- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Lentiviral Barcode Labeling and Transplantation of Fetal Liver Hematopoietic Stem and Progenitor Cells
Published: Vol 7, Iss 8, Apr 20, 2017 DOI: 10.21769/BioProtoc.2242 Views: 10030
Reviewed by: Ivan ZanoniAnupam JhingranAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Cellular barcoding enables the dissection of clonal dynamics in heterogeneous cell populations through single cell lineage tracing. The labeling of hematopoietic stem and progenitor cells (HSPCs) with unique and heritable DNA barcodes, makes it possible to resolve donor cell heterogeneity in terms of differentiation potential and lineage bias at the single cell level, through subsequent transplantation and high-throughput sequencing. Furthermore, cellular barcoding allows for bona fide hematopoietic stem cells (HSCs) to be defined based on functional rather than immunophenotypic parameters.
This protocol describes the work flow of lentiviral cellular barcoding, tracking 14.5 days post coitum (d.p.c.) fetal liver (FL) Lineage-Sca+cKit+ (LSK) HSPCs following long-term reconstitution (Figure 1) (Kristiansen et al., 2016), but can be adapted to the cell type or time frame of choice.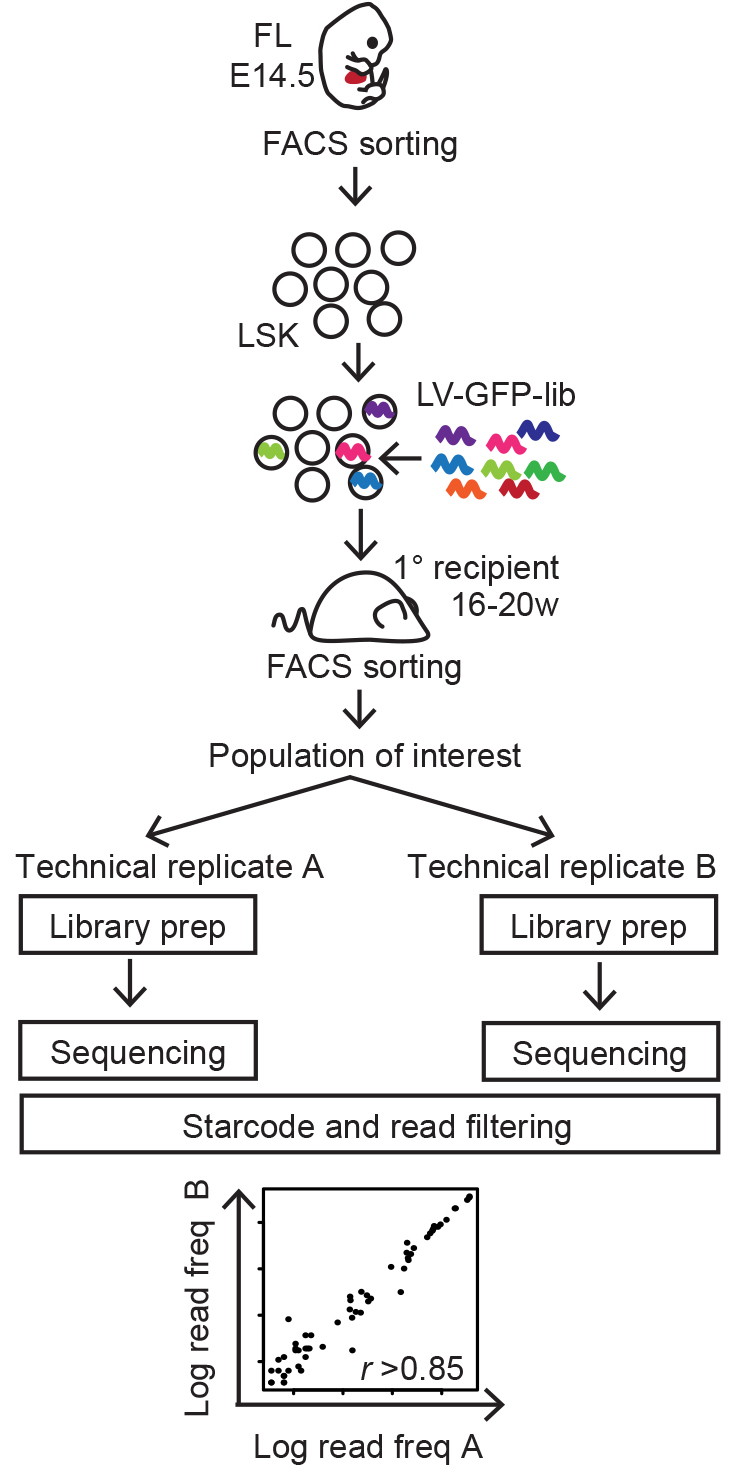
Figure 1. Summary of experimental workflow (Naik et al., 2013)
Background
The cellular barcoding technique was initially established to resolve single cell dynamics upon transplantation of hematopoietic cells in vivo and has in recent years contributed significantly to our appreciation of the functional heterogeneity within blood cell populations in a transplantation setting (Schepers et al., 2008; Gerrits et al., 2010; Lu et al., 2011; Naik et al., 2013; Verovskaya et al., 2013; Kristiansen et al., 2016). The generation and characterization of lentiviral barcode libraries, the importance of library complexity as well as the associated analytical challenges have been carefully reviewed (Bystrykh et al., 2014; Naik et al., 2014; Bystrykh and Belderbosv, 2016) and need to be considered before starting this protocol to ensure proper experimental design. The current protocol pertains our adaptation of the technology as seen in our recent article (Kristiansen et al., 2016), to trace the long-term reconstitution capacity of FL derived HSPCs.
Materials and Reagents
- 96-well non-treated plates (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 260836 )
- Tissue paper (KCWW, Kimberly-Clark, catalog number: 75512 )
- Autoclaved 1.5 ml Eppendorf tubes (Fisher Scientific, FisherbrandTM, catalog number: 05-408-129 )
- 50 ml tubes (Corning, Falcon®, catalog number: 352070 )
- 1 ml pipette tips (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 2179-05-HR )
- Cell strainers, 40 µm (Corning, catalog number: 431750 )
- 60 mm Petri-dishes (Corning, catalog number: 430166 )
- Cup filters, 50 µm (BD, BD Biosciences, catalog number: 340630 )
- 1 ml syringe with 29 gauge ½ inch needle (Terumo Medical, catalog number: BS-N1H2913 )
- FACS tubes (Corning, Falcon®, catalog number: 352058 )
- Mice: Males and females of appropriate experimental genotypes for timed pregnancies
Recipient mice of the appropriate experimental genotype
One mouse of the appropriate experimental genotype for support bone marrow cells
Note: This protocol was optimized using C57BL/6 mice. An example of animal usage is shown for a litter of 6 pups (Figure 2). If desired, donor derived cells can be distinguished from support and recipient derived cells using congenic mouse strains. However, this is not necessary since barcoded donor cells can be distinguished based on lentiviral GFP expression.
Optional: We recommend 2 recipients for each biological replicate containing HSPCs pooled from 3 pups (Figure 2), see discussion (Notes). - Virus: Concentrated lentiviral supernatants containing barcode library
- Retronectin (Takara Bio, Clontech, catalog number: T100A )
- Sterile PBS (GE Healthcare, catalog number: SH30028.02 )
- Antibodies
Ter119 biotin (Biolegend, catalog number: 116204 )
Ter119 PE-Cy5 (Biolegend, catalog number: 116210 )
CD3 PE-Cy5 (Biolegend, catalog number: 100310 )
Gr1 PE-Cy5 (Biolegend, catalog number: 108410 )
B220 PE-Cy5 (Biolegend, catalog number: 103210 )
Sca1 PE-Cy7 (Biolegend, catalog number: 108114 )
c-kit APC (Biolegend, catalog number: 105812 ) - StemSpanTM SFEM (SFEM) (STEMCELL Technologies, catalog number: 09650 )
- Penicillin/Streptomycin (P/S) solution (Nordic Biolabs, catalog number: SV30010 )
- Cytokines
SCF (PreproTech, catalog number: 250-03 )
FLT3 (PreproTech, catalog number: 300-19 )
IL3 (PreproTech, catalog number: 213-13 )
IL6 (PreproTech, catalog number: 216-16 )
IL7 (PreproTech, catalog number: 217-17 ) - Trypan blue (Sigma-Aldrich, catalog number: T6146 )
- Anti-Biotin MicroBeads (Miltenyi Biotec, catalog number: 130-090-485 )
- 7AAD (Sigma-Aldrich, catalog number: A9400 )
- 10% bovine serum albumin (BSA) in Iscove’s MDM (STEMCELL Technologies, catalog number: 09300 )
- Ciprofloxacin, 250 mg/tablet (Teva Pharmaceutical Industries, catalog number: 01 16 40 )
- Agencourt AMPure XP beads (Beckman Coulter, catalog number: A63880 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, catalog number: Q32854 )
- High Fidelity Taq polymerase (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11304011 )
- High Sensitivity DNA Bioanalyzer Kit (Agilent Technologies, catalog number: 5067-4626 )
- BSA ≥ 98% (Sigma-Aldrich, catalog number: A9647 )
- EDTA (Sigma-Aldrich, catalog number: E5134 )
- HBSS without MgCl2 (Thermo Fisher Scientific, GibcoTM, catalog number: 14175053 )
- Sodium chloride (NaCl)
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 74255 )
- Proteinase K
- Staining buffer (see Recipes)
- Lysis buffer (see Recipes)
Equipment
- Dissection tools: fine tipped tweezers and scissors for FL dissection
- Pestle and mortar
- Pipette
- Centrifuge with temperature control (for 50 and 15 ml tubes) (Sigma Laborzentrifugen, model: 3-18K , catalog number: 10290)
- Centrifuge with temperature control (for Eppendorf tubes) (Beckman Coulter, model: Microfuge® 20R centrifuge , catalog number: B31607)
- MACS columns (Miltenyi Biotec, catalog number: 130-042-401 )
- MACS separator magnet (Miltenyi Biotec, model: QuadroMACSTM separator , catalog number: 130-090-976)
- Small animal heat lamp
- Cell culture room and equipment certified for lentivirus work
- Agilent 2100 Bioanalyzer Instrument (Agilent Technologies, model: 2100 Bioanalyzer Instrument )
- Qubit Fluorometer
- DynaMag 96 well plate magnet (Thermo Fisher Scientific, catalog number: 12331D )
- BD FACS Aria III (BD, model: FACS Aria III) or comparable sorter
- Deep sequencing facility and reagents (e.g., Illumina MiSeq system [Illumina, model: MiSeq system ] with MiSeq Reagent Kit v3 150-cycle or the Ion PGM System [Thermo Fisher Scientific, model: Ion PGM System ] with Ion 314 Chip Kit v2)
Procedure
- Lentiviral barcode library
The generation of barcode libraries has been previously reviewed (Bystrykh et al., 2014). The complexity and distribution of barcodes in the virus library impact the risk of labelling of two cells with the same barcode and therefore poses a constraint on the number of single cell fates that can be faithfully traced. Thus, these properties need to be empirically determined for each new barcode library and virus prep (Lu et al., 2011; Naik et al., 2014; Bystrykh and Belderbos, 2016). As a rule of thumb the number of cells traced per recipient should not exceed 10% of the library diversity (Naik et al., 2014). Furthermore, the virus library needs to be titrated using the donor cell type of interest prior to experiment as transduction efficiency can vary greatly from cell type to cell type. In our recent study, a previously characterized barcode library was used (Lu et al., 2011). - Timed pregnancy set-up
Day -14- In the afternoon, add two female mice into each cage containing one male mouse (Figure 2).
- The following morning, separate females from male and check for vaginal plugs.
Note: Detection of a vaginal plug at this time point is counted as 0.5 d.p.c. The presence of a vaginal plug is not a guarantee of the female being pregnant. Likewise, the lack of a vaginal plug does not mean the female is not pregnant. To increase chances of fertilization, more than one cage of timed pregnancy should be set up. After E12 females can be palpated for abdominal distention, bilateral bulging and weight increase to confirm pregnancy. However if the female is pregnant with low number of embryos it can be difficult to distinguish.
- In the afternoon, add two female mice into each cage containing one male mouse (Figure 2).
- Transduction and cell sorting preparations
Day 0- Transduction preparations
- Dilute Retronectin with sterile PBS to 40 μg/ml.
- Coat one well per biological replicate by adding 100 μl of the diluted Retronectin from step C1a to a non-culture treated 96-well plate.
Note: If more than one biological replicate is desired, a litter of 6 embryos could be split up into 2 equal biological replicates to be transduced in 2 separate wells (Figure 2).
Optional: Transduced LSKs from each biological replicate can later be divided into two equal parts and transplanted into two recipients to facilitate data interpretation (see Notes) (Figure 2). - Coat one additional well for an untransduction control.
- Store the plate at 4 °C overnight.
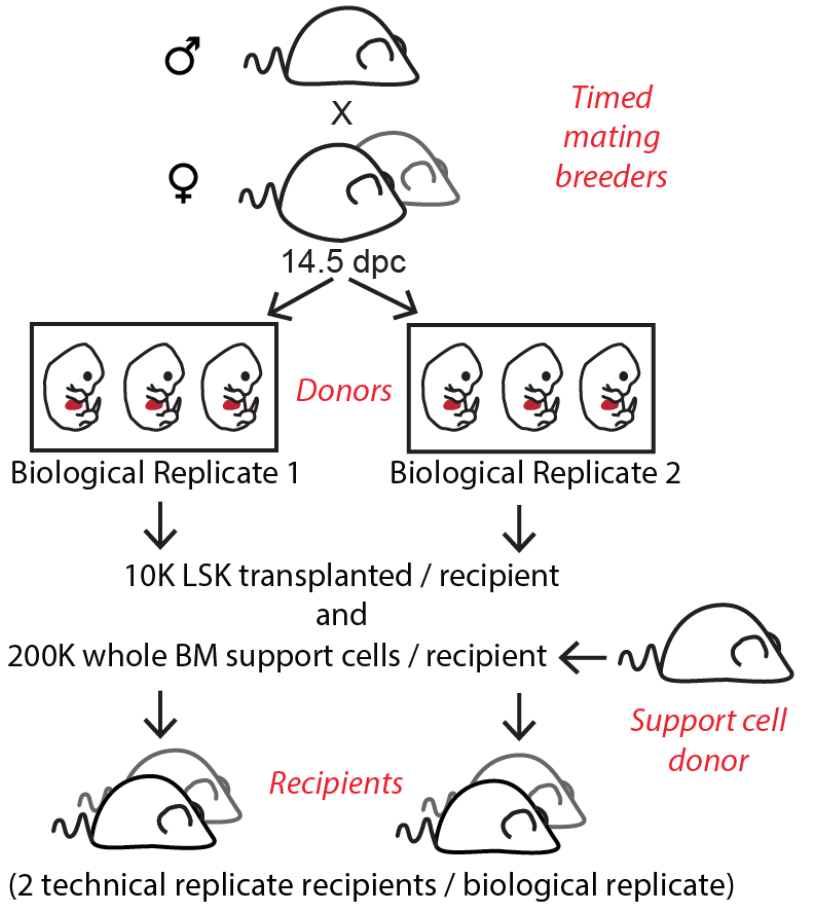
Figure 2. Example of experimental animal usage based on estimated number of pups and cell yield
- Dilute Retronectin with sterile PBS to 40 μg/ml.
- Tools
Autoclave dissection tools, pestle and mortar, tissue paper, Eppendorf tubes. - Cell sorting and culturing preparations
Optional: The following steps can be performed on Day 1.- Prepare LSK antibody cocktail for FACS (store at 4 °C in the dark until use).
Notes:- The fluorochromes listed here are suggestions and can be substituted with other colors.
- Lineage antibodies include: CD3 PE-Cy5, Gr1 PE-Cy5, Ter119 PE-Cy5, B220 PE-Cy5.
Antibody fluorochrome Dilution Final concentration (μg/100 μl) c-Kit APC 1:200 0.1 Sca1 PE-Cy7 1:200 0.1 Lineage Antibodies PE-Cy5 1:400 each 0.05 each - The fluorochromes listed here are suggestions and can be substituted with other colors.
- Prepare antibody dilutions for single stains and appropriate staining controls.
- Prepare cytokine mix in SFEM containing 1% P/S (store at 4 °C until use).
Cytokine Final concentration (ng/ml) SCF 50 IL6 50 FLT3 50 IL3 10 IL7 10
- Prepare LSK antibody cocktail for FACS (store at 4 °C in the dark until use).
- Transduction preparations
- Cell sorting and transduction
Note: All dissections should be performed in appropriate laboratory areas and cell preparations in sterile biosafety cabinets.
Day 1- Harvesting of total BM support cells
- In the morning, euthanize mouse for support cells (e.g., cervical dislocation or CO2 asphyxiation) (Figure 2).
- Dissect hind leg bones (femur, tibia and iliac crest) and collect in 50 ml tube with cold (4 °C) staining buffer (see Recipes). Crush bones using pestle and mortar.
- Make single cell suspension by gently pipetting up and down with a 1 ml pipette tip.
- Filter through a 40 µm cell strainer and adjust volume to 10 ml with staining buffer.
- Store at 4 °C until intravenous injections on Day 2.
- Harvesting of 14.5 d.p.c. FL donor cells
- Euthanize the pregnant female (e.g., cervical dislocation or CO2 asphyxiation) in the morning.
- Lift skin over the peritoneal cavity and make a small incision with scissors.
- Pull out the uterine horns and release the uterus by cutting the cervix.
- Dissect the embryos from the uterine membranes in a 60 mm Petri-dish filled with 5 ml cold (4 °C) staining buffer.
- Place an embryo on autoclaved tissue paper and use fine tipped tweezers to isolate the FL.
- Transfer the FL to a new 60 mm Petri-dish with 1 ml with cold staining buffer per FL and repeat for all embryos, combining only the FLs intended for the same biological replicate into the same Petri-dish.
- Remove any attached connective tissue and intestine prior to dissociating by gentle pipetting using a 1 ml pipette tip.
- Filter the resulting single cell suspension through a 50 µm cup filter and repeat for biological replicates.
- Count the cells and centrifuge cells at 350 x g for 5 min at 4 °C.
Note: Expect approximately 40 x 106/FL including red blood cells, although some variation in cell number can be expected (depending on loss of cells during dissection and size of the embryo). - Remove the supernatant and prepare for Ter119 depletion.
- Euthanize the pregnant female (e.g., cervical dislocation or CO2 asphyxiation) in the morning.
- Ter119 depletion using MACS
Ter119 is expressed by mature erythrocytes and erythroid progenitors which are the most abundant cells in E14.5 FL. Depletion of these cells enrich for the LSK population and shortens sorting time. - Resuspend the cell pellet to a volume of 50 μl staining buffer/107 cells and add Ter119 biotin antibody to a final concentration of 0.5 μg/ml.
- Incubate at 4 °C for 25 min.
- Add 10 ml cold staining buffer and centrifuge at 350 x g for 5 min at 4 °C, remove supernatant.
- Resuspend the pellet to a volume of 40 μl staining buffer/107 cells and add 10 μl of anti-Biotin MicroBeads/107 cells.
- Incubate at 4 °C for 15 min.
- Add 10 ml staining buffer and centrifuge at 350 x g for 5 min at 4 °C, remove supernatant.
- Resuspend cell pellet in 500 μl staining buffer.
- Put MACS columns in MACS separator magnet, placing a 50 µm cup filter on top of each column.
- Equilibrate the columns by running 3 ml buffer through each and discard the liquid flow through.
- Place labeled collection tubes under each column and add the 500 μl samples through the filter onto the column.
- Rinse the original sample containing tube with an additional 500 μl staining buffer and add onto the column through the same filter.
- Add 3 x 3 ml buffer onto each column to rinse off unbound cells.
Note: For each round of 3 ml rinse, wait until the existing buffer has almost entirely run through before adding the next 3 ml. - Count the cells in the 10 ml flow-through.
- Set aside approximately 200K cells for FACS staining controls.
- Centrifuge the remaining cells at 350 x g for 5 min at 4 °C and remove supernatant, the cell pellet should be white.
- Cell staining and sorting
Our workflow using the BD FACS Aria IIu or III is briefly described below.- Resuspend the cell pellets in the LSK antibody cocktail (step C3a) at a volume of 100 μl/107 Ter119 depleted cells.
- Stain the sample and appropriate staining controls in the dark at 4 °C for 30 min.
- For each sample prepare one Eppendorf tube containing 100 μl cold staining buffer for post-sort purity check and one containing 500 μl SFEM to collect the sorted cells.
- Rinse stained cells with staining buffer, centrifuge at 350 x g for 5 min at 4 °C and remove supernatant.
- Resuspend samples in staining buffer containing 7AAD to a final concentration of 1 μg/ml 7AAD.
Note: Volume of buffer to resuspend cells in for sorting depends on the cell number and sort settings (e.g., 7 x 106 cells can be resuspended in 700 μl when sorted on BD FACS Aria III at approximately 4,000 events/sec, flow rate < 4, purity mask 0-32-0, using a 70 μm nozzle). - Set-up staining compensation matrix using single stained controls.
- Set LSK gates as shown in Figure 3.
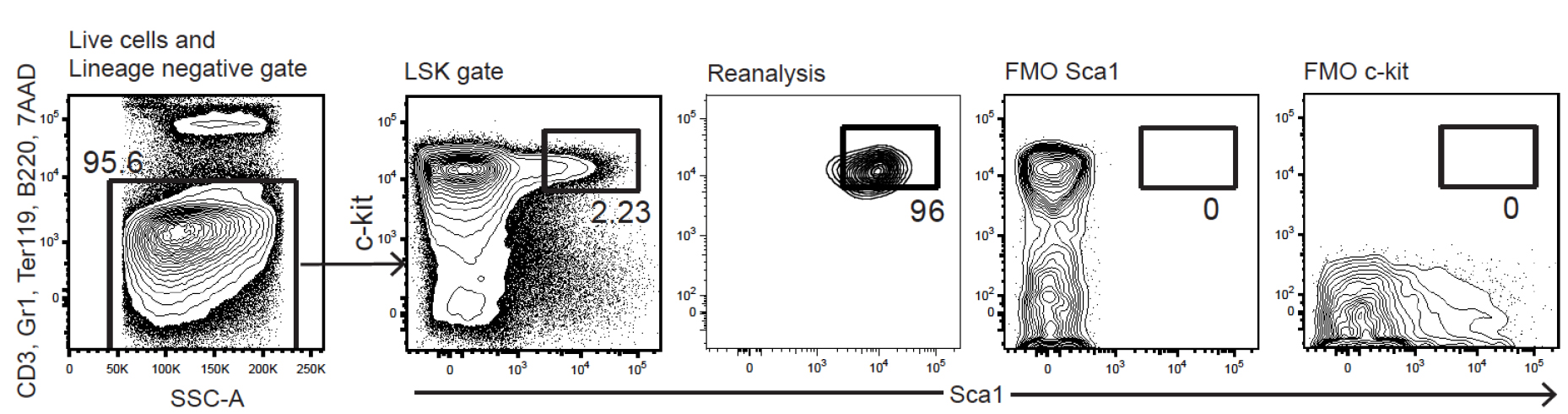
Figure 3. FACS plots for LSK HSPC gating during cell sorting. Fluorescence minus one (FMO) controls for Sca1 and c-Kit are shown to illustrate the stringent gating that we draw to maximize sort purity (reanalysis plot). The expected HSC content within the FL LSK gate varies from 4-10% in our experience. - Take appropriate precautions to ensure sorting purity of > 95%.
- Sort LSK cells for all samples into prepared collection tubes containing 500 μl cold SFEM.
Note: Sort at least 11,000 LSKs for each planned recipient to compensate for cell loss in steps E9-E10. It is possible to obtain enough LSKs from 3 FLs for 2 recipients if technical replicates are desired (see Notes). - Record the cell number yields from the sort and store tubes on ice.
- Resuspend the cell pellets in the LSK antibody cocktail (step C3a) at a volume of 100 μl/107 Ter119 depleted cells.
- Transduction
This step should be performed in an appropriate cell culture hood using procedures approved for lentiviral transduction.
Note: HSPCs from fetal liver and adult bone marrow differ in transduction efficiency with less virus required to achieve the same transduction efficiency in fetal HSPCs. Before starting the protocol titrate your lentiviral barcoding library specifically for the population of choice, determining the amount of virus needed to achieve a transduction efficiency that ensures that the number of transduced cells per recipient does not exceed 10% of the complexity of the lentiviral barcode library. For an estimated barcode complexity of 80K (Lu et al., 2011), we aimed for a transduction efficiency of 15-30% meaning that 1,500-3,000 cells out of the 10K cells injected per recipient mouse were barcoded (Figure 4). This routinely yielded hundreds of cell fates traced in each recipient out of which around 60 clones demonstrated functional characteristics of active HSCs and minimal instances in which the same barcode transduced more than one cell (Kristiansen et al., 2016) (see Notes).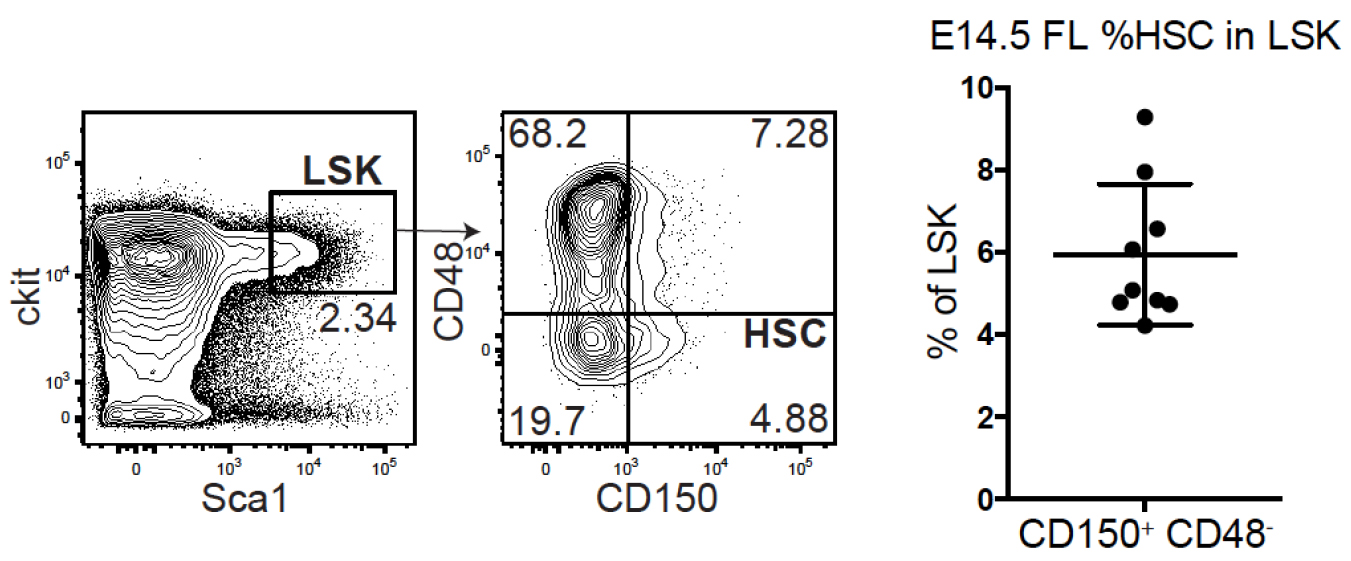
Figure 4. HSC frequency within the FL LSK population. (Left) representative FACS plots for FL HSCs defined by LSK CD48-CD150+. (Right) %frequency of HSCs within LSK compiled from 6+3 individual embryos from 2 litters in 2 separate experiments. - Remove Retronectin from the wells.
- Immediately add 200 μl 2% BSA (Note: BSA in Iscove’s MDM diluted in PBS.) to each of the coated wells.
- Incubate the plate for 30 min at room temperature.
- Set aside approximately 300 LSK cells in a separate tube for an untransduced control.
- Centrifuge tubes containing LSK cells at 350 x g for 5 min at 4 °C and remove supernatant.
Note: When working with low numbers of sorted cells, every precaution to minimize cell loss must be taken to get reproducible results. Therefore, omit further counting and filtering steps and always leave an appropriate volume when removing supernatant following centrifugation so as not to disturb the cell pellet. - Resuspend cell pellet to a final volume of 150 μl cytokine mix.
- Remove 2% BSA from the coated wells and plate the sorted cells from each biological replicate into a single well. Also plate the LSK cells set aside from step D5d into the well designated for the untransduced control (step C1c).
- Add the previously optimized amount of lentivirus to each well except for the untransduced control well.
- Gently rock the plate to disperse the virus.
- Incubate in humidified CO2 incubator at 37 °C and 5% CO2 overnight.
- Irradiation of recipient mice
- Irradiate recipients with 2 x 450 cGy with 3 h between the doses. Transplantation should be performed within 24 h of the first dose.
Note: Split dose irradiation is more gentle on the mice while still achieving the desired effects of lethal myeloablation. Recipient mice do not need treatment with antibiotics prior to irradiation. - Add Ciprofloxacin to a final concentration of 125 mg/L in the drinking water for 14 days.
- Irradiate recipients with 2 x 450 cGy with 3 h between the doses. Transplantation should be performed within 24 h of the first dose.
- Intravenous injections
The LSK population is heterogeneous containing stem and progenitor cells that survive and proliferate differently in culture. To minimize cell loss we omit additional cell counting steps and treat the donor cell number on Day 2 as equivalent to the number of sorted cells from Day 1.
Note: The following steps must strictly adhere to biosafety protocols for lentiviral handling and approved animal use protocols.
Day 2- In the morning, filter support cells from Day 1 through a 40 µm cell strainer as significant aggregation may have taken place over night. Count the cells.
Note: The cell count includes red blood cells. - Centrifuge support cells at 350 x g for 5 min at 4 °C and resuspend in cold sterile PBS to a concentration of 200K/100 μl.
- Collect each transduced LSK cell sample in autoclaved Eppendorf tubes containing 500 μl cold PBS.
Note: To minimize cell loss associated with filtering, donor cells are collected directly from the well for transplantation. - Rinse each well with at least 2 rounds of 200 μl PBS to recover all cells and combine in corresponding Eppendorf tubes.
Note: Check that all cells have been harvested from the wells using a microscope. - Transfer a small volume of each sample back to their original well for transduction efficiency readout on Day 3. Add cytokine mix to a final volume of 150 μl and incubate the cells at 37 °C in 5% CO2 overnight.
- Add an additional 500 μl cold PBS to each sample tube and centrifuge at 350 x g for 5 min at 4 °C.
- Dispose of lentiviral supernatant according to appropriate biosafety protocols.
- Add 200K support cells (100 μl from step E2)/10K sorted and transduced FL LSKs and add 1 ml cold PBS.
- Centrifuge samples at 350 x g for 5 min and discard supernatant according to appropriate biosafety protocols.
- Resuspend the pellet in 275 μl cold PBS/11,000 sorted and transduced FL LSKs (step D4i).
- Inject 250 μl (the equivalent of 10,000 sorted and transduced LSKs) of sample into the tail-vein of each recipient using a 29 gauge ½ inch 1 ml needle.
Note: We routinely warm up the recipient mice for 5 min under a heat lamp to facilitate intravenous injections.
- In the morning, filter support cells from Day 1 through a 40 µm cell strainer as significant aggregation may have taken place over night. Count the cells.
- Transduction efficiency readout
Day 3- Collect the transduced cells that were set aside on Day 2 for transduction efficiency readout (step E5) and the untransduced control into individual FACS tubes.
- Centrifuge samples at 350 x g for 5 min at 4 °C and remove supernatant.
- Resuspend each sample in 150 μl staining buffer containing 7AAD.
- Analyze by FACS the percentage GFP+ events among of live (7AAD-) cells (Figure 5).
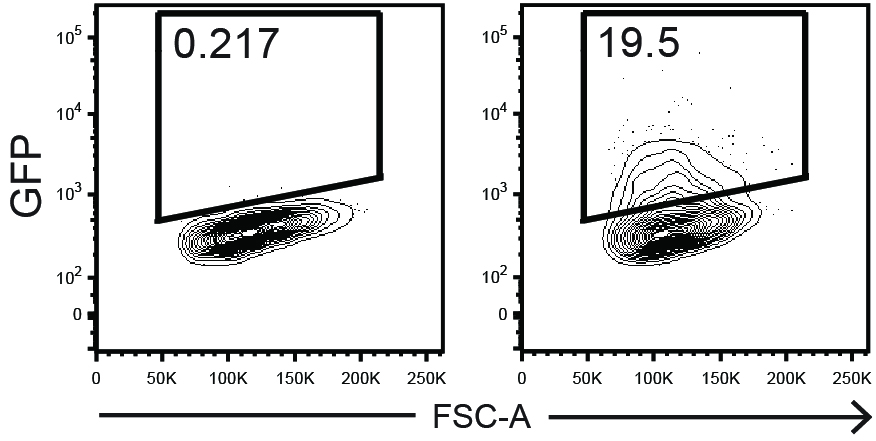
Figure 5. Example of GFP transduction efficiency readout on Day 3. 7AAD- singlet LSK cells from untransduced control (left) and transduced sample (right) are shown.
- Collect the transduced cells that were set aside on Day 2 for transduction efficiency readout (step E5) and the untransduced control into individual FACS tubes.
- Peripheral blood analysis
4-8 weeks post transplantation
We recommend checking initial engraftment of GFP+ barcoded cells in the peripheral blood. This is done by tail vein bleeding of recipient mice and assessment the percentage GFP+ cells e.g., among granulocytes and B cells using FACS.
Note: We frequently observe a higher GFP% in the peripheral blood compared to the transduction efficiency readout on Day 3 with FL HSPCs. This is likely due to a preferential transduction of highly proliferating HSPCs that engraft better. - Barcode extraction
Clonal dynamics of long-term HSCs stabilize after approximately 12 weeks post transplantation (Verovskaya et al., 2013). Therefore, we recommend reading out barcode distribution no earlier than 16 weeks post transplantation.
16-20 weeks post transplantation- Cell sorting and lysis
- Cell populations of interest based on the specific questions posed are FACS sorted. Cells are sorted into 500 μl staining buffer in non-stick tubes as described for step D4 and can be stored at -80 °C as cell pellets initially.
Note: Granulocytes have a short turnover time of a few days and is therefore a good representation of recent HSC activity. Lymphocytes on the other hand can survive for weeks and even months and will therefore contain barcodes derived from both long and short term progenitors at the time of barcode labelling. - Prepare cell lysis buffer (see Recipes).
- Cell pellets are lysed for genomic DNA in 100 μl cell lysis buffer/sample (2 h at 56 °C + 10 min at 95 °C to inactivate the proteinase K) within a week of sorting and can subsequently be stored at -20 °C.
Note: With a smaller number of cells it is recommended to sort directly into lysis buffer for immediate lysis. Cell numbers are a limiting factor to getting a good sequencing coverage of the population.
- Cell populations of interest based on the specific questions posed are FACS sorted. Cells are sorted into 500 μl staining buffer in non-stick tubes as described for step D4 and can be stored at -80 °C as cell pellets initially.
- Library preparation and barcode deep sequencing
- Genomic DNA is purified using Ampure XP beads according to manufacturer’s recommendations.
- Measure genomic DNA concentration using a high sensitivity method e.g., the Qubit Fluorometer and the Qubit dsDNA HS Assay Kit.
- Up to 100 ng DNA from each sample is divided equally into two technical replicate PCR reactions (50 ng each) using the appropriate primers and a high fidelity DNA Taq polymerase (e.g., Q5 Taq DNA polymerase, NEB) for amplification of the barcode fragment (Figure 1). The amplified fragments are purified using Ampure XP beads according to manufacturer’s recommendations.
Optional: Depending on the design of PCR primers and the choice of sequencing platform, a second step of PCR amplification can be performed to add multiplexing adapters. - Amplicons are quantified using the Bioanalyzer HS DNA Analysis Kit and according to manufacturers’ instructions. A product of the correct size without contamination of primer dimers is critical for successful sequencing. The presence of primer dimers can be removed with another round of Ampure XP bead purification according to manufacturer’s recommendations.
- Pool equivalent amounts of amplified and indexed barcode libraries for sequencing–the required concentration of product depends on the sequencing platform of choice.
Note: The extent of multiplexing should be informed by the read capacity of the sequencing platform of choice and the expected complexity of your barcoded cells. - Sequencing is performed by experienced technical personnel according to manufacturer’s instructions.
Note: We have thus far had good success using the Illumina MiSeq system (MiSeq Reagent Kit v3 150-cycle, Illumina) and the Ion PGM (Ion 314 Chip Kit v2, Thermo Scientific) platforms.
- Genomic DNA is purified using Ampure XP beads according to manufacturer’s recommendations.
- Cell sorting and lysis
Data analysis
The initial steps of cellular barcode analysis aims at generating a list of reliable barcodes for each sorted sample coupled with the barcode read frequency reflecting the abundance of each clone within the population. The most crucial part of this analysis is filtering out barcodes introduced by e.g., sequencing errors, which would otherwise bias the results. The filtering steps are customized for the barcode library of choice and experimental design. For example, if a reference library exists for the barcode library of choice, the sequenced barcodes can be mapped back to the reference and the filtering step can be streamlined. Here, we briefly provide an example for the analysis of randomly generated barcode libraries lacking a known reference (Figure 1; Kristiansen et al., 2016). For in depth recommendations, we have provided a number relevant references.
- Barcodes are isolated by the identification of flanking sequences using custom R scripts and the R package ShortReads.
- Correct for sequencing errors by subjecting each barcode list to e.g., the Starcode algorithm (Zorita et al., 2015). Based on Levenshtein distances, daughter barcodes generated by sequencing errors are identified and removed while their read frequencies are merged with that of the parent barcode.
- The cut-off for low frequency barcodes is individually defined for each sorted population based on the total read number retrieved and the cell number sampled such that barcodes that fall below the expected read representation of a single cell are excluded from further analyses.
- The barcode lists obtained from the two technical PCR replicates of each population are assessed for correlation of read frequencies with a criterion of r > 0.85 as previously described (Naik et al., 2014). Only barcodes that pass all filtering criteria in both technical replicates are considered for further analysis.
Note: These stringent filtering criteria produce a conservative barcode list for downstream analyses. However, it is important to note that these filtering steps may exclude true barcodes of very low representation (Naik et al., 2014) (Figure 1). - Barcode lists can be further analyzed for overlap, read frequency distributions etc. using custom scripts.
Notes
- For kinship analysis of cells of different lineages, qualitative barcode overlap is a critical readout. To avoid false positive barcode overlap readout, it is particularly important to control that the same barcode does not enter more than one cell. This can be done by comparing the filtered barcode lists from two technical replicate recipients (Figure 2) that received LSK cells transduced in the same well (Naik et al., 2014). Since the short virus incubation time is not likely to permit HSC division events, any overlap between the two recipients can be considered as noise. The current protocol is designed to accommodate two technical replicate recipients per biological replicate (Figure 2) by simply sorting (step D4i) and plating double the number of LSK cells per Retronectin coated well. In choosing populations for barcode content readout, we also recommend including two populations with expected barcode overlap as a positive control. These controls will allow for proper validation of the signal versus noise ratio in your experimental set up (Kristiansen et al., 2016).
- If the experimental set up is sound, and the appropriate controls and barcode filtering steps are applied, reproducibility between individual experiments should be high. However, donor HSPCs of different ages vary significantly in their propensity to become transduced and will yield age specific barcode distributions.
Recipes
- Staining buffer
0.5% BSA
Note: Use BSA ≥ 98%.
2 mM EDTA
in HBSS without MgCl2 - Lysis buffer
0.1 M Tris-HCl pH 8.5
0.5 mM EDTA pH 8
0.2 M NaCl
0.2% SDS
0.1 mg/ml proteinase K
Note: To be added to lysis buffer immediately before use.
Acknowledgments
This protocol was originally described in and adapted from Kristiansen et al. (2016). This work was supported by grants from the Swedish Cancer Foundation, the Swedish Research Council, StemTherapy, and The Knut and Alice Wallenberg Foundation.
References
- Bystrykh, L. V. and Belderbos, M. E. (2016). Clonal analysis of cells with cellular barcoding: When numbers and sizes matter. Methods Mol Biol 1516: 57-89.
- Bystrykh, L. V., de Haan, G. and Verovskaya, E. (2014). Barcoded vector libraries and retroviral or lentiviral barcoding of hematopoietic stem cells. Methods Mol Biol 1185: 345-360.
- Gerrits, A., Dykstra, B., Kalmykowa, O. J., Klauke, K., Verovskaya, E., Broekhuis, M. J., de Haan, G. and Bystrykh, L. V. (2010). Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood 115(13): 2610-2618.
- Kristiansen, T. A., Jaensson Gyllenback, E., Zriwil, A., Bjorklund, T., Daniel, J. A., Sitnicka, E., Soneji, S., Bryder, D. and Yuan, J. (2016). Cellular barcoding links B-1a B cell potential to a fetal hematopoietic stem cell state at the single-cell level. Immunity 45(2): 346-357.
- Lu, R., Neff, N. F., Quake, S. R. and Weissman, I. L. (2011). Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat Biotechnol 29(10): 928-933.
- Naik, S. H., Perie, L., Swart, E., Gerlach, C., van Rooij, N., de Boer, R. J. and Schumacher, T. N. (2013). Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 496(7444): 229-232.
- Naik, S. H., Schumacher, T. N. and Perie, L. (2014). Cellular barcoding: a technical appraisal. Exp Hematol 42(8): 598-608.
- Schepers, K., Swart, E., van Heijst, J. W., Gerlach, C., Castrucci, M., Sie, D., Heimerikx, M., Velds, A., Kerkhoven, R. M., Arens, R. and Schumacher, T. N. (2008). Dissecting T cell lineage relationships by cellular barcoding. J Exp Med 205(10): 2309-2318.
- Verovskaya, E., Broekhuis, M. J., Zwart, E., Ritsema, M., van Os, R., de Haan, G. and Bystrykh, L. V. (2013). Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood 122(4): 523-532.
- Zorita, E., Cusco, P. and Filion, G. J. (2015). Starcode: sequence clustering based on all-pairs search. Bioinformatics 31(12): 1913-1919.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kristiansen, T. A., Doyle, A. and Yuan, J. (2017). Lentiviral Barcode Labeling and Transplantation of Fetal Liver Hematopoietic Stem and Progenitor Cells. Bio-protocol 7(8): e2242. DOI: 10.21769/BioProtoc.2242.
Category
Immunology > Animal model > Mouse
Cell Biology > Cell engineering > Barcoding
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.