- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Live Imaging of Myogenesis in Indirect Flight Muscles in Drosophila
Published: Vol 7, Iss 13, Jul 5, 2017 DOI: 10.21769/BioProtoc.2377 Views: 9856
Reviewed by: Antoine de MorreeKanika GeraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







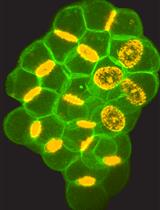
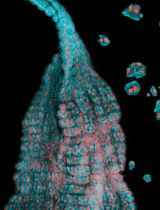
Abstract
The indirect flight muscles (IFMs) are the largest muscles in the fly, making up the bulk of the adult thorax. IFMs in Drosophila are generated during pupariation by fusion of hundreds of muscle precursor cells (myoblasts) with larval muscle templates (myotubes). Prominent features, including the large number of fusion events, the structural similarity to vertebrate muscles, and the amenability to the powerful genetic techniques of the Drosophila system make the IFMs an attractive system to study muscle cell fusion. Here we describe methods for live imaging of IFMs, both in intact pupae, and in isolated IFMs ex-vivo. The protocols elaborated upon here were used in the manuscript by (Segal et al., 2016).
Keywords: Myoblast fusionBackground
While Drosophila embryonic muscles have long been an established model system for the study of muscle development (Volk, 1999; Chen and Olson, 2004; Abmayr et al., 2008; Richardson et al., 2008) the adult Drosophila indirect flight muscles (IFMs), which form during pupal stages, have emerged in recent years as a complementary system to address cell-biological processes during myogenesis (Dutta, 2006; Oas et al., 2014; Weitkunat et al., 2014; Shwartz et al., 2016). Their large size, ample fusion events, structural similarity to vertebrate muscles, and amenability to powerful genetic techniques of the Drosophila system make the IFMs an attractive system to study muscle development. Historically, study of IFM development has been limited compared to embryonic muscle for several reasons. First, classic genetic approaches are difficult to implement in IFMs due to functional requirements earlier in development for many of the genes potentially involved in muscle development in the adult and to the syncytial nature of muscles, which restricts the usefulness of clonal analysis. Relatively recent advances in the available tools utilizing the GAL4-UAS system allow circumvention of these limitations, by expressing RNAi in a tissue specific manner. The power of this approach was demonstrated in a comprehensive screen for genes involved in pupal myogenesis (Schnorrer et al., 2010), and has been successfully implemented in several recent studies of myoblast fusion in IFMs (Mukherjee et al., 2011; Gildor et al., 2012; Dhanyasi et al., 2015; Segal et al., 2016). In addition, the technical challenges associated with dissection, accessibility, and visualization of IFMs have been overcome by advances in techniques and technology (Weitkunat and Schnorrer, 2014; Segal et al., 2016). This protocol is an expanded version of the methods used in the manuscript by (Segal et al., 2016), and is intended to contribute to the growing repertoire of techniques for study of IFMs. Here we describe methods for live imaging of IFMs, both in intact pupae, and in isolated IFMs ex-vivo. While previous work focused on stages of myotube growth via fusion (18-22 h after puparium formation [APF], 25 °C), these methods are readily applicable to other stages of IFM myogenesis, starting at 12 h APF onwards. Imaging of intact pupae can be suitable for studies of developmental processes which span several hours, while imaging of ex-vivo cultures is intended to better visualize finer structural details and dynamic behaviors over shorter time periods (e.g., 1 h). Parts of this protocol are variations on (Weitkunat and Schnorrer, 2014).
Materials and Reagents
- Glass single frosted microscope slides (Thermo Fisher Scientific, catalog number: 421-004T )
- Cover slip
- Custom-made plexiglass slide with round opening in center (will serve as basis for viewing chamber)
Note: The opening should be 1.25 cm in diameter. - Double-sided tape
- Kim-wipes
- Toothpick
- 8-well chamber slide (μ-slide) (ibidi, catalog number: 80826 )
- 200 μl and 1,000 μl pipette tips
- Sylgard silicone plates, stained with charcoal (Dow Corning, catalog number: 3097358-1004 )
- 1 cm Minutian pins for silicon plate (Fine Science Tools, catalog number: 26002-10 )
- Flies expressing muscle-specific fluorescent markers (e.g., mef2-GAL4> UAS-CD8-GFP)
- Schneider’s medium
- Fetal bovine serum (FBS)
- BD Matrigel matrix growth factor, reduced (Corning, catalog number: 354230 )
- Halocarbon oil
Equipment
- Thin paint brush
- Forceps (Dumont 55) (Fine Science Tools, catalog number: 11255-20 )
- Vannas Spring scissors, 2.5 mm Cutting Edge (Fine Science Tools, catalog number: 15001-08 )
- Confocal microscopy system (e.g., ZEISS, model: LSM 780 ) equipped with an inverted microscope and a 40x water immersion objective
- 200 μl and 1,000 μl pipette
- 125 ml Erlenmeyer flask
- 25 °C incubator to grow flies (Elcon company, custom-made)
- Vials with fly food to raise and collect flies
- 37 °C incubator for matrigel solidification (Thermo Fisher Scientific, Thermo ScientificTM, model: HeracellTM 150i )
- Oxygen tank with attached rubber tube
- Binocular dissecting microscope (Nikon Instruments, model: SMZ645 )
- Fluorescent binocular microscope (Leica, model: Leica MZ16F )
Software
- Zen Black software (ZEISS)
- Microsoft Excel
- R Statistical software (can be downloaded at: https://www.r-project.org/)
Procedure
- Preparation of intact pupae for live imaging
Note: IFM myoblast fusion is at its peak at 18-22 h APF when grown at 25 °C, but this method can be used to image IFMs before or after that time range. Pupae at 12 h APF or earlier are more difficult to work with, since head eversion has yet to occur, so the pupal case is still attached to the pupa.- Select fly line with IFMs expressing a fluorescent marker. Pupae are grown to the desired stage, by collecting white pre-pupae (which are 0-1 h old) into a new vial, and growing at 25 °C (e.g., for 18-20 h). Pre-pupae can be identified by their formed antennae and lack of any pigmentation (white color).
- Prepare viewing chamber, by attaching a cover slip to the custom-made plexiglass slide with opening, with tape on either side of the opening (so that the coverslip is positioned over the opening) (Figure 1A).
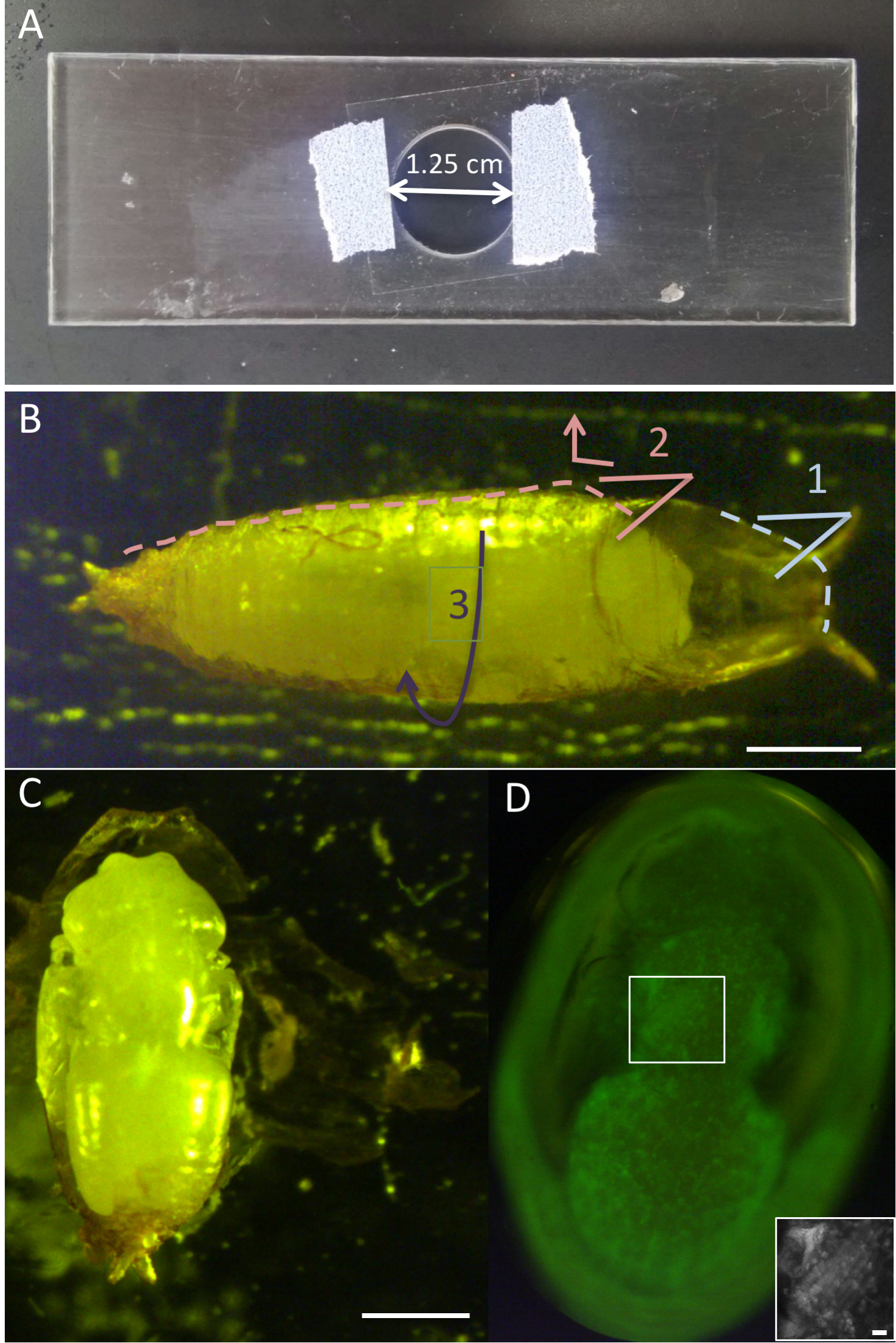
Figure 1. Intact pupae. A. Custom-made slide for imaging of intact pupae. Coverslip is taped from both sides over opening. B. Pupa adhered to double-sided tape on glass slide. For dissection, first remove the ‘lid’ of the pupal case with forceps (1). Then, place one side of the forceps between the pupa and the pupal case, and move downward and outward to tear the pupal case open (2). Lift the formed ‘flap’ to other side to expose pupa (3). C. Pupal case is peeled from side and opened to double-sided tape beneath to expose pupa. D. GFP fluorescent filter view, via a dissection microscope and through a cover-slip, showing the dorsal side of an intact pupa, immersed in halocarbon oil. Boxed area enlarged on bottom right showing the 6 IFM fibers. Scale bars = 500 μm (B and C), 50 μm (D). - On a glass microscope slide, place a strip of double-sided tape.
- Carefully, lift staged pupae with brush from vial and place them onto a Kim-wipe. Carefully brush pupae against Kim-wipe to remove possible food residue.
- Using brush, place a pupa dorsal side up (flat side down) on the double-sided tape strip. Press down lightly on the pupa with a brush to ensure it is well adhered, without injuring the pupa.
- The next few steps are designed to break open the pupal case and remove/transfer the intact pupa to the viewing chamber. At these developmental stages head eversion should be complete, so that a gap exists between the pupal case and the head. With forceps, carefully capture the extreme anterior end of the pupal case, and lift upward to peel off the circular ‘lid’ of the pupal case (Figure 1B).
- Slide forceps gently into the anterior opening you created, so that one arm of forceps protrudes into the space between the pupa and the pupal case.
- Pull outward to tear the pupal case. Continue this movement until the pupal case is opened along its dorsal aspect all the way to the posterior end.
- The ‘flap’ that has formed on the upper portion of the pupal case can now be lifted/opened by the forceps and attached to the tape on the opposite side, thereby revealing the pupa (Figures 1B and 1C).
- Place the closed forceps under the pupa, and gently lift and transfer it to the cover slip attached to custom-made slide. The pupa should stick to the forceps. If not, lift by the forming legs of the pupa, underneath the abdomen.
- Using the forceps, orient the pupa so that the thorax (dorsal side) faces the cover-slip.
- Place a drop of halocarbon oil on a separate glass slide. From this drop, using a toothpick, pick up a smaller drop of oil and gently transfer onto the pupa, so that it is mostly immersed in oil. Put only enough oil to cover the pupa, as excessive oil can cause movement of the pupa during long-term imaging. Absorb excess oil with a Kim-wipe (Figure 1C).
- Orient the pupa so that it is slightly tilted to one side at about a 45° angle, for a better angle of imaging for one of the IFMs.
- Image slide immediately using the inverted confocal microscope system. Live intact pupae have been successfully imaged for up to 12 h. See more details for image acquisition below.
- Select fly line with IFMs expressing a fluorescent marker. Pupae are grown to the desired stage, by collecting white pre-pupae (which are 0-1 h old) into a new vial, and growing at 25 °C (e.g., for 18-20 h). Pre-pupae can be identified by their formed antennae and lack of any pigmentation (white color).
- Dissection and isolation of IFMs for ex-vivo live imaging
- Prepare an ibidi μ-slide coated with Matrigel as follows. Matrigel should be diluted 1:10 in PBS from stock and thawed at 4 °C overnight, as described by manufacturer here. With a micropipette, transfer 100 μl into a single well, and spread with tip so that it covers the entire surface of the well. Re-pipette and remove the entire 100 μl Matrigel. The small amount that necessarily remains will create the Matrigel coat. For preparation of multiple wells, the same 100 μl can be re-used. Discard remaining liquid. Place ibidi μ-slide at 37 °C for 30 min in order to allow the Matrigel to harden.
- Prepare medium for live imaging. First, oxygenate Schneider’s medium as follows. Place 20-50 ml Schneider’s medium in 125 ml Erlenmeyer flask. Attach 1,000 μl micropipette tip to the end of a tube attached to an oxygen tank. Tilt the flask slightly (e.g., by placing bottom on another micropipette tip, see Figure 2). Place tube so that the tip is immersed in the medium, and gradually increase oxygen pressure until bubbles are seen. Oxygenate for 10 min. After oxygenating, add 10% FBS to desired amount of medium. Adding FBS before oxygenating is not recommended as it could create foam.

Figure 2. Oxygenation of Schneider’s medium. See text for details. In this set-up, the tube is taped to the wall for convenience of placement. - Stage and isolate a pupa from its pupal case, as in steps A1-A10 above. Transfer the pupa to a darkened Sylgard silicon plate, dorsal (curved) side down. Place 200 μl Schneider’s medium + 10% FBS, oxygenated, on pupa.
- Insert pin between head and thoracic segments of the pupa, angling ~135° away from thoracic segment, so as to not hinder the view of the thorax. Pin should be sharp to smoothly puncture the pupa (Figure 3A).
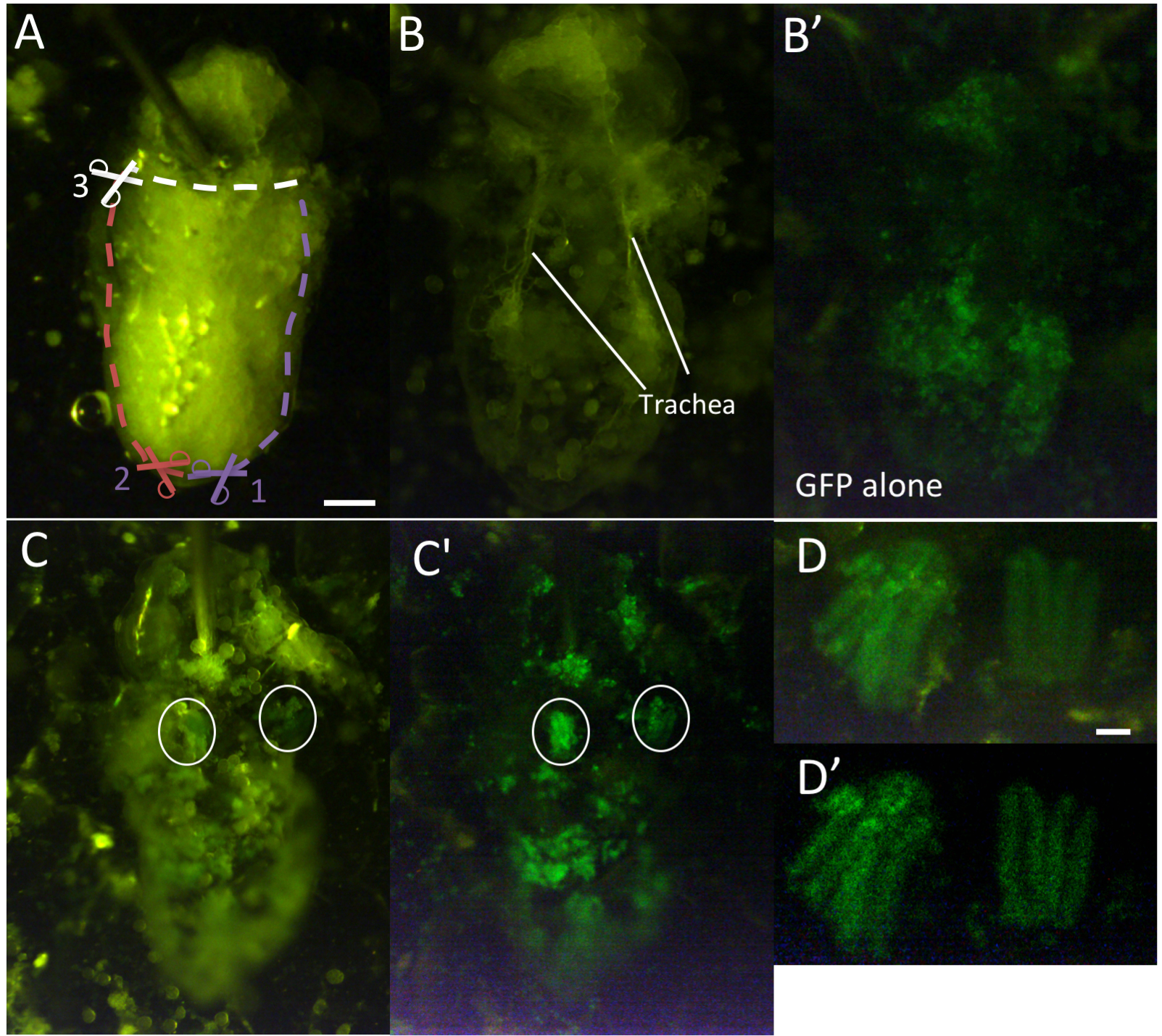
Figure 3. Isolation of IFMs for ex-vivo live imaging. A. Pinned pupa. First, cut posterior tip of abdomen (1). Then, cut ventro-lateral cuticle on either side (2 and 3), and finally cut horizontally to remove ventral cuticle (4). (B-B’) Opened pupa before removing fat and other irrelevant tissue viewed by transmitted (B) and flourescent (B’) light. Note that IFMs cannot be seen at this stage, as they are lightly adhered to the dorsal cuticle and are obscured by surrounding tissue. (C-C’) Pupa after clearing of other tissues viewed by transmitted and fluorescent light (C) or fluorescent light alone (C’). IFMs are circled. (D-D’) Isolated IFM pairs viewed by transmitted and fluorescent light (D) or fluorescent light alone (D’). Scale bars = 200 μm (A), 50 μm (D). - Under binocular, with microdissection scissors, snip off the posterior tip of the abdominal cuticle.
- With scissors, carefully slide up and cut the lateral sides of the cuticle, on both sides. Cut across the pupa below pin, allowing to remove and discard the ventral portion of the cuticle.
- Under fluorescent binocular, place blue light filter to visualize GFP. With forceps, gently remove fat bodies, intestines, and other tissues, avoiding contact with the dorsal cuticle and attached tissues. At these stages, the IFMs should be lightly adhered to the dorsal cuticle (Figures 3B and 3B’). Avoid clearing trachea at this stage.
- Depending on the specific fluorescent line used, there may be other tissues that also express GFP. At these stages, the IFMs should be recognizable morphologically in addition to GFP fluorescence, as 6 elongated fibers, lightly attached to both sides of the dorso-lateral cuticle, in the mid-anterior portion of the thoracic segment. Visualizing the IFMs without GFP fluorescence is more challenging.
- Gently clear remaining fat and trachea with forceps to find and expose the IFMs. In some cases, IFMs are lightly attached to trachea and must be separated with forceps (Figures 3C and 3C’, see ‘Notes’ section for some additional tips).
- Once IFMs are exposed, pinch with forceps underneath IFMs to detach them from the cuticle.
- Isolate the IFMs by gently pushing them with side of forceps to a region of the medium that is outside the dissected pupa (Figures 3D and 3D’).
- Transfer 200 μl Schneider’s medium + 10% FBS, oxygenated to a Matrigel-coated well in ibidi plate.
- With micropipette, wet 200 μl micropipette tip with clean medium. In the silicon plate, lift a small amount of liquid into the tip, and with micropipette still pressed, lift up the isolated IFMs into 50 μl liquid.
- Gently transfer the IFMs and liquid into the medium-filled well. The IFMs should sink to the surface of the well.
- With side of forceps, gently press, flatten, and orient the IFMs for imaging in the ibidi slide well.
Note: Optionally, chemical inhibitors (e.g., LatrunculinA) or dyes (e.g., SiR Tubulin) may be added to the medium at relevant concentrations. - Take immediately for imaging using the confocal microscope system (e.g., ZEISS 780). Isolated IFMs can be imaged ex-vivo for up to 1.5 h. Time-lapse movies were taken at 40x water immersion objective, optimal for thick biological samples. The entire muscle is approximately 35 μm thick. Analysis was often done on maximum intensity projections of acquired z-stacks or subsets of z-stacks, using the Zen software (ZEISS). The following acquisition settings allowed imaging most of the depth of the IFMs at a time resolution of approximately 1.5 min per stack: 33 slices of 1.1 μm thickness, 512 x 512 resolution, line averaging of 4, 2x digital zoom. For better time resolution, decrease averaging and/or number of slices.
- Prepare an ibidi μ-slide coated with Matrigel as follows. Matrigel should be diluted 1:10 in PBS from stock and thawed at 4 °C overnight, as described by manufacturer here. With a micropipette, transfer 100 μl into a single well, and spread with tip so that it covers the entire surface of the well. Re-pipette and remove the entire 100 μl Matrigel. The small amount that necessarily remains will create the Matrigel coat. For preparation of multiple wells, the same 100 μl can be re-used. Discard remaining liquid. Place ibidi μ-slide at 37 °C for 30 min in order to allow the Matrigel to harden.
Data analysis
In general, visualization over time of fluorescent markers of interest can provide insight of dynamic behavior simply by observation of time-lapse of the acquired data. Time-lapse movies of single slices or maximum intensity projections of the data were created using Zen software (ZEISS). For example, a time-lapse movie of a single optical slice from an ex-vivo culture of dissected IFMs expressing Lifeact-GFP to label actin fibers (Video 1) displays myoblasts with actin-based protrusions emanating from their edges, which appear to be entangled within one another. The myoblasts are surprisingly immobile over time.

Quantification of length, frequency, etc. of specific features may also be performed in this system. For example, Segal et al. 2016 measured the lengths of filopodia in IFMs expressing the membrane marker Gap-GFP (Ritzenthaler et al., 2000). In this case, dynamics over time were not necessary, so a z-stack at a single timepoint was acquired (Figure 4). Using Zen Black software, a subset of z-slices were selected to include the filopodia of the myotube but exclude background from neighboring myotubes. Then, a maximum intensity projection (MIP) of the subset of slices was made. An area of interest was selected and cropped (Figure 4, red box), and brightness was adjusted to highlight the filopodia. The cropped raw data was transferred to Zen Blue software, for measurement of filopodia lengths. The output of lengths of filopodia was transferred to Excel for analysis. Statistical analysis of differences in filopodia length between experimental groups was done in R software, but could be done on any statistical software.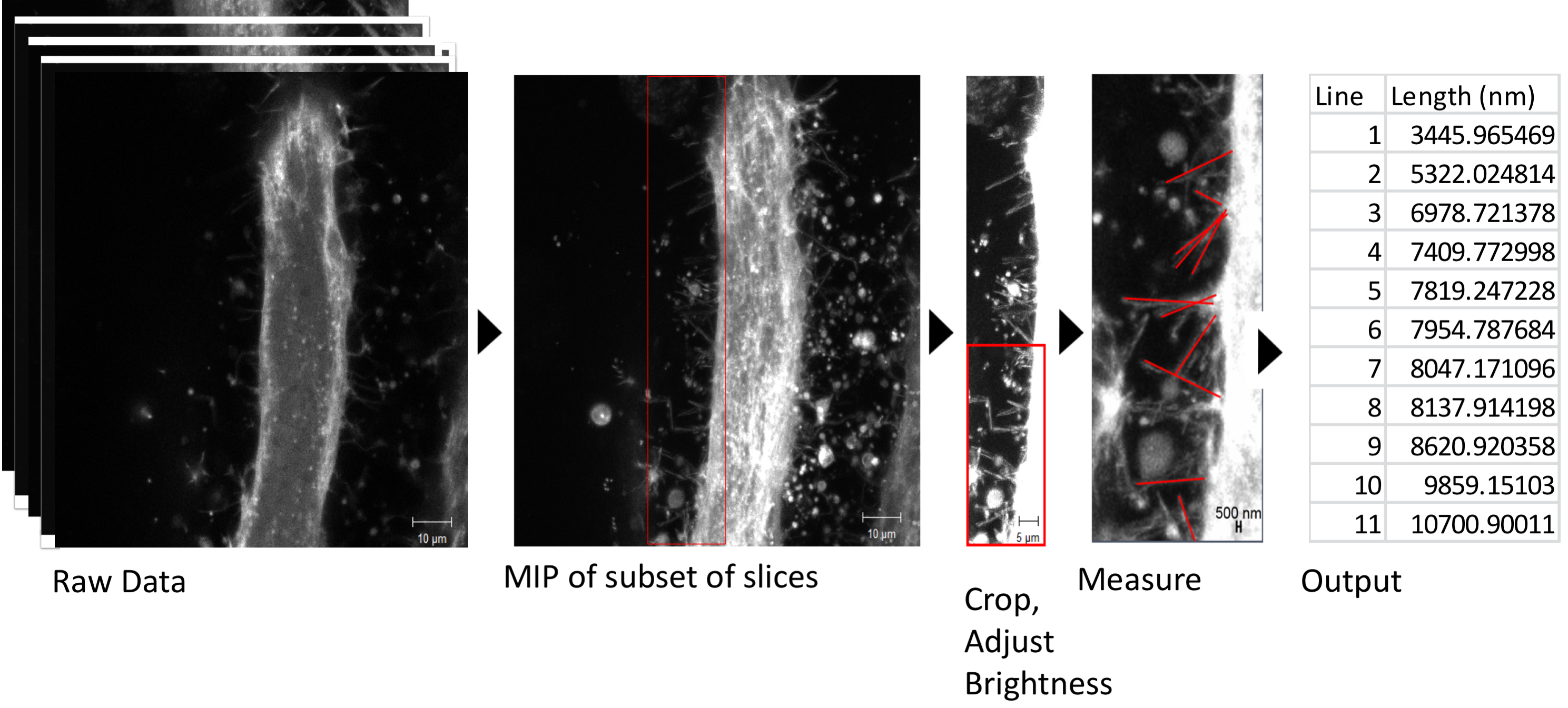
Figure 4. Analysis of filopodia length as in Segal et al., 2016. See text for details.
Notes
- Dorso-longitudinal muscles (DLMs) and dorso-ventral muscles (DVMs), the two types of IFMs, remain attached. DLMs can be distinguished as the larger of the two muscle groups. Previous work imaged the DLMs specifically (Segal et al., 2016).
- During development the DLMs split from three fibers to 6 fibers, between 16-18 h APF. Therefore, IFMs may be in the midst of splitting if dissected during this time range.
- Ideally, an intact DLM will include 3 or 6 fibers (see above) and the surrounding myoblasts. However, it is not uncommon for individual myofibers to be lost during dissection. In general, myoblasts will still remain associated with the remaining myofibers, and these samples can still be imaged, as long as the individual myofibers remain intact.
Acknowledgments
This work was done under the supervision of and with guidance from Prof. Benny Shilo and Dr. Eyal Schejter, at Weizmann Institute of Science in Rehovot, Israel. This work was supported by a grant from the Israel Science Foundation. Parts of this protocol were adapted from (Weitkunat and Schnorrer, 2014). This protocol was used in (Segal et al., 2016).
References
- Abmayr, S. M., Zhuang, S. and Geisbrecht, E. R. (2008). Myoblast fusion in Drosophila. Methods Mol Biol 475: 75-97.
- Chen, E. H. and Olson, E. N. (2004). Towards a molecular pathway for myoblast fusion in Drosophila. Trends Cell Biol 14(8): 452-460.
- Dhanyasi, N., Segal, D., Shimoni, E., Shinder, V., Shilo, B. Z., VijayRaghavan, K. and Schejter, E. D. (2015). Surface apposition and multiple cell contacts promote myoblast fusion in Drosophila flight muscles. J Cell Biol 211(1): 191-203.
- Dutta, D. and VaijayRaghavan, K. (2006). Metamorphosis and the formation of the adult musculature. In: Muscle Development in Drosophila. Springer 125-142.
- Gildor, B., Schejter, E. D. and Shilo, B. Z. (2012). Bidirectional Notch activation represses fusion competence in swarming adult Drosophila myoblasts. Development 139(21): 4040-4050.
- Mukherjee, P., Gildor, B., Shilo, B. Z., VijayRaghavan, K. and Schejter, E. D. (2011). The actin nucleator WASp is required for myoblast fusion during adult Drosophila myogenesis. Development 138(11): 2347-2357.
- Oas, S. T., Bryantsev, A. L. and Cripps, R. M. (2014). Arrest is a regulator of fiber-specific alternative splicing in the indirect flight muscles of Drosophila. J Cell Biol 206(7): 895-908.
- Richardson, B., Beckett, K. and Baylies, M. (2008). Visualizing new dimensions in Drosophila myoblast fusion. Bioessays 30(5): 423-431.
- Ritzenthaler, S., Suzuki, E. and Chiba, A. (2000). Postsynaptic filopodia in muscle cells interact with innervating motoneuron axons. Nat Neurosci 3(10): 1012-1017.
- Schnorrer, F., Schonbauer, C., Langer, C. C., Dietzl, G., Novatchkova, M., Schernhuber, K., Fellner, M., Azaryan, A., Radolf, M., Stark, A., Keleman, K. and Dickson, B. J. (2010). Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464(7286): 287-291.
- Segal, D., Dhanyasi, N., Schejter, E. D. and Shilo, B. Z. (2016). Adhesion and fusion of muscle cells are promoted by filopodia. Dev Cell 38(3): 291-304.
- Shwartz, A., Dhanyasi, N., Schejter, E. D. and Shilo, B. Z. (2016). The Drosophila formin Fhos is a primary mediator of sarcomeric thin-filament array assembly. Elife 5.
- Volk, T. (1999). Singling out Drosophila tendon cells: a dialogue between two distinct cell types. Trends Genet 15(11): 448-453.
- Weitkunat, M., Kaya-Copur, A., Grill, S. W. and Schnorrer, F. (2014). Tension and force-resistant attachment are essential for myofibrillogenesis in Drosophila flight muscle. Curr Biol 24(7): 705-716.
- Weitkunat, M. and Schnorrer, F. (2014). A guide to study Drosophila muscle biology. Methods 68(1): 2-14.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Segal, D. (2017). Live Imaging of Myogenesis in Indirect Flight Muscles in Drosophila. Bio-protocol 7(13): e2377. DOI: 10.21769/BioProtoc.2377.
Category
Neuroscience > Peripheral nervous system > Flight muscle
Cell Biology > Tissue analysis > Tissue isolation
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.