- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Transient Transfection-based Fusion Assay for Viral Proteins

Published: Vol 7, Iss 5, Mar 5, 2017 DOI: 10.21769/BioProtoc.2162 Views: 12497
Reviewed by: Yannick DebingMarielle CavroisBalasubramanian Venkatakrishnan
Original research article
The authors used this protocol in:

Mar 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Membrane fusion is vital for entry of enveloped viruses into host cells as well as for direct viral cell-to-cell spread. To understand the fusion mechanism in more detail, we use an infection free system whereby fusion can be induced by a minimal set of the alphaherpesvirus pseudorabies virus (PrV) glycoproteins gB, gH and gL. Here, we describe an optimized protocol of a transient transfection based fusion assay to quantify cell-cell fusion induced by the PrV glycoproteins.
Keywords: HerpesvirusBackground
Membrane fusion is essential for entry and spread of enveloped viruses. Many enveloped viruses require only one or two viral proteins to mediate attachment to host cells and membrane fusion, and the molecular mechanisms are well understood (Harrison, 2015). In contrast, herpesviruses use a more complex mechanism requiring a receptor-binding protein and the core fusion machinery composed of gB and the heterodimeric gH/gL complex for infectious entry. The mechanism leading to fusion of herpesvirus envelopes with cellular membranes is only incompletely understood. Detailed knowledge of the molecular basis of herpesvirus entry and spread is important for efficient countermeasures against a variety of diseases. A better understanding is aided by studying the cell fusion activity of cells transiently expressing the relevant proteins. Different model systems, whereby fusion is induced with a minimal set of the core fusion machinery represented by glycoproteins gB and gH/gL and receptor-binding gD, in the absence of infection, have been developed, for example, for herpes simplex viruses type 1 and 2 (HSV-1 and 2 [Turner et al., 1998; Muggeridge, 2000; McShane and Longnecker, 2005]). Unlike HSV-1 and 2, PrV does not require signaling of gD for membrane fusion during direct cell-to-cell spread, reducing the number of relevant proteins to three (Schmidt et al., 1997). These systems are also used to quantify membrane fusion. However, the evaluation or quantitation of fusion activity, which is often based on counting the number of nuclei of a formed syncytium, is very time consuming. Our incentive to develop the present protocol was to improve the current protocol to facilitate and accelerate evaluation, and make results more robust and comparable by combining important factors like size and number of formed syncytia. Here, we describe an optimized protocol for an in vitro transient transfection based cell-cell fusion assay to quantify membrane fusion induced by the PrV glycoproteins gB and gH/gL (Schröter et al., 2015). However, we know that this assay also is functional with other fusion-active glycoproteins, not just those of pseudorabies virus.
Materials and Reagents
- 1.5 ml tubes (e.g., Fisher Scientific, catalog number: S348903 )
- 24-well cell culture plate (e.g., Corning, Costar®, catalog number: 3527 )
- Pipette tips 10 µl (TipOne) (STARLAB INTERNATIONAL, catalog number: S1110-3000 )
- Pipette tips 1 ml (Greiner Bio One International, catalog number: 740290 )
- Pipette tips 100 µl (Greiner Bio One International, catalog number: 739290 )
- pcDNA3 (Thermo Fisher Scientific, Invitrogen)
Note: pcDNA3 from Invitrogen is no longer available. Alternatively, pcDNA3.1(+) (Thermo Fisher Scientific, catalog number: V79020 ) can be used. - Rabbit kidney (RK13) cells (Collection of Cell Lines in Veterinary Medicine-RIE 109)
- MEM Eagle (Hank’s salts and L-glutamine) (Sigma-Aldrich, catalog number: M4642 )
- MEM (Earle’s salts) (Thermo Fisher Scientific, catalog number: 61100061 )
- Sodium bicarbonate (NaHCO3) (Carl Roth, catalog number: 6885.1 )
- NEA (nonessential amino acids) (Biochrom, catalog number: K 0293 )
- Na-pyruvate (EMD Millipore, catalog number: 106619 )
- Tris-HCl (pH 8.5)
- Fetal bovine serum (FBS) (Biowest, catalog number: S181G )
- Lipofectamine® 2000 reagent (Thermo Fisher Scientific, catalog number: 11668027 )
- Opti-MEM® reduced serum medium (Thermo Fisher Scientific, catalog number: 31985062 )
- Sodium chloride (NaCl) (Carl Roth, catalog number: 9265.1 )
- Potassium chloride (KCl) (Carl Roth, catalog number: 5346.1 )
- Dextrose (Sigma-Aldrich, catalog number: D9434 )
- Trypsin (1:250) powder (Thermo Fisher Scientific, catalog number: 27250018 )
- Ethylenediaminetetraacetic acid (EDTA) (SERVA Electrophoresis, catalog number: 11280.01 )
- Paraformaldehyde (Carl Roth, catalog number: 0335.1 )
- 10% growth media (see Recipes)
- 3% paraformaldehyde (see Recipes)
- 10 mM Tris HCl (pH 8.5) (see Recipes)
- Alsever’s-Trypsin Versen (ATV-) solution (see Recipes)
Note: Solutions and media #6, 11, 14, 15, 16, 21, 22, 23, 24 and 26 should be kept at 4 °C.
Equipment
- T75 flask (e.g., Corning, catalog number: 430725U )
- Incubator (e.g., Panasonic, Sanyo, model: MCO-19AIC or Fisher Scientific, catalog number: 12826756 )
- Fluorescence microscope (e.g., Nikon Instruments, model: Eclipse Ti-S)
- Super high pressure mercury lamp power supply (e.g., Nikon Instruments, model: C-SCH1 )
- Light source (e.g., Nikon Instruments, model: LH-M100C-1 )
- Vortexer (e.g., Fisher Scientific, catalog number: S96461A )
- Laminar flow hood (Thermo Fisher Scientific, Thermo ScientificTM, model: Safe 2020 Class II , catalog number: 51026637)
- Centrifuges for 1.5 ml tubes (e.g., Eppendorf, model: 5415 D )
- Pipette controller (e.g., pipetboy acu 2, VWR, catalog number: 37001-856 )
- Nanophotometer (e.g., VWR, IMPLEN, model: P330, catalog number: CA11027-294 )
Software
- Computer running software NIS-Elements V 4.00.01 (Nikon, Düsseldorf, Germany)
Procedure
- Preparation of plasmids
- Dilute the plasmids expressing the putative fusion proteins to 200 ng/µl with Tris-HCl pH 8.5 (see Recipes). Include a plasmid expressing an autofluorescent protein (e.g., GFP or mCherry) as a marker to facilitate evaluation of the assay. Here, a concentration of 100 ng/µl is sufficient.
Note: The expression plasmids we use for our experiments are based on pcDNA3, a mammalian expression vector with constitutive transgene expression under control of the human cytomegalovirus immediate-early 1 promoter/enhancer complex and a neomycin-resistance pEGFP-N1 as marker to facilitate evaluation of the assay (Schröter et al., 2015).
- Dilute the plasmids expressing the putative fusion proteins to 200 ng/µl with Tris-HCl pH 8.5 (see Recipes). Include a plasmid expressing an autofluorescent protein (e.g., GFP or mCherry) as a marker to facilitate evaluation of the assay. Here, a concentration of 100 ng/µl is sufficient.
- Preparation of cells
- Grow rabbit kidney (RK13) cells in Eagle’s minimum essential medium supplemented with 10% FBS. Trypsinize cells with ATV (Recipe 3) and seed ~1.8 x 105 cells per well onto 24-well cell culture dishes.
- On the following day, use cells for transfection. Cells should have a confluency of 80-90%.
Note: Cell confluency for optimal transfection efficiency varies depending on the cell type. For RK13 and Vero cells 80-90% confluency is suggested.
- Grow rabbit kidney (RK13) cells in Eagle’s minimum essential medium supplemented with 10% FBS. Trypsinize cells with ATV (Recipe 3) and seed ~1.8 x 105 cells per well onto 24-well cell culture dishes.
- Transfection
- Prepare DNA-mixture and Lipofectamine-mixture in separate tubes.
- DNA-mixture:
Use 200 ng of each of the expression plasmids and dilute the DNA-mixture in 50 µl Opti-MEM. - Lipofectamine-mixture:
Use 1 µl Lipofectamine 2000TM in 50 µl Opti-MEM per well. Add Lipofectamine drop-wise to 50 µl Opti-MEM and mix gently. Incubate Lipofectamine-mixture for 5 min at room temperature.
Note: The optimal amount of Lipofectamine varies depending on the cell type. For RK13 and Vero cells 1 µl per 24-well is suggested. - Add Lipofectamine-mixture drop-wise into DNA-mixture, gently tap the solution and incubate for 20 min at room temperature (RT) (Video 1).
Note: It is important to mix gently but thoroughly to allow formation of the DNA-lipid complex.
 Video 1. Tutorial: How to add Lipofectamine-mixture drop-wise to DNA-mixture
Video 1. Tutorial: How to add Lipofectamine-mixture drop-wise to DNA-mixture - Remove media from the cells and add the transfection-solution drop-wise directly to the cell-monolayer (Video 2).
Note: To allow equal distribution of the transfection-solution add it circularly drop-by-drop onto the monolayer (Video 2).
Video 2. Tutorial: How to add transfection solution to the cell-monolayer - Add an additional 200 µl Opti-MEM to the cells to prevent drying of cells.
- Incubate cells at 37 °C and 5% CO2 for 3 h.
- After 3 h, remove the transfection-solution from the cells and add fresh MEM supplemented with 2% FBS to the cells and incubate them 18-24 h at 37 °C and 5% CO2. For an overview of the whole procedure see Figure 1.
Note: To achieve comparable results, use exactly the same incubation time in each experiment before fixing the cells.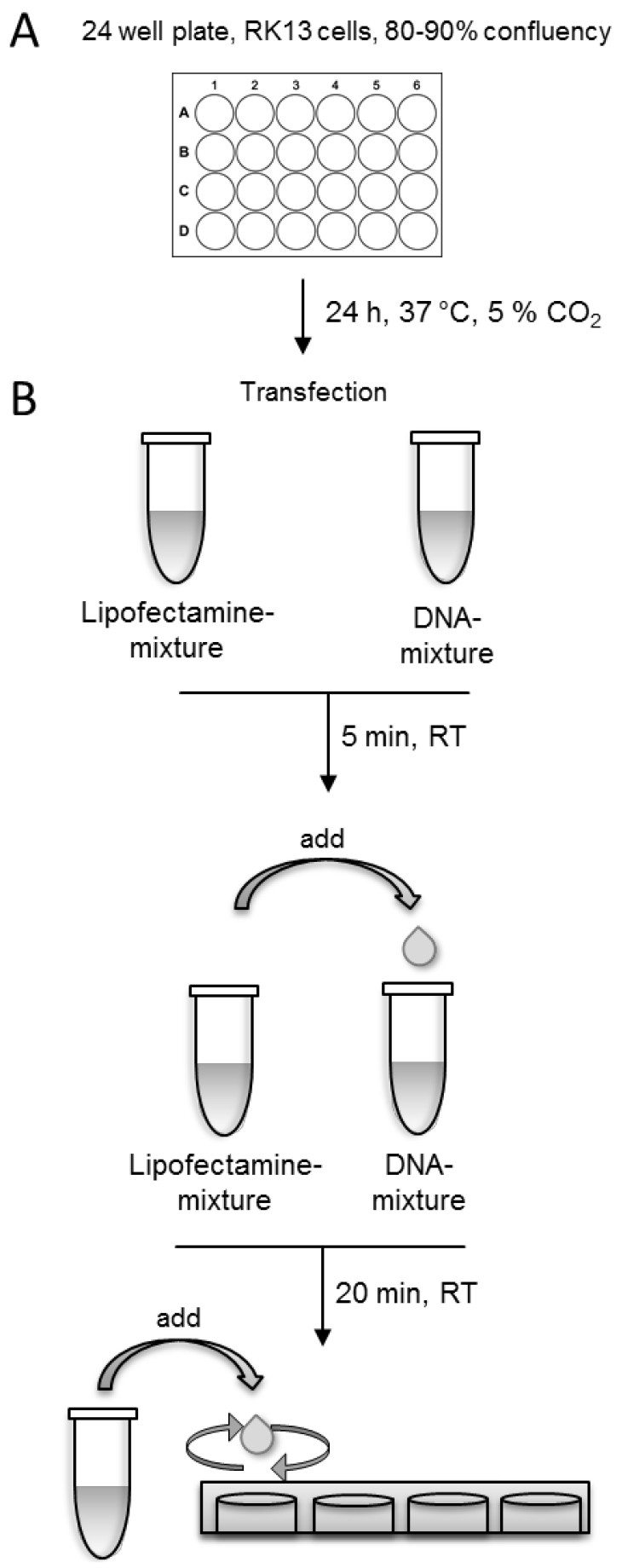
Figure 1. Schematic presentation of the transfection procedure
- Fixation
- Prepare 3% PFA (see Recipes).
- Remove media and wash cell monolayer for 2 min with ~1 ml PBS per well using a rocker at RT.
- Remove PBS and fix cells with 200 µl 3% PFA per well. Fix cells for 20 min at RT.
- Wash cells 2 x with 1 ml PBS per well.
- Add 1 ml PBS per well and store cells at 4 °C.
- Prepare 3% PFA (see Recipes).
- Determination of fusion activity
Note: To quantify fusion activity the number and the area of green-fluorescing syncytia with three or more nuclei is measured using a Nikon Eclipse Ti-S fluorescence microscope and Nikon NIS-Elements imaging software.- Use fixed cells for determination of fusion activity by fluorescence microscopy.
- Open NIS-Elements imaging software (Nikon) and select corresponding type of fluorescence and objective (e.g., FITC, 10x) (see Figure 2 red boxes).
- Select Measure Manual Measurement Area (Figure 2A, Video 3).
Note: NIS-Elements supports units including: pixels, nanometers, micrometers, millimeters, centimeters, decimeters, meters, inches, and mils. For our measurements we use micrometers.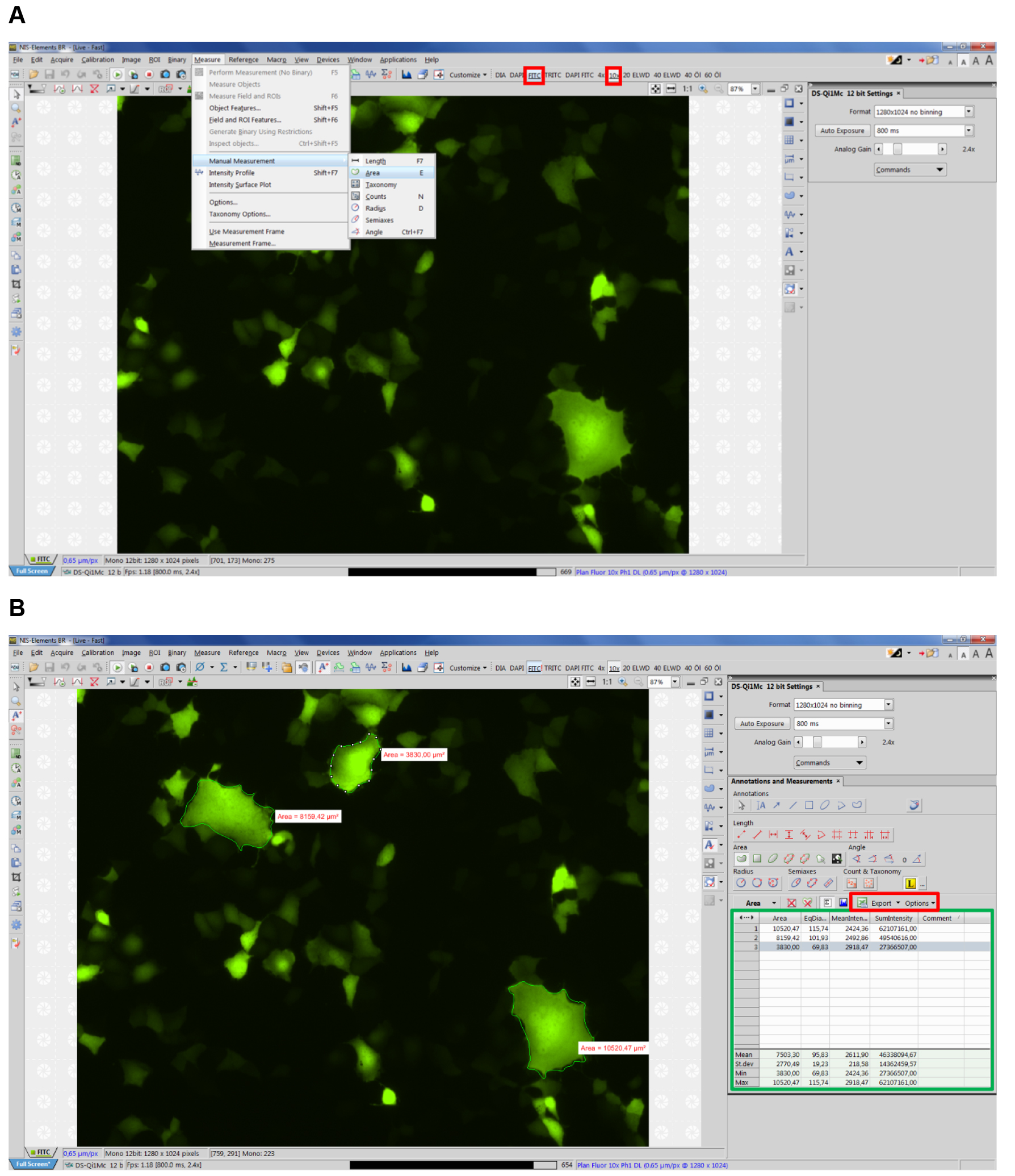
Figure 2. Example view of NIS Elements software. A. Selecting manual measurement; B. Exporting data.
Note: First, measure the area of syncytia on the right-hand corner of the screen, since each syncytium will be marked with a white icon on its right site, which can hinder the view over syncytia to the right of it (see Figure 2B). - Measure all apparent syncytia in 10 fields of view (~5.5 mm2 each), each time starting in the top right-hand corner of the corresponding image. The data, including number of each syncytium as well as mean syncytia area will appear in a table on the lower right-hand corner (see Figure 2B green box, Video 3).
Video 3. Tutorial: How to measure syncytia with NIS Elements software - Export the data directly to an Excel file or a text file by clicking the export options button (see Figure 3 red box). (Select whether to export the data to an Excel file or a text file.) You can modify the name and path of the file via the browse button.
- Repeat the entire experiment at least three times on three different days to calculate the relative fusion activity and corresponding standard deviations (SD).
- Use fixed cells for determination of fusion activity by fluorescence microscopy.
Data analysis
- To calculate the total fusion activity for each sample, corresponding to a given well, multiply the number of syncytia with the mean syncytia area of the 10 fields of view.
E.g., you counted 107 syncytia and the mean syncytia area was 7,048 µm2, the fusion activity would be 107 x 7,048.6 = 754,200.2. - Calculate the standard deviation (SD) of the total fusion activity of each sample by comparing the fusion activities estimated for each of the three independent experiments.
- The relative fusion activity of the different samples is determined by comparing their total fusion activity to the positive control (e.g., wild-type gB, gH and gL), which should be set as 100%.
E.g., if the total fusion activity of the positive control is 754,200.2 and the fusion activity of the test sample was 431,708.6, the relative fusion activity of the test sample would be (100/754,200.2) x 431,708.6 = 57% (Figure 3).
Note: Use Excel/Prism or comparable tools to facilitate calculations and create graphs. - Representative data
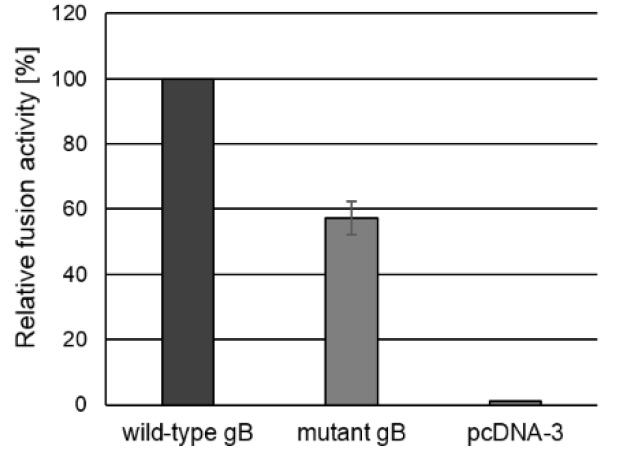
Figure 3. Fusion activity. To test for fusion activity RK13 cells were cotransfected with expression plasmids encoding wild-type gB or mutated gB, and gH, gL and gD as well as EGFP to facilitate evaluation of the assay by fluorescence microscopy. Assays conducted with a plasmid encoding wild-type gB in combination with wild-type gH, gL and gD were set as 100%. Assays with the empty expression vector pcDNA3 served as negative control. One day after transfection, the areas of cells containing three or more nuclei were measured and multiplied by the number of syncytia to determine the fusion activity. Corresponding standard deviations for three independent assays were calculated.
Recipes
- 10% growth media (1 L), pH 7.2
The following reagents should be dissolved in double distilled water to a final volume of 1 L:
5.32 g MEM Eagle (Hank’s salt)
4.76 g MEM (Earle’s salt)
1.25 g NaHCO3
10 ml NEA
120 mg Na-pyruvate - 10 mM Tris-HCl, pH 8.5
Prepare 2 M stock solution (500 ml):
121.14 g Tris HCl
Adjust pH to 8.5 with ~50 ml fuming HCl
Dilute stock solution 1:20 with ddH2O - Alsever’s-Trypsin Versen (ATV-)solution, pH 7.2
8.5 g NaCl
0.4 g KCl
1.0 g dextrose
0.58 g NaHCO3
0.5 g trypsin 1:250
0.2 g EDTA
Ad 1 L ddH2O - 3% PFA (100 ml)
3 g paraformaldehyde
100 ml PBS
Let PFA dissolve at 65 °C
Acknowledgments
This protocol was adapted and modified from a previous study (Schroter et al., 2015). This work was supported by grants from DFG (ME 854/11).
References
- Harrison, S. C. (2015). Viral membrane fusion. Virology 479-480: 498-507.
- Turner, A., Bruun, B., Minson, T. and Browne, H. (1998). Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol 72(1): 873-875.
- Muggeridge, M. I. (2000). Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J Gen Virol 81(Pt 8): 2017-2027.
- McShane, M. P. and Longnecker, R. (2005). Analysis of fusion using a virus-free cell fusion assay. Methods Mol Biol 292: 187-196.
- Schmidt, J., Klupp, B. G., Karger, A. and Mettenleiter, T. C. (1997). Adaptability in herpesviruses: glycoprotein D-independent infectivity of pseudorabies virus. J Virol 71(1): 17-24.
- Schröter, C., Vallbracht, M., Altenschmidt, J., Kargoll, S., Fuchs, W., Klupp, B. G. and Mettenleiter, T. C. (2015). Mutations in pseudorabies virus glycoproteins gB, gD, and gH functionally compensate for the absence of gL. J Virol 90(5): 2264-2272.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Vallbracht, M., Schröter, C., Klupp, B. G. and Mettenleiter, T. C. (2017). Transient Transfection-based Fusion Assay for Viral Proteins. Bio-protocol 7(5): e2162. DOI: 10.21769/BioProtoc.2162.
Category
Microbiology > Microbe-host interactions > Virus
Microbiology > Microbe-host interactions > In vitro model > Cell line
Biochemistry > Lipid > Lipid binding
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.