- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Senescence Associated β-galactosidase Staining

Published: Vol 2, Iss 16, Aug 20, 2012 DOI: 10.21769/BioProtoc.247 Views: 75365
Original research article
The authors used this protocol in:

Dec 2011
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Detection of senescent cells using a cytochemical assay was first described in 1995 (Dimri et al., 1995). The identification of senescent cells is based on an increased level of lysosomal β-galactosidase activity (Kurz et al., 2000). Cells under normal growth condition produce acid lysosomal β- galactosidase, which is localized in the lysosome. The enzymatic activity can be detected at the optimal pH 4.0, using the chromogenic substrate 5-bromo-4-chloro-3-indolyl β D-galactopyranoside (X-gal) (Miller, 1972). In comparison, upon senescence, the lysosomal mass is increased, leading to production of a higher level of β-galactosidase, termed senescence-associated β-galactosidase (SA-β-gal) (Kurz et al., 2000). The abundant senescence-associated enzyme is detectable over background despite the less favorable pH conditions (pH 6.0) (Dimri et al., 1995). The SA-β gal positive cells stain blue-green, which can be scored under bright-field microscopy. In this assay it is best to avoid over-confluency of the cells, or cells that have undergone too many passages, as these conditions can cause false positive results.
Keywords: SenescenceMaterials and Reagents
- Paraformaldehyde (PFA) (Sigma-Aldrich)
- 5-bromo-4-chloro-3-indolyl β D-galactopyranoside (X-gal) (Sigma-Aldrich)
- Potassium ferrocyanide (Sigma-Aldrich, catalog number: B4252 )
- Potassium ferricyanide (Sigma-Aldrich, catalog number: P9387 )
- Phosphate buffered saline (PBS)
- Sodium hydroxide
- Dimethylformamide
- Sodium chloride
- Magnesium chloride
- Dibasic sodium phosphate
- Citric acid
- Sodium phosphate
- 4% paraformaldehyde (PFA) (see Recipes)
- Senescence associated β-galactosidase (SA-β-gal) staining solution (see Recipes)
Equipment
- Inverted microscope [e.g. Olympus 1 x 71 inverted microscope (Olympus)]
- 24-well plate
- p1000 pipette
- 37 °C incubator
- Hot plate
Procedure
- The manipulation of cells for the SA-β-gal staining assay may be performed in a 24-well plate format.
- Prepare each sample in triplicate.
- At 120 h post-transfection or after the cell manipulation, aspirate the cell culture medium and wash the cells with PBS (500 μl per well) twice, using a p1000 pipette.
- After the last rinse, replace the PBS with 250 μl of 4% PFA for fixation.
- Incubate the cells for 5 min at room temperature.
- Aspirate the 4% PFA and wash the cells two times for 5 min each at room temperature with gentle shaking with 500 μl PBS.
- Add 250 μl SA-β-gal staining solution to each well.
- Incubate the cells in the dark in a 37 °C incubator.
- Terminate the reactions when the cells are stained blue-green, as visualized under an inverted bright-field microscope.
- To terminate the reaction, aspirate the staining solution and replace with distilled water.
- Wash the cells a second time in distilled water.
- After the last wash add 500 μl of distilled water to each well and observe the plate under an inverted bright-field microscope.
- Capture images of cells in each well using a 10x objective.
- Images may be printed for counting the total cell number, or count the stained cells on a computer monitor, which may give better distinction between the unstained and stained cells.
- Represent the SA-β-gal positive cells as a percentage of the total cell number.
Note: A trial may need to be carried out to determine the optimal length of incubation with SA-β-gal staining solution for each of the cell lines to be studied. Cells may be observed every 4 h during the first 12 h, and subsequently every 12 h. For example, the optimal incubation period in our hands was determined based on the visibility of the stained cells in test samples (e.g. PAX8siRNA treated samples) but not in the control samples (i.e. untreated sample or/and control siRNA treated sample). It is important to remember that the detection principle of this assay is based on the cellular abundance of the lysosomal β-galactosidase, which varies between cell lines. A table showing the length of optimal incubation time for several cell lines in our hands is as follows (Table 1).
Table 1. Example incubation times required for the appearance of the SA-β-gal activity in cell lines
| Cell line | Incubation time (h) |
| A498 (renal cell carcinoma) | 24 |
| 786-O (renal cell carcinoma) | 24 |
| TK-10 (renal cell carcinoma) | 12 |
| K1 (thyroid carcinoma) | 12 |
Figure 1A illustrates the appearance of increased SA-β-gal activity detected in response to PAX8 knockdown in four cell lines. Bright-field images show SA-β-gal positivecells in blue-green (insets). The colors of the images were inverted usingAdobe Photoshop (version 10.0) to aid the visibility of the positive (pink) cells. SA-β-gal positive cells were undetectable (or at a very low frequency) in control siRNA(SN) treated samples (highest detection was 3%, in TK-10 cells, Figure 1B). In comparison, adistinct elevation of SA-β-gal positive cells was observed in all PAX8 siRNA (S8) treated samples (highestdetection was 38%, in TK-10 cells).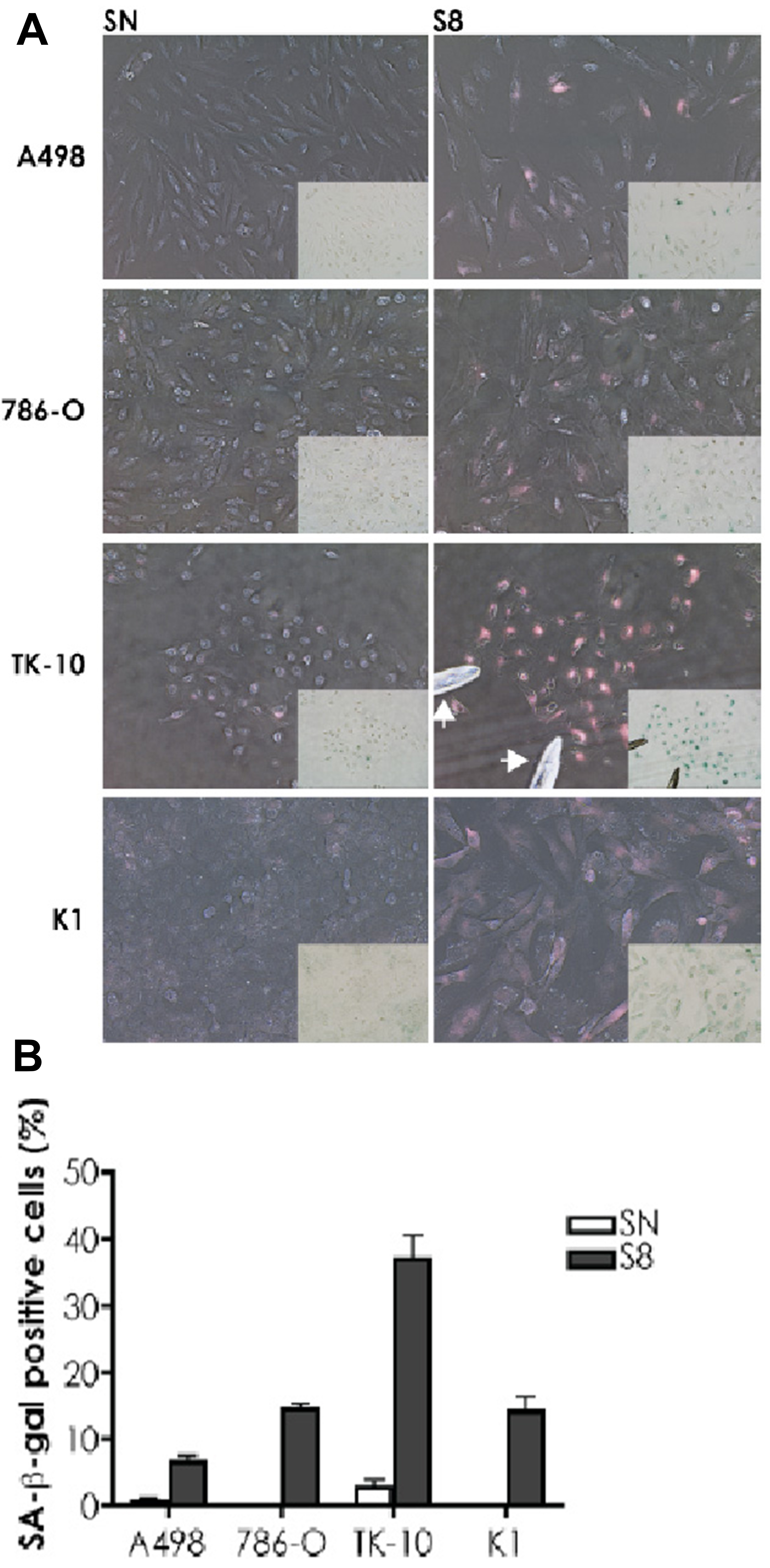
Figure 1. Identification of senescent cells with the SA-β-gal staining assay. Cells treated with a control siRNA (SN), or PAX8 siRNA (S8) were assayed for SA-β-gal activity at 120 h post-siRNA treatment. A. Bright-field images are shown in the insets. These images were inverted using Adobe Photoshop to aid visibility of the positive cells (pink). The white arrows indicate precipitates from the staining solution. Magnification100x. B. Graph showing the percentage of positive cells (of the total cell number) in the treated samples.
Recipes
- 4% paraformaldehyde (PFA)
To make 100 ml 4% PFA, dissolve 4 g PFA in 100 ml of PBS with continuous stirring on a hot plate (with the solution not exceeding 60 °C). Add 20 μl 1 M sodium hydroxide to dissolve the residual PFA. Aliquot and freeze at -20 °C for long-term storage. - Senescence associated β-galactosidase (SA-β-gal) staining solution
Firstly, prepare the following stock solutions.- To prepare 10% X-gal in dimethylformamide (DMF) dissolve 1 g X-gal in 10 ml DMF. Store the stock X-gal solution at -20 °C.
- 400 mM citric acid/sodium phosphate solution add 36.85 ml 0.1 M citric acid to 63.15 ml 0.2 M dibasic sodium phosphate. Verify the pH and adjust to pH 6.0 with 0.1 M citric acid, if necessary.
- 0.5 M potassium ferrocyanide and 0.5 M potassium ferricyanide, Store stock solutions of potassium ferrocyanide and potassium ferricyanide in the dark at 4 °C.
Acknowledgments
This protocol is based on the same protocol as published in Li et al. (2011). MRE wishes to acknowledge support from the University of Otago Leading Thinkers Advancement Campaign, and the New Zealand Institute for Cancer Research Trust. CGL wishes to acknowledge scholarship support from the Health Research Council, the University of Otago International Fees Scholarship and Postgraduate Publishing Bursary and the Dunedin School of Medicine Finishing Your PhD Scholarship. Research grants supporting the implementation of this protocol were from the Health Research Council of New Zealand and the University of Otago Faculty of Medicine Trust Fund.
References
- Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., Medrano, E. E., Linskens, M., Rubelj, I., Pereira-Smith, O. and et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92(20): 9363-9367.
- Kurz, D. J., Decary, S., Hong, Y. and Erusalimsky, J. D. (2000). Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113 (Pt 20): 3613-3622.
- Li, C. G., Nyman, J. E., Braithwaite, A. W. and Eccles, M. R. (2011). PAX8 promotes tumor cell growth by transcriptionally regulating E2F1 and stabilizing RB protein. Oncogene 30(48): 4824-4834.
- Miller, J. H. (1972). Experiments in molecular genetics. Cold Spring Harbor Laboratory xvi, 466 p.pp.
Article Information
Copyright
© 2012 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Eccles, M. and Li, C. G. (2012). Senescence Associated β-galactosidase Staining. Bio-protocol 2(16): e247. DOI: 10.21769/BioProtoc.247.
Category
Cancer Biology > Cell death > Cell biology assays > Cell cycle
Cell Biology > Cell viability > Cell death
Cell Biology > Cell staining > Whole cell
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.