click on “Adjust” and go to “Threshold” (Step 1 in Figure 6). In the window that will appear, the histogram represents the distribution of pixel intensities in the image, from 0 (black) to 255 (white). Set the threshold from 0 to ~240, and the pixels in the image which fall in the selected range will be highlighted in red (Step 2 in Figure 6). When satisfied with the choice of the threshold, click “Apply” (Step 3 in Figure 6).
Notes:
- Between 230 and 256 there will be the light grey and white pixels belonging to the background. The setting of the threshold will be image specific, but between images taken with the same parameters (pinhole, contrast, intensity), we experienced differences in maximum 5 intensity graduations. If the threshold is set too high, red pixels will appear in the background, in that case, decrease the threshold until pixels in the neurites and in the cell body will be highlighted in red.
- The transgenic mouse used for this protocol has been extensively studied and the specificity of mitochondrial localization has been previously assessed (Chandrasekaran et al., 2006). Therefore we considered mitochondria every particle localized in the neurite that has a pixel value >1 and < of the threshold indicated.
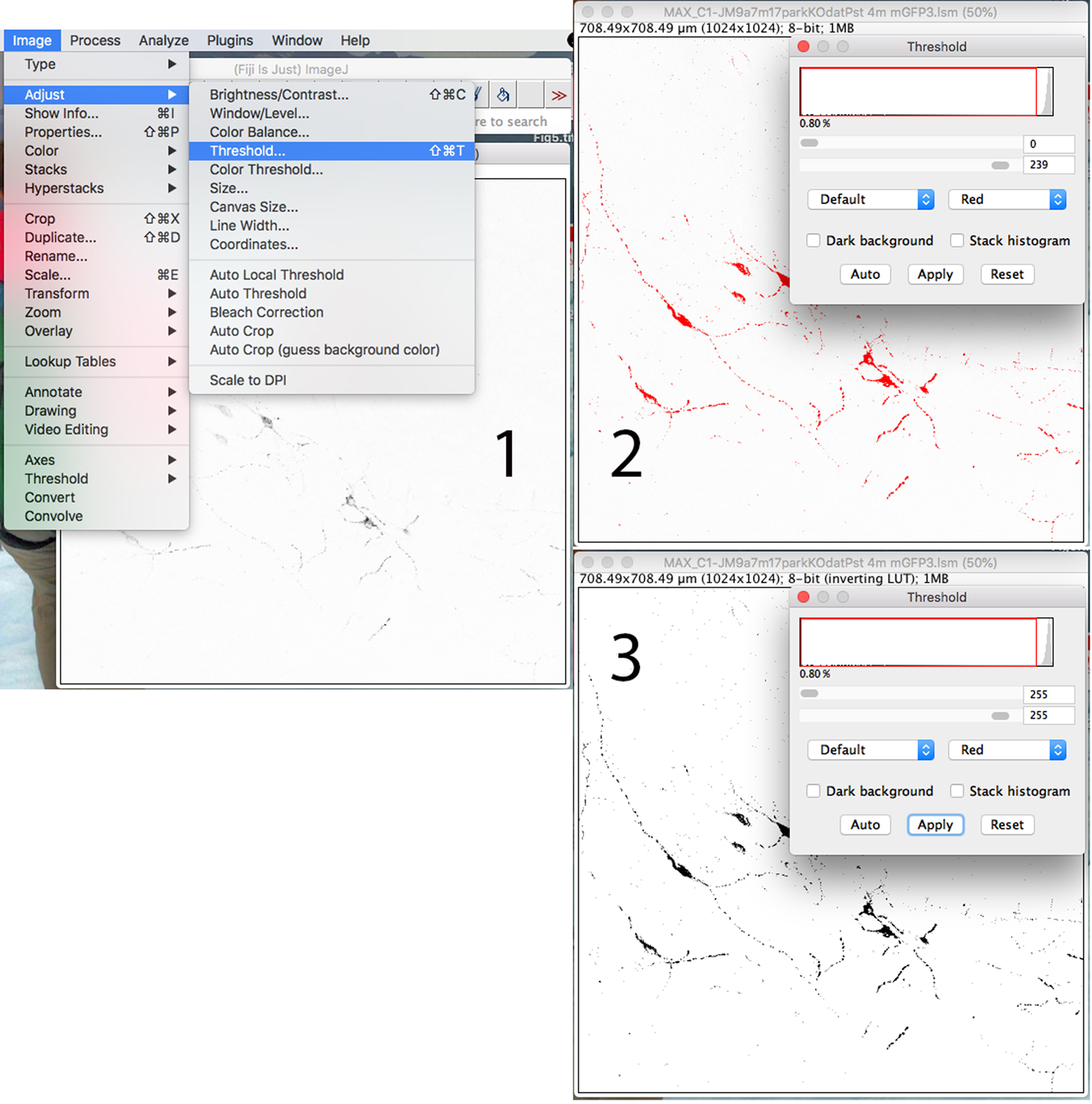
Figure 6. Adjusting channel threshold to see the axons and neurites better
- To analyze the mitochondria in a neurite, select “Freehand Selection” and trace the entire length of the neurite to be analyzed. When tracing the neurite to be analyzed, the line does not need to be “tight” along the neurite, but must not include mitochondria from other neurites (Figure 7).
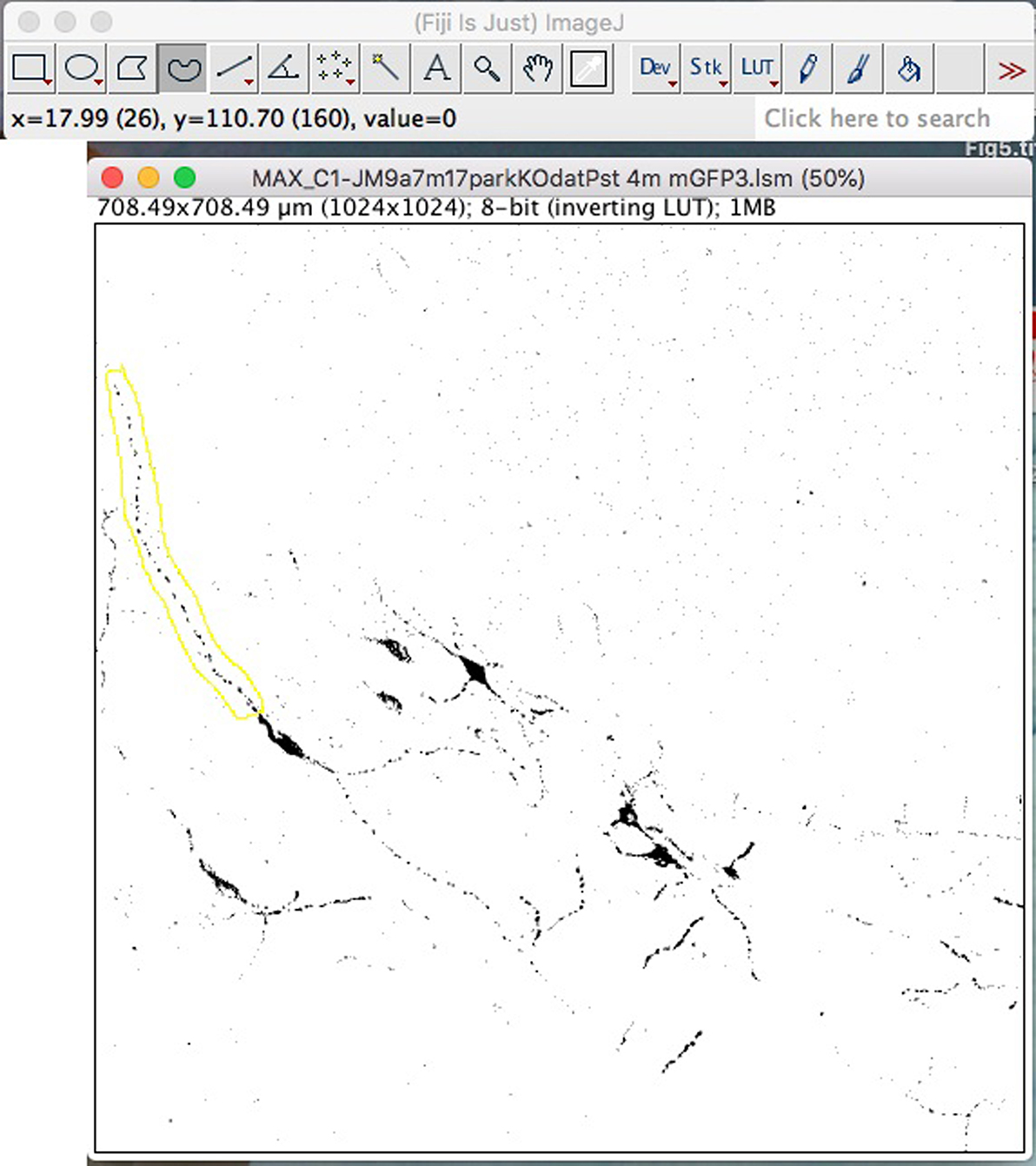
Figure 7. Freehand selection to trace axons/neurites for analyses
- To analyze the mitochondria in the selected neurite, click on the “Analyze” drop-down menu: Analyze particles → Set from “1 to Infinity” → Check “Display results” Click OK → Copy the first two columns of the results to Excel (Figure 8).
Note: The typical size of mitochondria is very dependent on the cell type/compartment analyzed. With our microscope settings and image size, the mean size for mitochondria is 4.1 ± 0.8 pixels (1.95 μm2), matching what is commonly reported across species (0.75 to 3 μm2) (Bereiter-Hahn et al., 1990; Wiemerslage and Lee, 2016; Xu et al., 2016).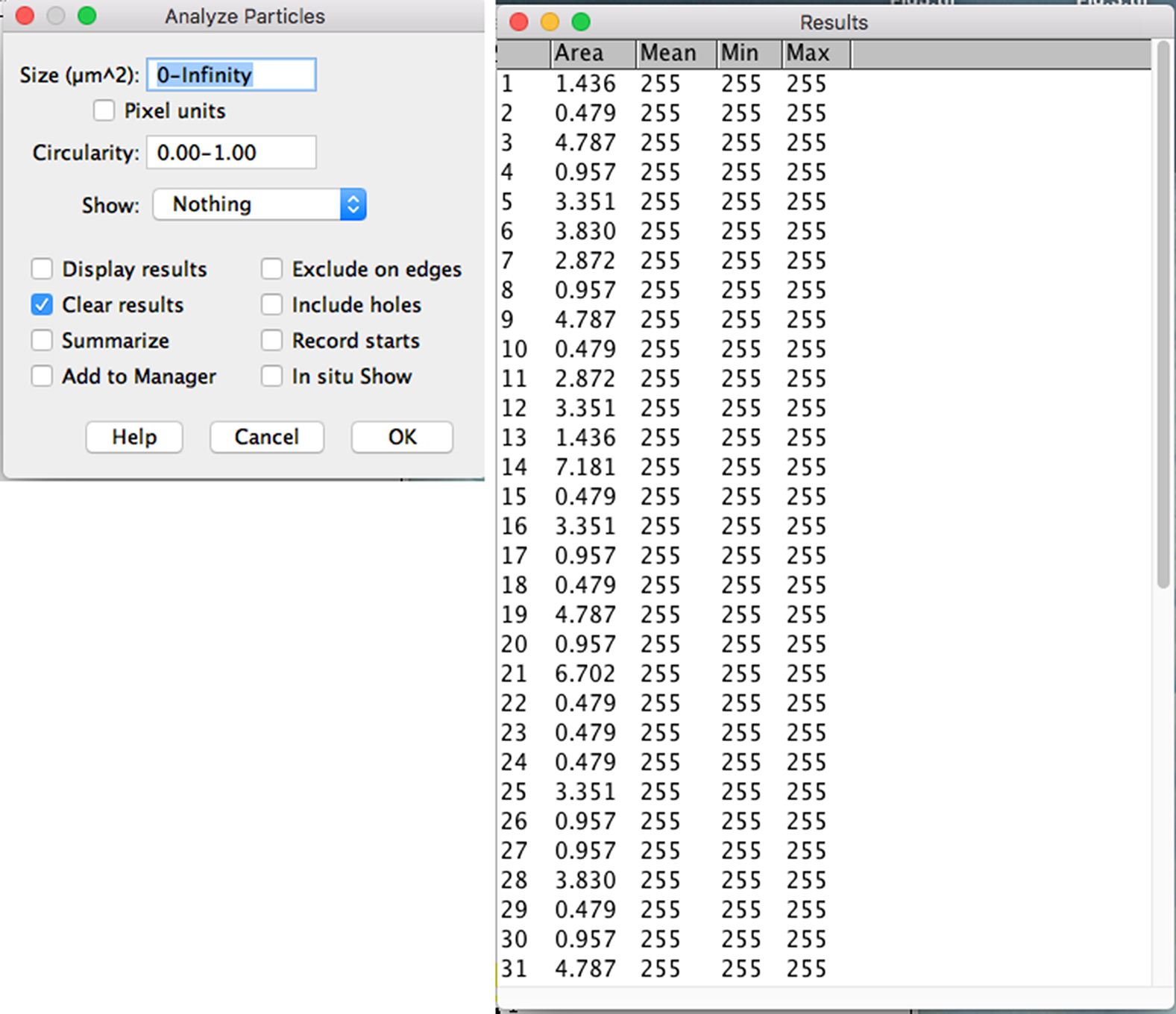
Figure 8. Analyses of the individual mitochondrial area in selected axon/neurite
- To find the total length of the chosen neurite, click on the “Plugins” drop-down menu: Segmentation → Simple Neurite Tracer → Click on start of neurite → Proceed to extend the neurite path, clicking yes (y) if the segment, or no (n) if the segment is not correct (if for example the program traces a neurite different than the chosen one) → When finished withdrawing neurite length, click finish (f) → Copy neurite length data to Excel (Figure 9).
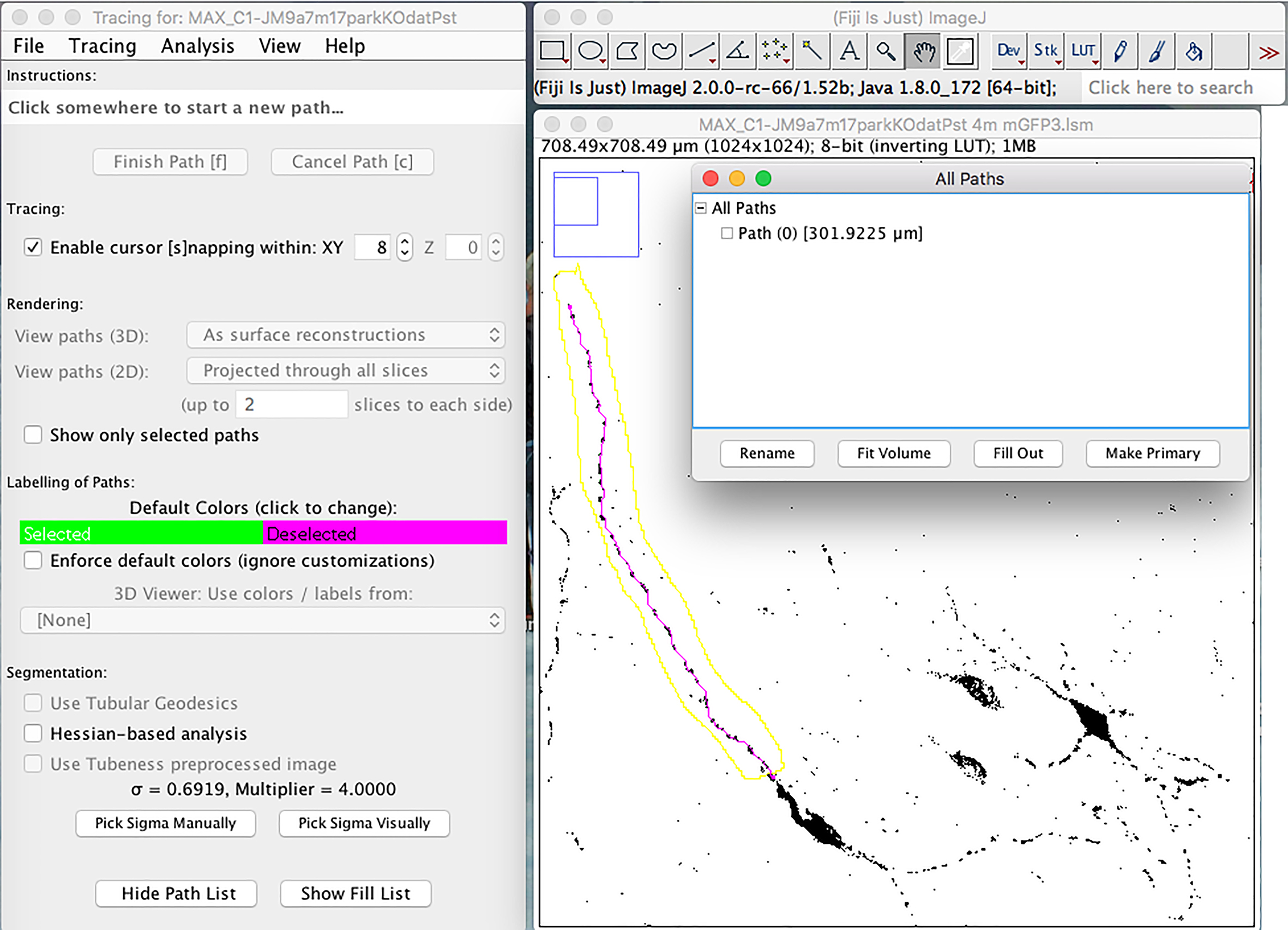
Figure 9. Determination of selected axon/neurite length
- Repeat these steps for 3-5 neurites per picture and 3-5 pictures per mouse.
Data analysis
In Figure 10 we included an example of an excel file with the described calculations:
- Number of mitochondria per μm
Divide the number of mitochondria for each neurite (Figure 8) and divide by neurite length (Figure 9).
Example: Cell G3 in Figure 10 was calculated by dividing the value in B31 (# of particles) by the value in A2 (neurite length) [=B31/A2] - Average mitochondrial size
Average the values obtained for the areas of each mitochondrion (Figure 8).
Example: Cell G6 in Figure 10 is the average of the particles size [=AVERAGE(C2:C31)] - Mitochondrial size distribution
- Using the Excel sort function, sort the mitochondrial size data from smallest to largest (column D in Figure 10).
- Individualize the particles that belong to the different bins (column E in figure 10) and count the number of mitochondria which fall within the predetermined bin ranges (values in H9, H10 and H11 in Figure 10).
- Divide the number of mitochondria which fall within in each bin by the total number of mitochondria counted for each animal and multiply by 100 to get the percentage of each binned group.
Example: In Figure 10, cell I9 is calculated by dividing H9 (number of particles in bin#1) by B31 (total number of particles) multiplied by 100 [=H9/B31*100]. Values in cells I10 and I11 are calculated in the same way for bin #2 and bin #3. - Using a stacked bar graph, create a graph of the mitochondrial size distribution. Example: In figure 10 graph was created using values in cells I8, I11
Notes:
- For our purpose, we chose the bin sizes of 0-5 μm, 5-10 μm, 10-20 μm, and > 20 μm arbitrarily. Once the mitochondrial size data has been sorted from smallest to largest, we must manually separate the “binned groups” to generate the mitochondrial size distribution graphs.
- Analysis between the groups has been done using one-way ANOVA. Each group should have an n = 3 animals, with 9-25 individual neurites being analyzed from each animal (3-5 slices per animal, and 3-5 neurites per slice).
Example: in our study (Pinto et al., 2018), each group had an n = 3 animals, with 9-19 individual neurites being analyzed from each animal (3 to 5 pictures per animal, 3 to 5 neurites per picture).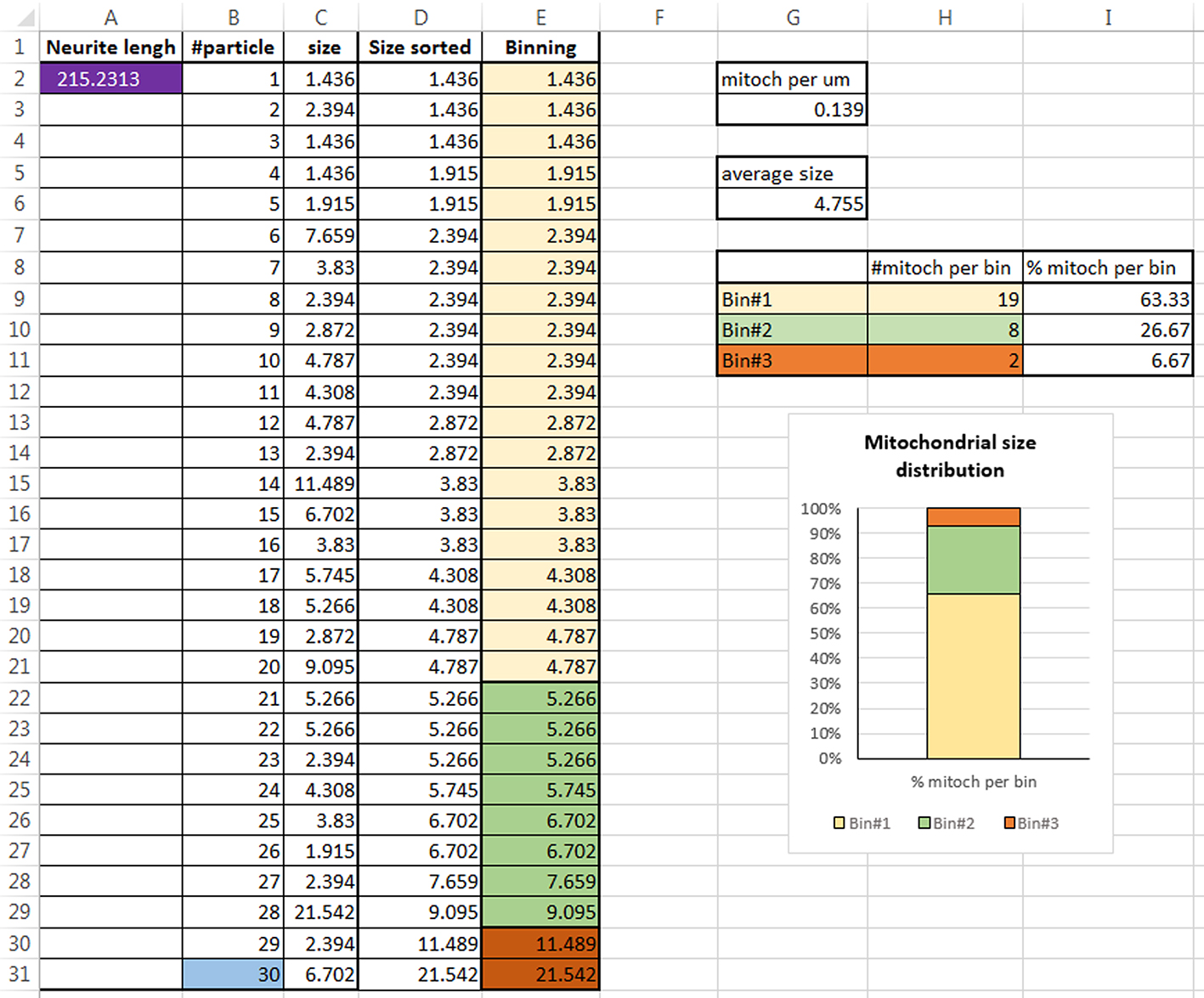
Figure 10. Screenshot of an Excel file with the Data analysis for one neurite
Recipes
- PBS 1x
Add 100 ml of 10x PBS to 900 ml of MilliQ water
Note: PBS solution does not need to be made fresh, and can be stored at room temperature up to 24 months. - 4% Paraformaldehyde (100 ml)
- Add 80 ml of 1x PBS to a glass beaker on a stir plate in a ventilated hood
- Heat while stirring to approximately 60 °C
- Take care that the solution does not reach 70 °C
- Add 4 g of paraformaldehyde powder to the heated PBS solution
- Add 1-2 drops of 1 N NaOH
- The solution will be clear in about 20-30 min
- Once the paraformaldehyde is dissolved, the solution is cooled and filtered
- Adjust the volume of the solution to 100 ml with 1x PBS
- Check the pH, and adjust it with NaOH to 7.4
- 0.02% (w/v) Sodium azide
Dissolve 0.2 g in 1 L of PBS 1x
Note: Sodium azide is highly toxic, weigh under a chemical hood. 0.02% Sodium azide solution does not need to be made fresh and can be stored at room temperature.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants 1R01AG036871, 1R01NS079965, and 5R01EY010804 (CTM), and Parkinson’s Disease Foundation Fellowships PDF-FBS-1316 (MP). We acknowledge support from the NEI center grant P30-EY014801 from the National Institutes of Health (NIH).
Competing interests
The authors have no conflicts of interest or competing interests to declare.
Ethics
All animal procedures were performed in accordance with a protocol approved by the University of Miami Institutional Animal Care and Use Committee.
References
- Bereiter-Hahn, J. (1990). Behavior of mitochondria in the living cell. Int Rev Cytol 122: 1-63.
- Braak, H., Ghebremedhin, E., Rub, U., Bratzke, H. and Del Tredici, K. (2004). Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res 318(1): 121-134.
- Chandrasekaran, K., Hazelton, J. L., Wang, Y., Fiskum, G. and Kristian, T. (2006). Neuron-specific conditional expression of a mitochondrially targeted fluorescent protein in mice. J Neurosci 26(51): 13123-13127.
- Chang, D. T. and Reynolds, I. J. (2006). Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol 80(5): 241-268.
- Dauer, W. and Przedborski, S. (2003). Parkinson's disease: mechanisms and models. Neuron 39(6): 889-909.
- Devine, M. J. and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci 19(2): 63-80.
- Gage, G. J., Kipke, D. R. and Shain, W. (2012). Whole animal perfusion fixation for rodents. J Vis Exp 65: Pii 3564.
- Hamanaka, R. B. and Chandel, N. S. (2010). Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 35(9): 505-513.
- Koh, H. and Chung, J. (2010). PINK1 and Parkin to control mitochondria remodeling. Anat Cell Biol 43(3): 179-184.
- Misgeld, T. and Schwarz, T. L. (2017). Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 96(3): 651-666.
- Narendra, D. P. and Youle, R. J. (2011). Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal 14(10): 1929-1938.
- Neupert, W. and Herrmann, J. M. (2007). Translocation of proteins into mitochondria. Annu Rev Biochem 76: 723-749.
- Picard, M., Shirihai, O. S., Gentil, B. J. and Burelle, Y. (2013). Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol 304(6): R393-406.
- Pinto, M., Nissanka, N. and Moraes, C. T. (2018). Lack of Parkin anticipates the phenotype and affects mitochondrial morphology and mtDNA levels in a mouse model of Parkinson's disease. J Neurosci 38(4): 1042-1053.
- Sheng, Z. H. (2017). The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol 27(6): 403-416.
- Shutt, T. E. and McBride, H. M. (2013). Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta 1833(2): 417-424.
- Wiemerslage, L. and Lee, D. (2016). Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters. J Neurosci Methods 262: 56-65.
- Xu, C. C., Denton, K. R., Wang, Z. B., Zhang, X. and Li, X. J. (2016). Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis Model Mech 9(1): 39-49.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Nissanka, N., Moraes, C. T. and Pinto, M. (2018). Image-Based Analysis of Mitochondrial Area and Counting from Adult Mouse Dopaminergic Neurites. Bio-protocol 8(16): e2471. DOI: 10.21769/BioProtoc.2471.
- Pinto, M., Nissanka, N. and Moraes, C. T. (2018). Lack of Parkin anticipates the phenotype and affects mitochondrial morphology and mtDNA levels in a mouse model of Parkinson's disease. J Neurosci 38(4): 1042-1053.
Category
Neuroscience > Cellular mechanisms > Mitochondria
Cell Biology > Cell imaging > Fixed-tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.