- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Polysome Fractionation to Analyze mRNA Distribution Profiles

Published: Vol 7, Iss 3, Feb 5, 2017 DOI: 10.21769/BioProtoc.2126 Views: 32315
Reviewed by: Antoine de MorreeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Eukaryotic cells adapt to changes in external or internal signals by precisely modulating the expression of specific gene products. The expression of protein-coding genes is controlled at the transcriptional and post-transcriptional levels. Among the latter steps, the regulation of translation is particularly important in cellular processes that require rapid changes in protein expression patterns. The translational efficiency of mRNAs is altered by RNA-binding proteins (RBPs) and noncoding (nc)RNAs such as microRNAs (Panda et al., 2014a and 2014b; Abdelmohsen et al., 2014). The impact of factors that regulate selective mRNA translation is a critical question in RNA biology. Polyribosome (polysome) fractionation analysis is a powerful method to assess the association of ribosomes with a given mRNA. It provides valuable information about the translational status of that mRNA, depending on the number of ribosomes with which they are associated, and identifies mRNAs that are not translated (Panda et al., 2016). mRNAs associated with many ribosomes form large polysomes that are predicted to be actively translated, while mRNAs associated with few or no ribosomes are expected to be translated poorly if at all. In sum, polysome fractionation analysis allows the direct determination of translation efficiencies at the level of the whole transcriptome as well as individual mRNAs.
Keywords: mRNA translationBackground
Gene expression is regulated at many steps, including gene transcription, pre-mRNA splicing, and mRNA export to the cytoplasm, turnover and translation. Given the robust impact of post-transcriptional gene regulatory mechanisms on overall protein expression patterns in the cell, there is great interest in elucidating the processes that control these events. In particular, the steady-state mRNA levels of one-half of the transcriptome show poor correlation with the level of proteins translated from these mRNAs, indicating that protein levels in the cell are potently regulated at the level of mRNA translation and/or protein stability (Schwanhausser et al., 2011). A number of assays can be used to study how translation is regulated in response to different conditions – both at the transcriptome level and at the level of single mRNAs. Traditionally, Western blot analysis, puromycin labeling, and 35S-methionine/cysteine labeling assays have been used to measure the efficiency mRNA translation.
The method discussed here focuses on analyzing the sizes of polysomes that form on a given mRNA. The premise of this analysis is that mRNAs found in larger polysomes are expected to be translated robustly, while mRNAs present in smaller polysomes or devoid of ribosome components are expected to be translated poorly or remain untranslated. This protocol allows the capture of actively translating mRNAs by ‘freezing’ translating ribosomes and thus permitting the measurement of the relative size of polysomes forming on given mRNAs. This method has been successfully used in dozens of studies to analyze how RBPs and microRNAs affect the translation of target mRNAs and can be used to explore the role of polysome-associated proteins and noncoding RNAs on global translation and the translation of specific mRNAs.
Materials and Reagents
- Tube, thin-wall, polypropylene, 13.2-ml (Beckman Coulter, catalog number: 331372 )
- 9” Pasteur pipet (Kimble Chase Life Science and Research Products, catalog number: 883350-0009 )
- 15 ml tube
- Posi-Click 1.7-ml microcentrifuge tube (Denville Scientific, catalog number: C2171 )
- ThermoGridTM rigid strip 0.2-ml PCR tubes [(Denville Scientific, catalog number: C18064 (1000859) ]
- MicroAmp® optical 384-well reaction plate (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4309849 )
- MicroAmp® optical adhesive film (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4311971 )
- Piercing needle
- 100-mm dish
- Bromophenol blue (BPB) (Sigma-Aldrich, catalog number: B0126 )
- Cycloheximide (CHX) (Sigma-Aldrich, catalog number: C7698 )
- Dimethyl sulfoxide (DMSO)
- Dulbecco’s phosphate-buffered saline (DPBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 20012027 )
- RiboLock RNase inhibitor (40 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EO0381 )
- TRIzol® reagent (Thermo Fisher Scientific, AmbionTM, catalog number: 15596018 )
- Chloroform
- Isopropanol
- GlycoBlueTM coprecipitant (15 mg/ml) (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9515 )
- Ethanol (Sigma-Aldrich, catalog number: E7023 )
- Nuclease-free water (Thermo Fisher Scientific, AmbionTM, catalog number: AM9930 )
- Random primers (150 ng/µl) (Sigma-Aldrich, catalog number: 11034731001 )
- dNTP mix (10 mM each) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0193 )
- Maxima reverse transcriptase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0741 )
- KAPA SYBR® FAST ABI prism 2x qPCR master mix (Kapa Biosystems, catalog number: KK4605 ), or SYBR Green from other vendors
- EDTA
- Sucrose (Sigma-Aldrich, catalog number: S1888 )
- NaCl
- Tris-HCl
- MgCl2
- KCl
- Nonidet P-40
- DTT
- 5x RT buffer (250 mM Tris-HCl [pH 8.3 at 25 °C], 375 mM KCl, 15 mM MgCl2, 50 mM DTT, provided with Maxima Reverse Transcriptase)
- cOmplete EDTA-free protease inhibitor cocktail (Sigma-Aldrich, catalog number: 11873580001 )
- 2.2 M sucrose (MW 342.3) (see Recipes)
- 10x salts solution (see Recipes)
- Chase solution (60% sucrose) (see Recipes)
- Cycloheximide (CHX) (1,000x) (see Recipes)
- 25x protease inhibitors (see Recipes)
- Polysome extraction buffer (PEB) (see Recipes)
Equipment
- SW 41 Ti rotor package (Beckman Coulter, catalog number: 331336 )
- Manual pipettor (SP Scienceware - Bel-Art Products - H-B Instruments, catalog number: F37911-1010 )
- Cell scraper
- Vortexer
- Refrigerated centrifuge (Eppendorf, model: 5430 R )
- NanoDrop spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: ND-ONE-W )
- OptimaTM XE 90K - preparative ultracentrifuge (Beckman Coulter, catalog number: A94471 )
- Spectrophotometer
- PCR strip tube rotor, mini centrifuge C1201 [Denville Scientific, catalog number: C1201-S (1000806) ]
- Veriti® 96-Well thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4375786 )
- Eppendorf ThermoMixer® F1.5 (Eppendorf, catalog number: 5384000012 )
- MPS 1000 mini plate spinner (Next Day Science, catalog number: C1000 )
- Density gradient fractionation system (Brandel, catalog number: BR-188 )
- QuantStudio 5 Real-Time PCR System, 384-well (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: A28140 )
Procedure
- Preparation of sucrose gradients (Table 1) (Notes 3 and 4)
Table 1. Preparation of sucrose stock solutions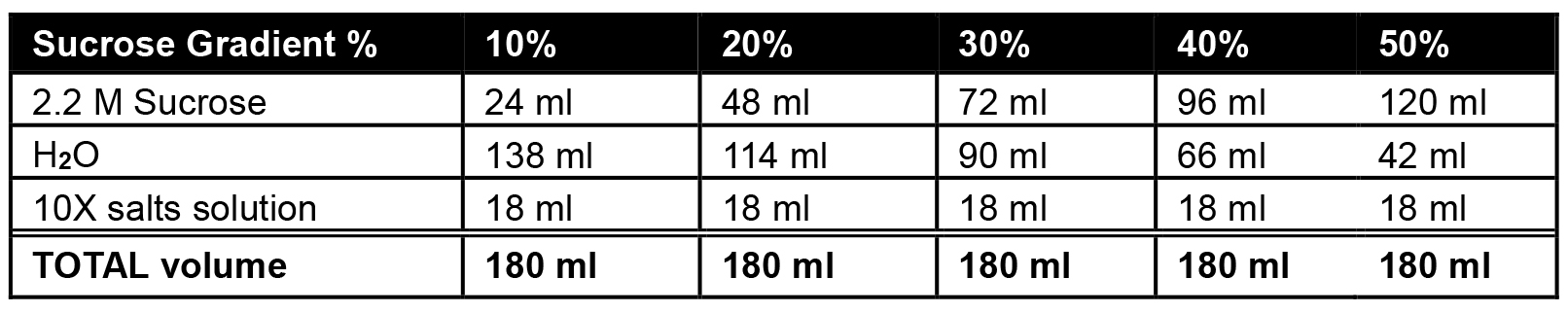
- Prepare stock solutions for 10%, 20%, 30%, 40% or 50% sucrose as mentioned in the table and store it at 4 °C until use.
- Prepare the sucrose gradients a day before the polysome fractionation experiment and pre-cool the SW41Ti rotor and buckets at 4 °C overnight.
- Using prepared solutions containing 10%, 20%, 30%, 40% or 50% sucrose (see Table 1), begin by adding 2.2 ml of 10% sucrose gradient to the bottom of the thin-wall ultracentrifuge tube.
- Underlay each subsequent layer (2.2 ml each of 20%, 30%, 40%, then 50%) using a 9” Pasteur pipet and manual pipettor (Figure 1; Note 1).
- Leave gradients at 4 °C overnight to allow the gradient to become linear (Figure 2; Notes 6 and 7)
- Make chase solution (see Recipes), a high-density sucrose solution which will push the sample through the absorbance detector, with a speck of bromophenol blue (BPB) powder added for slight color (optional step to observe the transition between samples and the chase solution) and store at 4 °C.

Figure 1. Schematic of sucrose gradient preparation - Prepare stock solutions for 10%, 20%, 30%, 40% or 50% sucrose as mentioned in the table and store it at 4 °C until use.
- Cytoplasmic lysate preparation
- Add 10 µl of 100 mg/ml cycloheximide (CHX) in DMSO to the cells containing 10 ml culture media resulting in 100 µg/ml CHX and incubate for 10 min at 37 °C (Note 2).
- Remove media and wash the cells three times with ice cold PBS supplemented with 100 µg/ml CHX.
- Trypsinize for 3 min or long enough for the cells to come off the plate or scrape the cells with cell scraper and transfer the cells to a 15-ml tube using cold PBS with CHX to rinse the plate (Note 8).
- Pellet the cells by centrifugation for 5 min at 500 x g.
- Cell pellets can be stored at -80 °C or immediately used for cytoplasmic lysate preparation.
- Disrupt the cell pellet by pipetting 0.5 to 1.0 ml polysome extraction buffer (PEB) containing 100 µg/ml CHX, 1x protease inhibitors and 1:1,000 dilution of RiboLock RNase inhibitor.
- Transfer the cell suspension to a 1.7-ml microfuge tube and incubate on ice for 10 min with occasional inverting every two minutes (do not vortex).
- Centrifuge at 12,000 x g for 10 min at 4 °C to pellet the nuclei and debris.
- Transfer approximately 9/10 of the total volume of the supernatant (cytoplasmic lysate) to a fresh tube.
- Measure the protein concentration by Bradford assay and/or the total RNA concentration by Nanodrop.
- This supernatant can also be stored at -80 °C and fractionated another time or proceed to polysome fractionation.
- Add 10 µl of 100 mg/ml cycloheximide (CHX) in DMSO to the cells containing 10 ml culture media resulting in 100 µg/ml CHX and incubate for 10 min at 37 °C (Note 2).
- Polysome fractionation
- Carefully layer equal amounts of cytoplasmic lysate (1 ml or less containing 1.0-1.5 mg protein or 100-300 µg RNA) to the top of a 10-50% sucrose gradient. Be sure all tubes are equally balanced.
- Using a Beckman ultracentrifuge, centrifuge the gradients for 90 min in a SW41Ti swinging bucket rotor at 190,000 x g (~39,000 rpm) at 4 °C with maximum acceleration and brake (Figure 2).
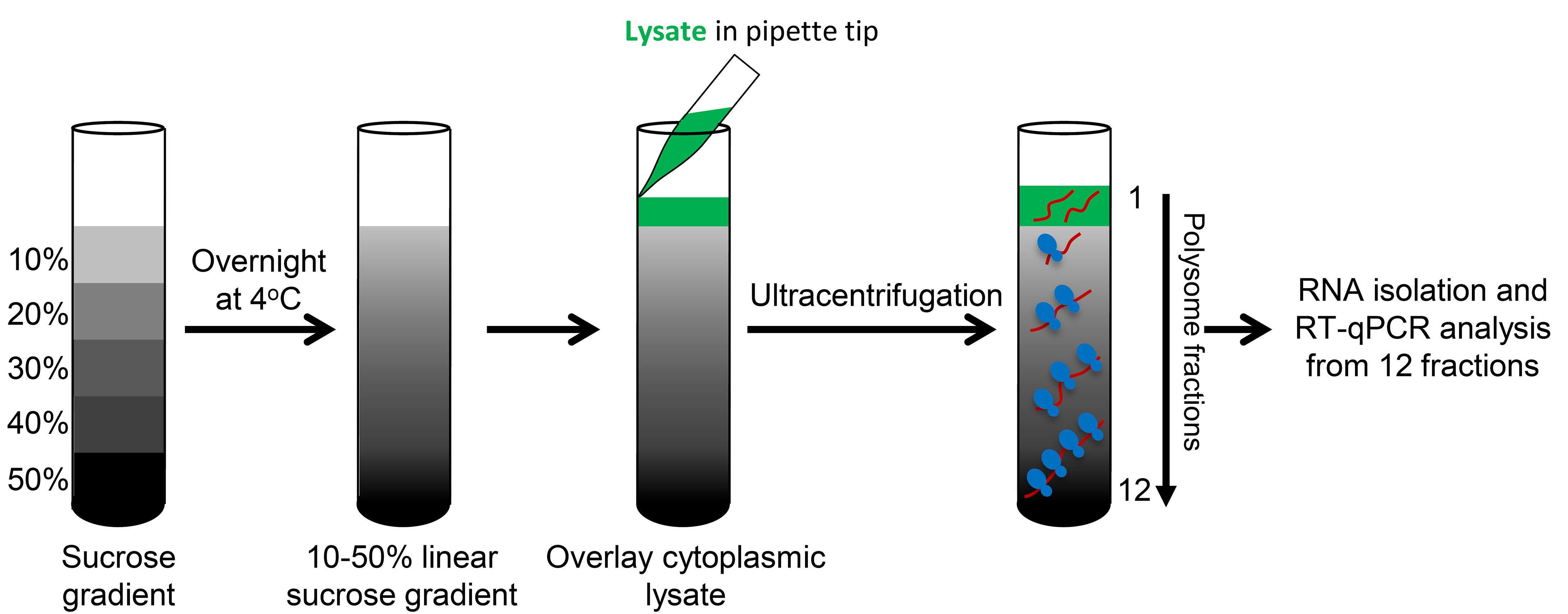
Figure 2. Schematic of polysome fractionation - Keep the gradients on ice after centrifugation. To begin, place the gradient onto the fractionating system - the piercing needle will puncture the bottom of the tube.
- Collect 1 ml/min fractions into 12 tubes per gradient and immediately transfer the fractions to an ice bucket.
- The 254 nm spectrophotometer within the optical unit of the fractionation system reads the RNA amount in each fraction and plots a chart which displays the polysome profile of the gradient (Figure 3A; Note 5).
- Analyze the fractions as desired or store the fractions at -80 °C until use.
- Carefully layer equal amounts of cytoplasmic lysate (1 ml or less containing 1.0-1.5 mg protein or 100-300 µg RNA) to the top of a 10-50% sucrose gradient. Be sure all tubes are equally balanced.
- RNA and cDNA preparation from sucrose gradient
- Take 0.5 ml of each sucrose fraction in a fresh 1.7-ml tube, and add 0.5 ml TRIzol and 200 µl chloroform.
- Vortex or shake vigorously for 10 sec and centrifuge at 13,000 x g for 15 min at 4 °C.
- Transfer 500 µl of the upper layer to a fresh microfuge tube containing 1 ml isopropanol and 2 µl of 15 mg/ml Glycoblue.
- Precipitate the RNA by incubating overnight at -20 °C followed by centrifugation at 13,000 x g for 15 min at 4 °C.
- Discard the supernatant and wash pellet once with 1 ml ice-cold 70% ethanol.
- Centrifuge at 13,000 x g for 15 min at 4 °C and discard the supernatant.
- Centrifuge for few seconds at 4 °C to collect the remaining ethanol at the bottom of the tube.
- Discard the ethanol and air dry RNA pellet for 5-10 min at 25 °C.
- Dissolve RNA in 12 µl nuclease-free water (Note 9).
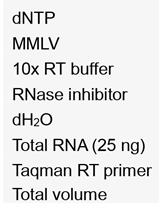
- Use the whole RNA solution for reverse transcription by preparing a 20-µl reaction containing 12 µl of prepared RNA, 1 µl random hexamers, 4 µl 5x RT buffer, 1 µl RiboLock, 1 µl dNTP mix and 1 µl Maxima reverse transcriptase.
- Mix and centrifuge for a few seconds to settle the reaction mixture at the bottom of the tube.
- The cDNA synthesis was performed using the thermal cycler set at 25 °C for 10 min, 50 °C for 30 min and 5 min at 85 °C.
- The cDNA product can be stored at -20 °C or -80 °C, or used immediately for RT-qPCR analysis.
- Take 0.5 ml of each sucrose fraction in a fresh 1.7-ml tube, and add 0.5 ml TRIzol and 200 µl chloroform.
- RT-qPCR analysis of mRNA distribution over the sucrose gradient
- Dilute the cDNAs (20 µl volume) with nuclease-free water to 1,000 µl final volume.
- To amplify specific transcripts, prepare the gene-specific forward and reverse primer mix at a final concentration of 1 µM in nuclease-free water. Use primer sets for mRNAs of interest as well as for 1 or 2 mRNAs encoding housekeeping proteins (e.g., ACTB mRNA or GAPDH mRNA).
- Prepare 20 µl qPCR reactions in a 384-well plate, add 10 µl 2x SYBR Green PCR mix, 5 µl cDNA, and 5 µl primer mix.
- Cover the plate with optical adhesive film and vortex at maximum speed for 10 sec to mix the reaction.
- Centrifuge the plate for 30 sec using Plate Spinner to settle the reactions at the bottom of the wells.
- Use a QuantStudio 5 Real-Time PCR System for qPCR with a cycle set up of 3 min at 95 °C and 40 cycles of 5 sec at 95 °C plus 20 sec at 60 °C.
- It is recommended to analyze dissociation curves to verify that the primer set is optimal.
- Using the cycle threshold (CT) values, the percent (%) distribution for the mRNAs across the gradients can be calculated using the ΔCT method (Tables 2 and 3).
Table 2. % of mRNA A distribution over sucrose gradient in control cells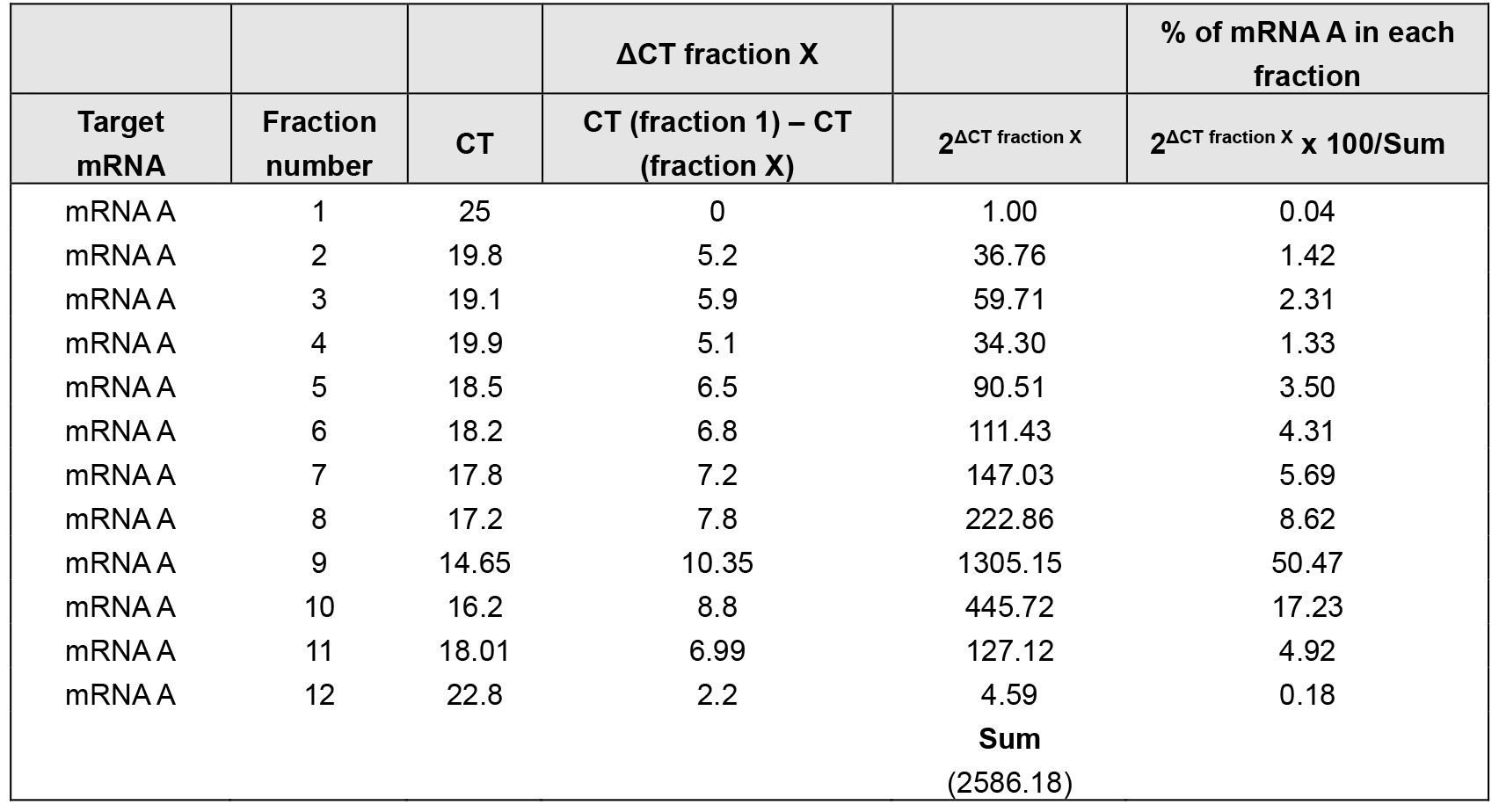
Table 3. % of mRNA A distribution over sucrose gradient in treated cells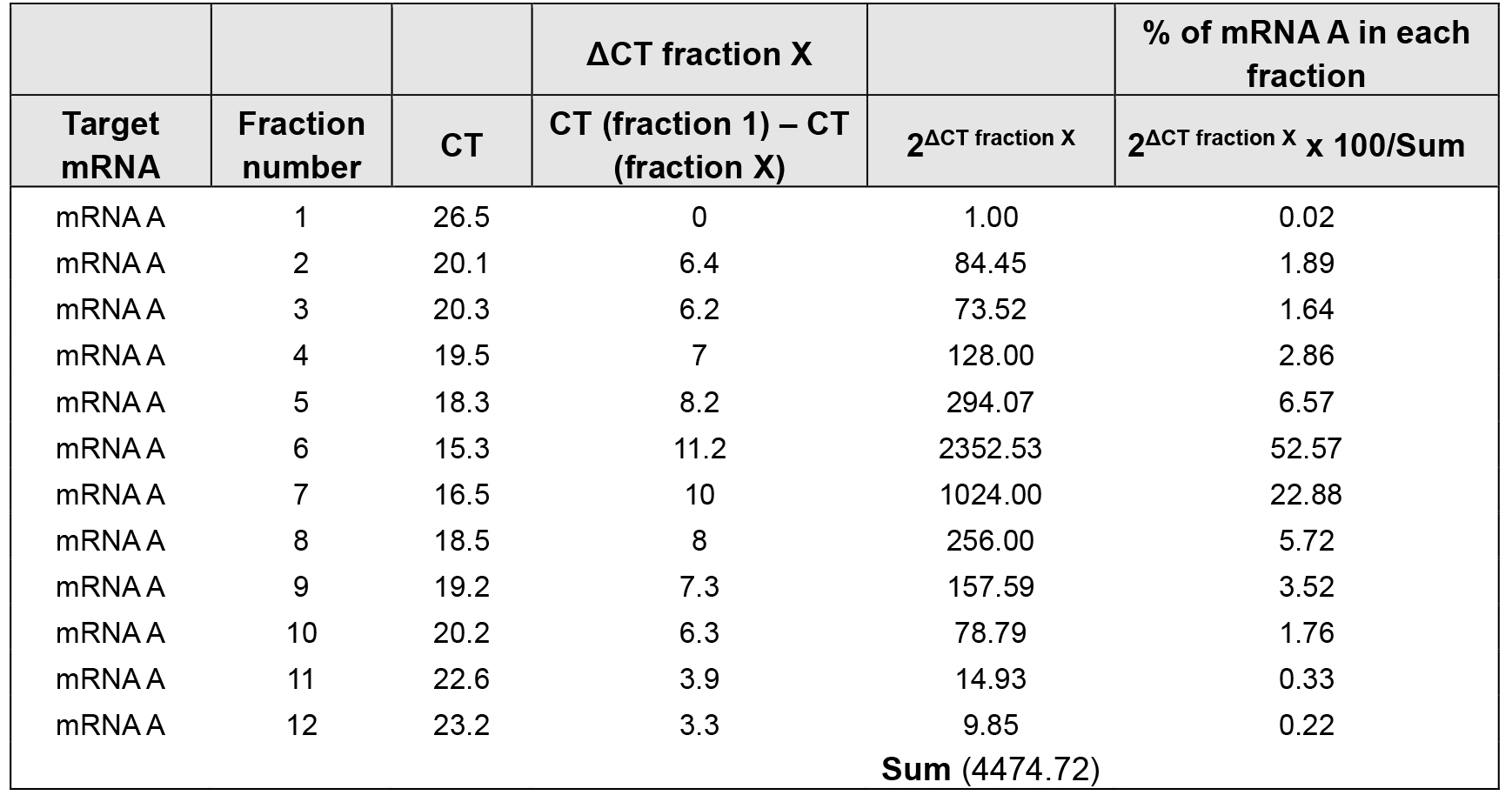
- Dilute the cDNAs (20 µl volume) with nuclease-free water to 1,000 µl final volume.
Data analysis
The density gradient fractionation system generates a polysome profile for each sample, which includes, from light to heavy fractions, fractions without ribosomal material, and fractions with 40S, 60S, 80S (monosomes), and low- and high-molecular-weight (LMW and HMW) polysomes. As these peaks did not appear to be different in the treated cells compared to control of the hypothetical example in Figure 3A, the treatment does not seem to have a major effect on the global mRNA translation. However, the % distribution of mRNA A across the gradient shows that this mRNA was most abundant in the actively translating fractions (fractions 8 to 10, spanning high-molecular-weight polysomes) of the gradient containing control sample (Table 2 and Figure 3B, solid line). In the treated cells, the distribution of mRNA A displayed a leftward shift on the gradient and was most abundant in fractions 5 to 7 (spanning low-molecular-weight polysomes) (Table 3 and Figure 3B, dotted line), indicating that mRNA A associated with smaller polysomes after treatment of cells. These data are consistent with the notion that translation of mRNA A is suppressed in treated cells without changes in global mRNA translation (Figures 3A and 3B). The distribution of mRNAs encoding housekeeping proteins (e.g., GAPDH mRNA or ACTB mRNA) should be measured and plotted similarly (Figure 3C; Note 10). The fact that GAPDH mRNA did not show significant change in distribution pattern supports the notion that the reduced sizes of mRNA A polysomes following treatment is specific (Figures 3B and 3C).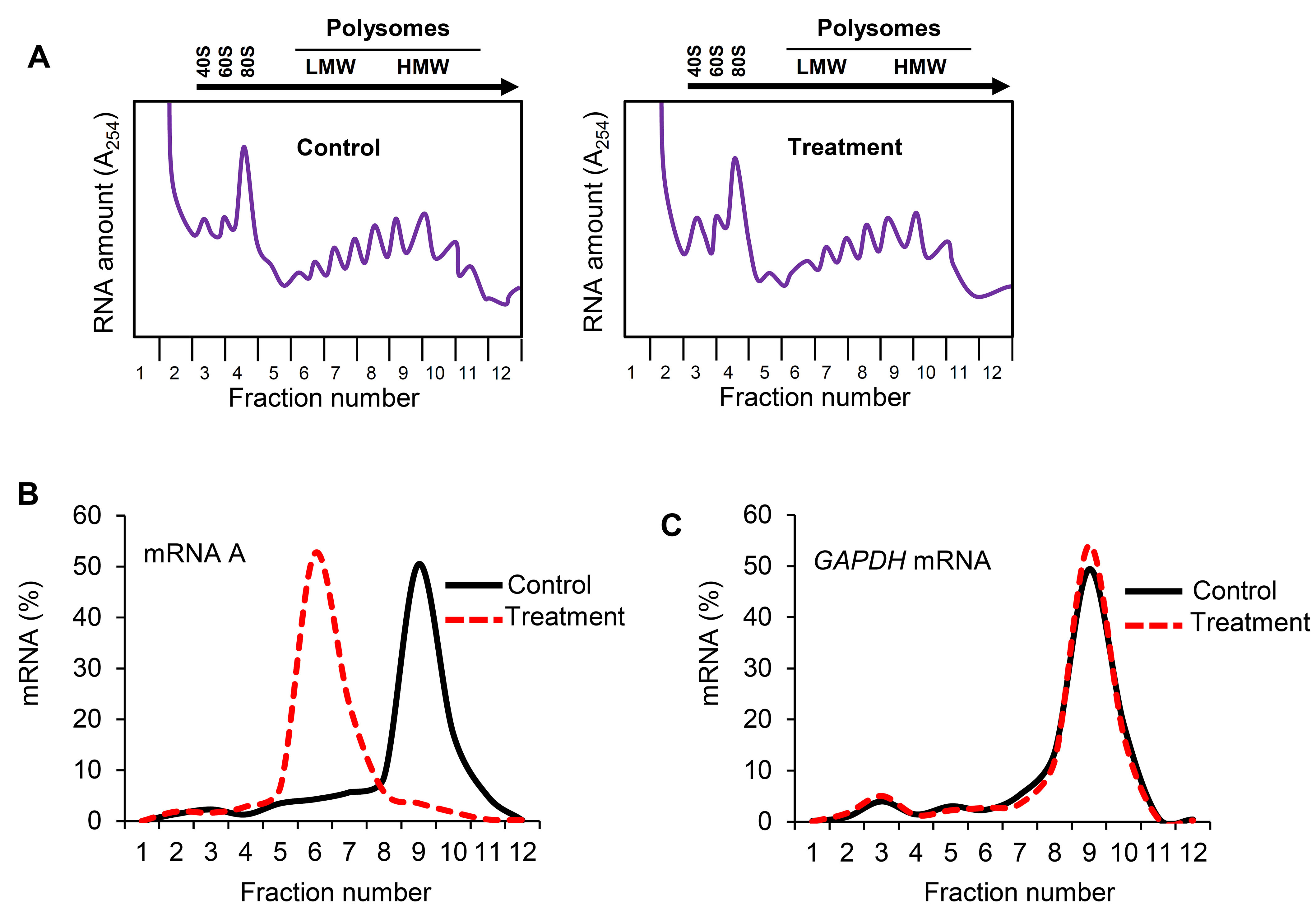
Figure 3. Effect of treatment on distribution of mRNA A across the sucrose gradient. A. Cytoplasmic lysates from control and treated cells were fractionated through sucrose gradients. Global RNA polysome profiles generated by the density gradient fractionation system are shown. B. The relative distribution of the % mRNA A (left) and GAPDH mRNA (right), encoding a housekeeping protein, over the sucrose gradient was studied by RT-qPCR analysis of the RNA in each of the 12 gradient fractions.
Notes
- Add the sucrose gradient layers slowly without introducing any air bubbles.
- For good polysome peaks, use fresh lysates from one 100-mm dish not more than 80% confluent. Too few cells will result in smaller peaks, perhaps below the detector sensitivity settings. Cells which are overly confluent may not be in a proliferative state with active translation, so the polysome peaks will not appear as robust.
- Use either nuclease-free water or DEPC-treated water for all buffers and solutions.
- Do not autoclave sucrose.
- Fractionation of a 10-50% sucrose gradient without cell lysate should yield an absorbance profile that is a straight, flat line. If the profile generates a sloped line, then the sucrose itself is contributing to the absorbance readings and may interfere with the resolution of polysome peaks in the experimental lysates.
- The gradients are OK to use within 16 h to 3 days (at 4 °C).
- The linear gradient can be frozen at -20 °C or -80 °C for 2 weeks (or more) and used immediately after thawing.
- For differentiated cells (neurons, muscle) or senescent cells, it is better to add PEB directly to the plate and scrape the cells.
- Do NOT measure RNA concentrations in the polysome fractions as this will vary between fractions, with some fractions having very little RNA.
- There is no normalization of the RT-qPCR values for mRNA A to the control mRNA. The mRNAs encoding housekeeping proteins (e.g., GAPDH mRNA or ACTB mRNA) are processed side by side simply to test if generic mRNAs are distributed similarly across the gradients for different treatment conditions.
- To confirm that mRNAs fractionated along the gradients are indeed associated with polysomes, the cell lysates can be incubated with 20 mM EDTA for 10 min on ice before loading on the sucrose gradient. Unlike the control sample, the EDTA-treated sample should show no polysome peaks and increased peaks for monosomes and ribosomal subunits. In the EDTA-treated sample, the distribution of mRNAs encoding housekeeping proteins and the mRNA of interest should not peak in the polysome fractions and instead should accumulate in the unbound and monosome fractions, showing an overall shift toward lighter fractions of the gradient.
Recipes
- 2.2 M sucrose (MW 342.3)
753.06 g of sucrose in 1,000 ml of nuclease-free water(Notes 3 and 4)
Store at 4 °C - 10x salts solution
1,000 mM NaCl
200 mM Tris-HCl (pH 7.5)
50 mM MgCl2
Store at 4 °C - Chase solution (60% sucrose)
40 ml 2.2 M sucrose
5 ml H2O
5 ml 10x salts solution
Speck of bromophenol blue powder (BPB) - optional
Store at 4 °C - Cycloheximide (CHX) (1,000x)
Stock solution is 100 mg/ml in dimethyl sulfoxide (DMSO)
Aliquot and store at -20 °C - 25x protease inhibitors
Dissolve 1 tablet of cOmplete protease inhibitors in 2 ml RNase-free water
Aliquot and store at -20 °C - Polysome extraction buffer (PEB)
20 mM Tris-HCl (pH 7.5)
100 mM KCl
5 mM MgCl2
0.5% Nonidet P-40
Store at 4 °C
Add protease inhibitors, RNase inhibitor, and 100 µg/ml CHX to an aliquot of PEB before use
Acknowledgments
This work was supported by the National Institute on Aging Intramural Research Program, National Institutes of Health.
References
- Abdelmohsen, K., Panda, A. C., Kang, M. J., Guo, R., Kim, J., Grammatikakis, I., Yoon, J. H., Dudekula, D. B., Noh, J. H., Yang, X., Martindale, J. L. and Gorospe, M. (2014). 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res 42(15): 10099-10111.
- Panda, A. C., Abdelmohsen, K., Martindale, J. L., Di Germanio, C., Yang, X., Grammatikakis, I., Noh, J. H., Zhang, Y., Lehrmann, E., Dudekula, D. B., De, S., Becker, K. G., White, E. J., Wilson, G. M., de Cabo, R. and Gorospe, M. (2016). Novel RNA-binding activity of MYF5 enhances Ccnd1/Cyclin D1 mRNA translation during myogenesis. Nucleic Acids Res 44(5): 2393-2408.
- Panda, A. C., Abdelmohsen, K., Yoon, J. H., Martindale, J. L., Yang, X., Curtis, J., Mercken, E. M., Chenette, D. M., Zhang, Y., Schneider, R. J., Becker, K. G., de Cabo, R. and Gorospe, M. (2014a). RNA-binding protein AUF1 promotes myogenesis by regulating MEF2C expression levels. Mol Cell Biol 34(16): 3106-3119.
- Panda, A. C., Sahu, I., Kulkarni, S. D., Martindale, J. L., Abdelmohsen, K., Vindu, A., Joseph, J., Gorospe, M. and Seshadri, V. (2014b). miR-196b-mediated translation regulation of mouse insulin2 via the 5'UTR. PLoS One 9(7): e101084.
- Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., Chen, W., Selbach, M. (2011). Global quantification of mammalian gene expression control. Nature 473; 337-342.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Panda, A. C., Martindale, J. L. and Gorospe, M. (2017). Polysome Fractionation to Analyze mRNA Distribution Profiles. Bio-protocol 7(3): e2126. DOI: 10.21769/BioProtoc.2126.
Category
Cancer Biology > General technique > Biochemical assays > RNA
Molecular Biology > RNA > RNA-protein interaction
Cell Biology > Organelle isolation > Polyribosome
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.