- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Examination of the Interaction between a Membrane Active Peptide and Artificial Bilayers by Dual Polarisation Interferometry




Published: Vol 7, Iss 1, Jan 5, 2017 DOI: 10.21769/BioProtoc.2087 Views: 8406
Reviewed by: Marc-Antoine SaniFilipa Vaz
Original research article
The authors used this protocol in:

Jun 2016
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Examining the interaction of peptides with lipid bilayers to determine binding kinetics is often performed using surface plasmon resonance (SPR). Here we describe the technique of dual polarisation interferometry (DPI) that provides not only information on the kinetics of the peptide binding to the bilayer, but also how the peptide affects the lipid order of the bilayer.
Keywords: Membrane interactionBackground
The search for and development of new drugs to effectively treat resistant infections is a serious challenge. One group of molecules, the antimicrobial peptides, shows promise as effective new therapeutics due to their range of activity against bacterial, fungal and cancer cells (Mader and Hoskin, 2006; van der Weerden et al., 2013). The cell killing ability of antimicrobial peptides often involves interaction with the membrane (Brogden, 2005; Zasloff, 2002). To develop these peptides as effective therapeutics we need to understand the nature of the interaction of the peptide with the cell membrane and why this interaction results in cell death. That is, we need to know the full sequence of events that occurs between the peptide and the membrane in real time; from the initial electrostatic interaction of the peptide with the bilayer, to phospholipid selectivity, to final disruption of the membrane including changes in lipid order.
Most techniques that examine peptide-lipid interactions have a limited capacity to provide information on the entire process. For example, surface plasmon resonance (SPR) provides binding data in real time but does not reveal how peptide binding affects membrane structure (Green et al., 2000; Mozsolits and Aguilar, 2002). Other techniques such as quartz crystal microbalance (QCM) and atomic force microscopy (AFM) provide very little time resolved data as they provide information on the overall state of the system. In contrast, dual polarisation interferometry (DPI) provides real-time changes and enables quantification of the thickness, mass/density and birefringence of the membrane during peptide binding. Birefringence quantifies the degree of alignment and uniaxial packing of the lipid molecules. The changes in birefringence that occur relative to the amount of peptide bound provide information on the rate that membrane order changes, which is not available with other techniques. DPI provides unique insights into the mechanism of peptide binding, including how the peptide destabilizes the membrane, by following the dynamic changes that occur in real time as peptide binding disrupts the packing of the lipids.
This method describes DPI measurements used to examine the interaction between peptides and lipid bilayers using the Analight BIO200 (Farfield Group Ltd, Manchester, UK). The interaction between the bilayer and the interacting peptide occurs on a dual slab waveguide sensor chip that is illuminated with two alternating polarized laser beams (He-Ne, wavelength 632.8 nm). The sensor chip has four layers of silicon oxynitride deposited on a silicon wafer surface with an upper sensing waveguide that supports the lipid bilayer, and a lower optical reference waveguide. Two orthogonal polarizations are passed through the sensor chip creating two different waveguide modes; the transverse electric (TE) and transverse magnetic (TM). Both of these modes generate a field spanning from the top sensing waveguide surface to the materials coming into contact with the sensor surface. The molecules that make contact with the surface change the refractive index. When this occurs, the phase difference between the sensing waveguide and the buried reference waveguide is altered and the position of the interference fringes changes. This interference fringe pattern for both the TE and TM illuminates a 1,024 x 1,024 element-imaging device and the data from this is transferred to the digital signaling processing unit. Data is collected every 2 milliseconds using a spatial Fourier transform method and is transferred to the computer for real time data display and further analysis of the data to reveal thickness, RI and birefringence values.
Materials and Reagents
Note: All solutions must be degassed prior to running on the DPI machine.
- Liposome preparation
- Round-bottom glass vials such as Kimble culture tubes 20 mm diameter, 125 mm length with screw caps (Kimble, catalog number: 73770-20125 )
- Parafilm
- Polycarbonate membrane; 19 mm diameter, 100 nm pore diameter (Sigma-Aldrich, catalog number: Z377419 )
- Syringes (50 ml for running buffer, 3 ml for sample loading)
- Wide gauge needle
- Lipids
- L-α-phosphatidylinositol (PI, bovine liver) (Avanti Polar Lipids, catalog number: 840042P )
- L-α-phosphatidylinositol-4,5-bisphosphate (PI[4,5]P2, porcine brain) (Avanti Polar Lipids, catalog number: 840046X )
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) (Avanti Polar Lipids, catalog number: 850757P )
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, catalog number: 850457P )
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS) (Avanti Polar Lipids, catalog number: 840034P )
- Chloroform (VWR, catalog number: 22707.320 )
- Methanol (Sigma-Aldrich, catalog number: 494291 )
- MilliQ water
- N2 gas
- MOPS (3-morpholinopropane-1-sulfonic acid) buffer (Sigma-Aldrich, catalog number: M1254 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 793566 )
- Round-bottom glass vials such as Kimble culture tubes 20 mm diameter, 125 mm length with screw caps (Kimble, catalog number: 73770-20125 )
- Dual polarization interferometry
- 10% Hellamanex II (Hellma, Müllhein, Germany)
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 74255 )
- 100% ethanol
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 746495 ) (see Recipes)
- Membrane interacting protein (for example NaD1)
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: EDS ) (see Recipes)
- Bovine serum albumin (BSA), fraction V, fatty acid free (Sigma-Aldrich, catalog number: 10775835001 )
- MilliQ water
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: S5881 )
- Bulk buffer (see Recipes)
- 2% SDS (see Recipes)
- 80% ethanol (see Recipes)
- 10% Hellamanex II (Hellma, Müllhein, Germany)
Equipment
- Liposome production
- Water bath
- Fume hood
- Vacuum pump and chamber
- Sonicator bath
- Shaking incubator
- Avestin lipofast extruder (Sigma-Aldrich, catalog number: catalog number: Z373400 )
- Water bath
- Dual polarization interferometery
- Silicone oxynitride AnaChip unmodified (Farfield Group Ltd)
- Gasket two-channel (Farfield Group Ltd)
- Analight® Bio200 DPI instrument (Farfield Group Ltd, UK)
- Harvard Apparatus PHD2000 programmable syringe pumps
- Silicone oxynitride AnaChip unmodified (Farfield Group Ltd)
Software
- Analight Explorer software
Procedure
- Preparation of liposomes for DPI
- Liposomes are prepared for DPI as described in Lee et al. (2010). This involves dissolving the dried phospholipids in chloroform to a 2 mM stock, and for PI(4,5)P2 a 2 mM stock is prepared in chloroform, methanol and MilliQ water (20:9:1). Avanti polar lipids provides information about the requirements for dissolving their lipids on their product sheets and website.
- Lipid mixtures of POPC: POPS: POPE: PI and POPC: POPS: POPE: PI(4,5)P2 in a ratio of 56:10:30:4 are prepared by adding each of the desired 2 mM lipid stocks to glass vials to a final volume of 410 μl.
- The lipids are then dried under a stream of nitrogen gas in a warm water bath (≈ 55 °C) in a fume hood. A thin lipid film that coats the bottom and sides of the round bottom tube is desired. This is created by constantly turning the tilted vial in the warm bath under the stream of nitrogen gas. The gas is at a flow rate that just causes a disruption to the surface of the lipid mixture. Once all the visible liquid is evaporated, the nitrogen gas stream is increased and the tube is given a final blow out to remove remaining organic solvent.
- Lipid films are vacuum dried overnight to ensure complete liquid removal. After this stage the vials are capped and sealed with Parafilm prior to storage at -20 °C until required.
- Lipids are resuspended to 1.14 mM (average lipid concentration) in degassed, freshly made 10 mM MOPS, pH 7 with 150 mM NaCl at 40 °C (720 μl). The lipids are rehydrated by incubating for 30 min in a shaking incubator at 40 °C. The temperature for this incubation should be above the gel-liquid crystal transition temperature (Tc or Tm), so that the lipids are rehydrated in their fluid phase. Avanti polar lipids provide information regarding the Tc/Tm of their lipids.
- The hydrated liposome mixture is then sonicated in a warm water bath (≈ 40 °C) for 20 min, or until clear. A sonicator bath is preferred over a sonicator probe for this procedure as probes are likely to overheat the lipid sample. Sonicator probes can also generate titanium fragments that need to be removed by centrifugation before using liposomes.
- The liposomes are made into a uniform size by extruding the mixture through a 100 μm polycarbonate membrane 21 times using a pre-warmed Avenstin lipofast extruder. Warming the assembled extruder helps to ensure the lipid mixture remains above the Tc of the lipids. Extruding lipid mixtures below the Tc will usually result in the membrane becoming clogged with rigid liposomes. To load the liposomes into the extruder syringe use a wide gauge needle. Remove all the air from the syringe prior to attaching to the extruder assembly. Once the liposomes are prepared they should be used immediately for DPI.
- Liposomes are prepared for DPI as described in Lee et al. (2010). This involves dissolving the dried phospholipids in chloroform to a 2 mM stock, and for PI(4,5)P2 a 2 mM stock is prepared in chloroform, methanol and MilliQ water (20:9:1). Avanti polar lipids provides information about the requirements for dissolving their lipids on their product sheets and website.
- Dual polarisation interferometry
- Dual polarisation interferometry (DPI) is conducted as described in Lee et al. (2010). Measurements are performed using an Analight® Bio200 DPI instrument. The flow rate is controlled by external Harvard Apparatus PHD2000 programmable syringe pumps, which are primed with degassed, double-distilled water. If using this setup, the flow rate must be changed on the pumps and set in the program Analight Inject that controls the DPI machine.
- A highly clean chip surface is a critical step in forming lipid bilayers with reproducible structure. The washing procedure is performed after the chip is loaded into the machine and thus the lines are cleaned at the same time. The silicone oxynitride AnaChip is cleaned using a three-step washing procedure involving an initial wash of 10% Hellamanex II, injected at 25 μl/min for 8 min. A second wash of 2% SDS injected at 50 μl/min for 4 min followed by the third wash of 100% ethanol at 50 μl/min for 4 min. A water rinse at 50 μl/min for 2 min is performed between all three wash steps.
- After cleaning the chip, the interference fringes for each channel are set up in the set channel window. To calibrate the channels on the chip, press the button that has three parallel vertical lines and snap a high resolution image to observe the quality of the chip. Each of the three channels should have distinct bands and a relatively even pattern corresponding to the valleys and the peaks of light (Figure 1). If the chip quality is okay, data acquisition begins by pressing the start button.
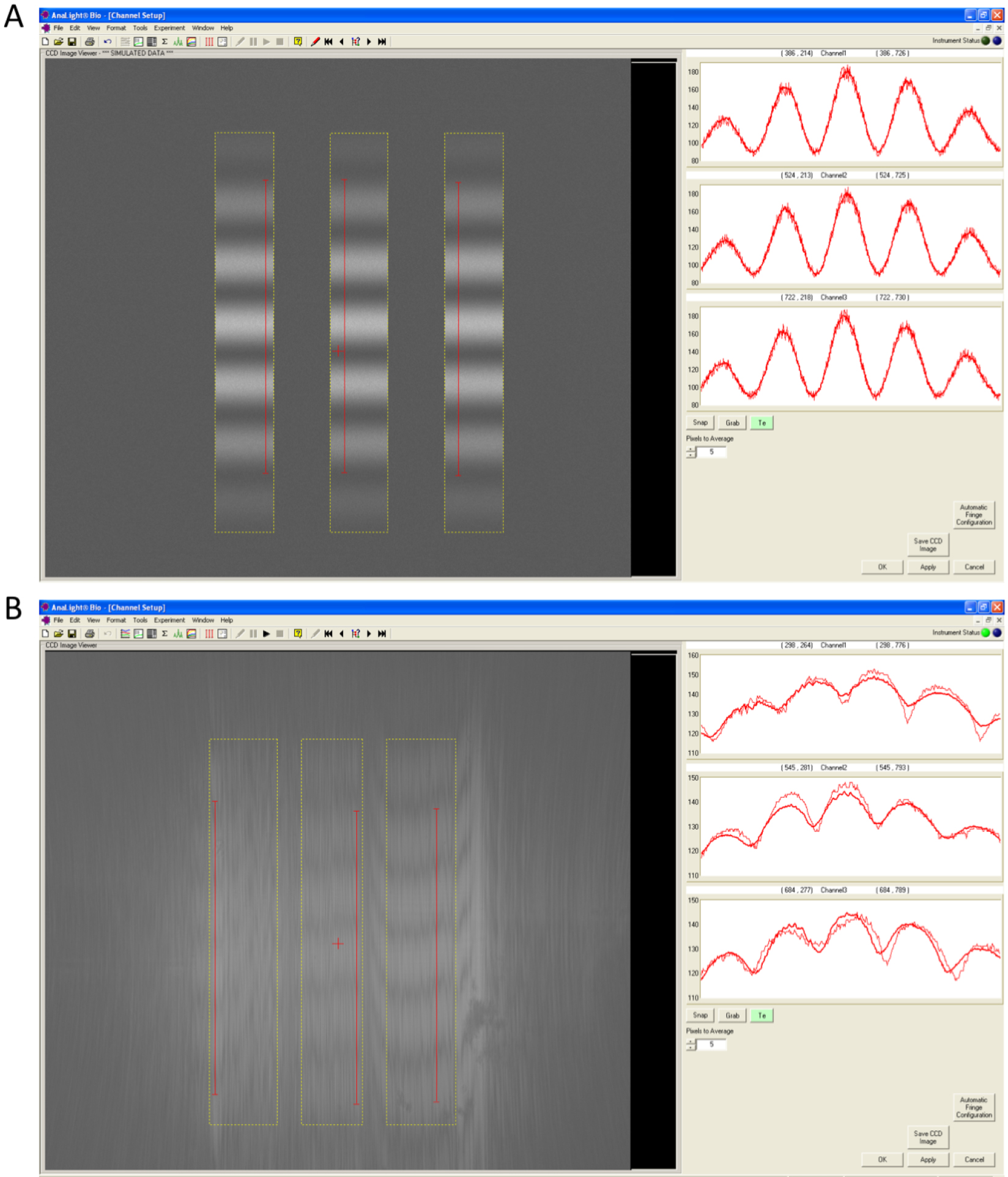
Figure 1. Quality of a chip can by observed through the Interference fringes of the three chip channels. A. A high quality chip surface will have distinct bands in each of the three channels that represent the valleys and peaks of light. B. These bands will become less distinct and uneven as the quality of the chip deteriorates. - Calibration of the chip is achieved by injecting 120 μl of 80% ethanol at 50 μl/min followed by a linear gradient of 100-0% of 80% ethanol to the bulk buffer (10 mM MOPS buffer, pH 7 with 150 mM NaCl) at 20 μl/min (Figure 2). A 140 μl water injection at 50 μl/min is then performed for the bulk calibration. The buffer baseline should remain stable throughout these calibration steps and should only change when the temperature is changed.
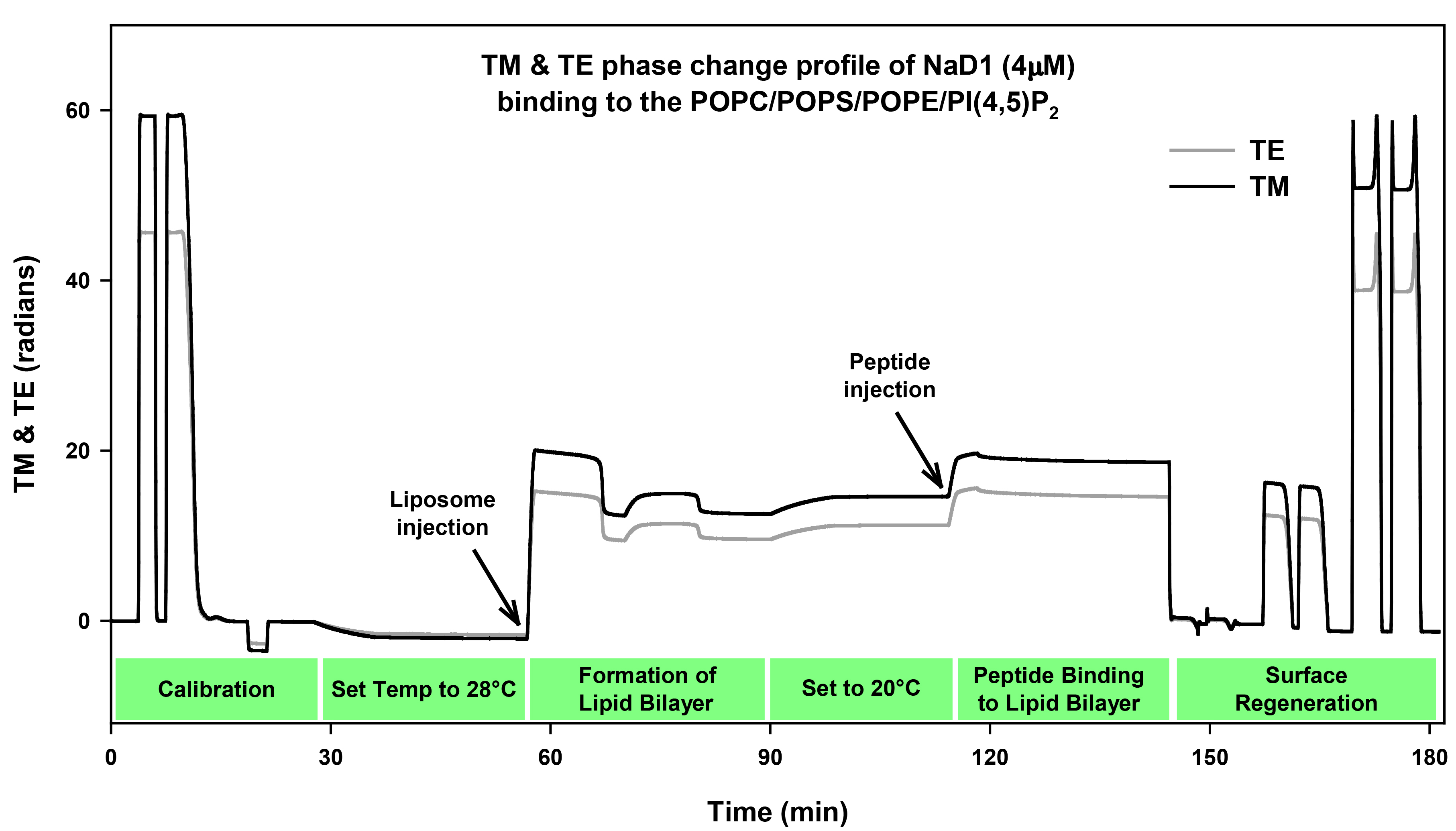
Figure 2. TM and TE phase change profile of NaD1 (4 µM) binding to bilayer. The first section of the profile contains the chip calibration data that consists of the initial 80% ethanol injection, followed by the linearization injection of 100 to 0% of 80% ethanol and then the bulk calibration of 140 μl water injection. The profile baseline decreases during the change of temperature from 20 to 28 °C. The lipid bilayer is then formed once the base line stabilizes by injecting liposomes (POPC: POPS: POPE: PI[4,5]P2) followed by a calcium injection. The baseline then increases due to the decrease in temperature back to 20 °C. Next the protein is injected (NaD1, 4 μM) for 4 min, followed by disassociation for 30 min. The chip surface is regenerated for future use by removing the lipid bilayer using a three-step wash procedure of two washes each of 10% Hellamanex II, 2% SDS, and 100% ethanol. - The chip readings are allowed to stabilise in the bulk buffer at both 20 °C and 28 °C. Once the baseline is stable at 28 °C, liposomes (0.1-0.2 mM) are injected in the presence of 2 mM CaCl2 at 20 µl/min for 10 min followed by an injection of 2 mM CaCl2 in 10 mM MOPS buffer pH 7 with 150 mM NaCl (200 μl) (Figure 2). The presence of calcium helps the formation and stabilization of the supported lipid bilayer. The concentration of calcium may need to be changed in the range of 1-5 mM depending on lipid composition. Liposomes with high PE and other negatively charged lipids will require greater amounts of calcium to form a stable bilayer. If necessary for protein interaction studies, the calcium ions can be removed from the lipid bilayer surface by injecting 2 mM EDTA in running buffer.
- The adsorbed lipid bilayers are further equilibrated in running buffer for an additional 20 min until a stable baseline is achieved. This is performed at both 28 °C and 20 °C. The stabilization of baseline takes approximately 20 min for each temperature.
- The coverage and presence of defects in the supported lipid bilayer can be checked with an injection of 50 μl BSA (50 μg/ml in running buffer). Complete coverage of the lipid bilayer is confirmed by the absence of BSA binding.
- The protein of interest, NaD1 is prepared for injection in running buffer. The sample (160 μl) with 16, 8, 4, 2 or 1 μM of NaD1 is degassed prior to injection at 40 μl/min for 4 min followed by a dissociation for 30 min in the running buffer (Figure 2).
- The chip surface is regenerated and cleaned and a new bilayer was deposited for each NaD1 injection. All solutions are degassed before injection.
- Dual polarisation interferometry (DPI) is conducted as described in Lee et al. (2010). Measurements are performed using an Analight® Bio200 DPI instrument. The flow rate is controlled by external Harvard Apparatus PHD2000 programmable syringe pumps, which are primed with degassed, double-distilled water. If using this setup, the flow rate must be changed on the pumps and set in the program Analight Inject that controls the DPI machine.
Data analysis
- Data processing using Analight Explorer
- Open the data file in the Analight Explorer software.
- First the calibration data is processed by clicking into the Resolver Configuration in the ‘Display Window’. To process the calibration data each aspect of the calibration (chip calibration, linearization, bulk at 20 °C and bulk at 28 °C) of the displayed profile needs to be defined. These calibration aspects are defined by placing the blue axis at the start and end of each section of the calibration (Figure 3).
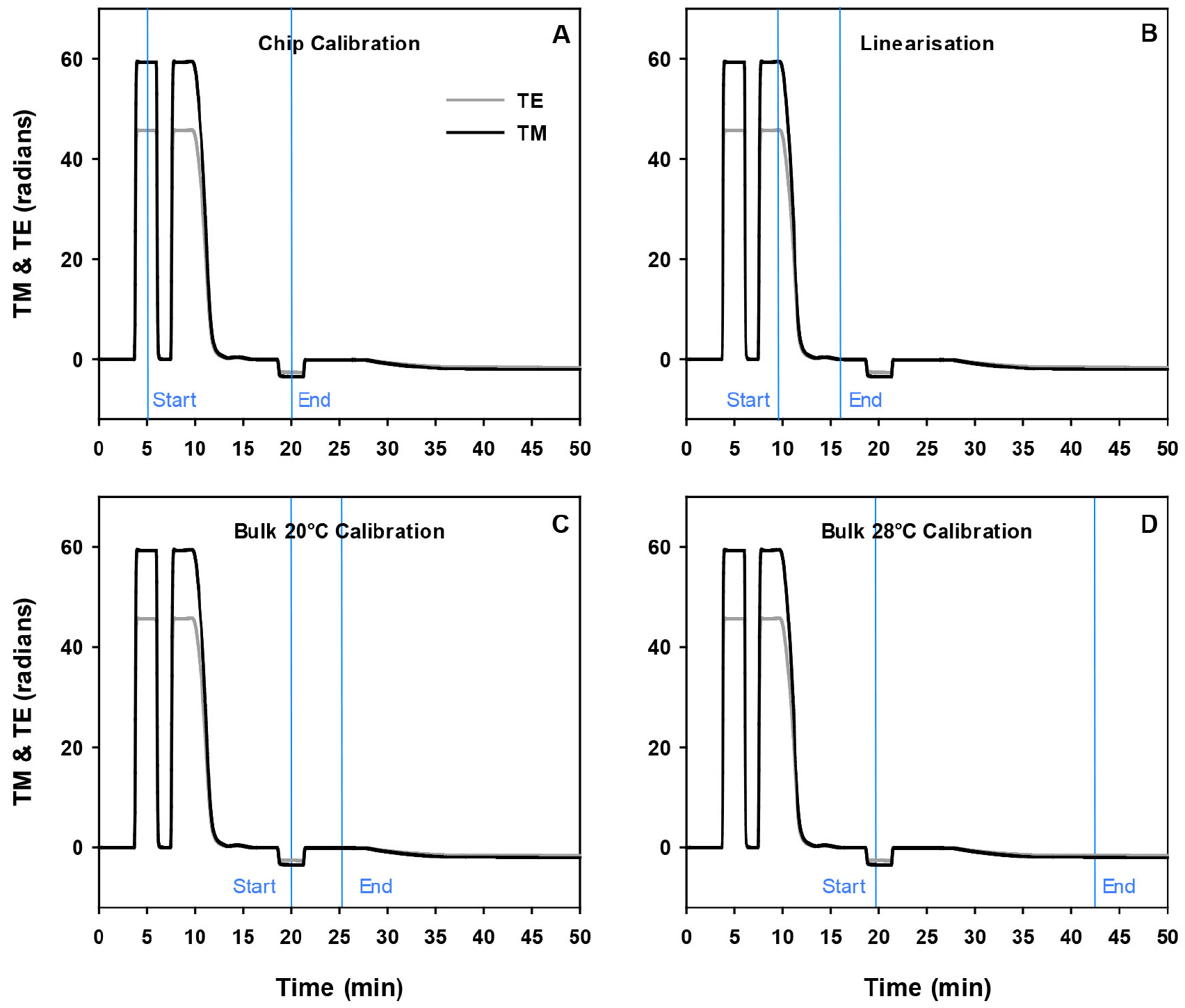
Figure 3. Processing of the DPI calibration data using the Analight Explorer software. Analysis of each of the 4 parts of calibration, chip calibration, linearisation, bulk at 20 °C and 28 °C, is performed by first defining the start and end of the calibration data on the profile. For the chip calibration this involves placing the first blue axis at the apex of the 80% ethanol peak and the end blue axis at the apex of the water peak (A). The periodic error is corrected through the linearization data by placing the starting blue axis at the beginning edge of the 80% ethanol-H2O gradient and the end blue axis on the end of the 80% ethanol-H2O gradient (B). The bulk calibration at 20 °C is obtained by placing the start blue axis at the apex of the H2O depression and the end blue axis at the bulk at 20 °C (C) while the bulk buffer at 28 °C is calibrated by placing the start blue axis at the apex of the H2O and the end blue axis at the bulk at 28 °C with a stable baseline prior to the liposome injection (D). - The values for the lipid and peptide layers are processed by clicking into the Resolver Discrete Data using the same method used for analysis of the calibration data in Figure 2. That is, a blue axis is placed at the buffer baseline just before the liposome injection when the profile is representing the bare chip and the second blue axis is placed at the point on the profile that represents the lipid layer just prior to the peptide injection at 20 °C.
- Finally, a last blue axis is placed on the profile at the end of the protein dissociation just before the regeneration of the peptide-lipid layers at 20 °C. The layer properties can be processed and presented in the ‘Analysis’ window. In this window the individual materials for each part of the profile need to be defined. That is, 80% ethanol and H2O are selected as the chip calibration materials and H2O is selected for both bulk materials at 20 °C and 28 °C. In the discrete data section, lipid is selected for the RII (refractive index increment), bulk 28 is selected for bare chip, and bulk 20 is selected for both lipid and peptide layers. All real-time values can be calculated by clicking on the ‘calculate all’. From these real-time values the birefringence values can be calculated. This is performed by selecting the RI (or thickness value) as a fixed value and then clicking the ‘calculate all’ button.
- Open the data file in the Analight Explorer software.
- Calculation of optical birefringence and mass of an anisotropic layer
- The lipid molecules self-assemble into uniaxial aligned bilayers creating an anisotropic system with an uniaxial optical axis with two principal refractive indices, namely, the extraordinary refractive index (ne) which denotes the electric vector polarised parallel to the optical axis, and the ordinary refractive index (no) which denotes the electric vector polarised perpendicular to the optical axis. The difference between these two refractive indices ne and no for a lipid film is defined as the birefringence:
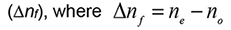
(Den Engelsen, 1976; Ramsden, 1999; Salamon and Tollin, 2001).
Birefringence is obtained from DPI by calculating the difference between the two effective refractive indices, namely the RI of the TM waveguide mode (nTM) and the RI of the TE waveguide mode (nTE). The birefringence values represent an averaged measurement of the degree of alignment and density of lipid molecules and are highly sensitive to small changes in these parameters. The effective birefringence (nTM - nTE) is determined by fixing the RI of the deposited layer to 1.47, which is indicative of a uniform layer coverage as determined by the absence of bovine serum albumin binding at 1 mg/ml in running buffer.
The de Feijter formula (De Feijter et al., 1978) is used to calculate the mass of adsorbed lipid and peptide layers from the DPI measurements. The mass of the lipid bilayer (mlipid) and the mass of the peptide (mpeptide) binding to the chip are calculated using: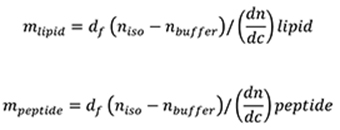
Where,
df is the thickness of the bilayer,
niso is the average isotropic RI of the attached bilayer,
nbuffer is the RI of the MOPS buffer,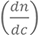 is the specific RI increment of the attached bilayer.
is the specific RI increment of the attached bilayer.
The De Feijter formula assumes that remains constant throughout the experiment. For the analysis, the standard values of 0.135 and 0.182 ml/g were used for lipids and peptides, respectively. The refractive index of the MOPS buffer (nbuffer) was measured as 1.3349 (T = 20 °C). Altering the salt concentration of the buffer will directly effect the RI value, for example increased salt concentration will result in an increased RI value. An average bilayer thickness of 46.1 with the standard deviation from ten experiments being 0.4 Å and a birefringence of 0.0198 ± 0.0005 was obtained for POPC: POPS: POPE: PI (n = 10), while an averaged bilayer thickness of 48.6 ± 0.5 Å and a birefringence of 0.0191 ± 0.0003 was obtained for the POPC: POPS: POPE: PI(4,5)P2 membrane (n = 10) in the absence of peptide. Data obtained from this method can be viewed in Payne et al. (2016).
- The lipid molecules self-assemble into uniaxial aligned bilayers creating an anisotropic system with an uniaxial optical axis with two principal refractive indices, namely, the extraordinary refractive index (ne) which denotes the electric vector polarised parallel to the optical axis, and the ordinary refractive index (no) which denotes the electric vector polarised perpendicular to the optical axis. The difference between these two refractive indices ne and no for a lipid film is defined as the birefringence:
Notes
Graphing data: DPI generates thousands of data points, initial calculations involving these data points can be done in Microsoft excel type programs. However, Graphpad Prism or Sigma plot is more efficient for graphing the entire data set due to the large number of data points that need to be graphed.
Recipes
- Bulk buffer (10 mM MOPS buffer pH 7 with 150 mM NaCl)
10 ml 0.1 M MOPS
90 ml degassed MilliQ
88 μl 5 N NaOH
876.6 mg NaCl
Dissolve salt and then degas the buffer - 2% SDS
4 g of SDS was dissolved in 200 ml of MilliQ and degassed - 80% ethanol
160 ml of 100% ethanol was added to 40 ml of MilliQ and degassed - CaCl2
2 mM CaCl2 was prepared in bulk buffer and degassed - EDTA
2 mM EDTA was prepared in bulk buffer and degassed - BSA
BSA was dissolved at 50 μg/ml in bulk buffer and then degassed
Acknowledgments
MAA is funded by the Australian Research Council (DP150104386). MIA acknowledges the support of the National Health & Medical Research Council (#1044327) and the Australian Research Council (DP1110101866). This protocol was adapted from the work of Lee et al. (2010) and Payne et al. (2016).
References
- Brogden, K. A. (2005). Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3(3): 238-250.
- De Feijter, J., Benjamins, D. J. and Veer, F. A. (1978). Ellipsometry as a tool to study the adsorption behavior of synthetic and biopolymers at the air–water interface. Biopolymers 17(7): 1759-1772.
- Den Engelsen, D. (1976). Optical anisotropy in ordered systems of lipids. Surface Science 56: 272-280.
- Green, R. J., Frazier, R. A., Shakesheff, K. M., Davies, M. C., Roberts, C. J. and Tendler, S. J. (2000). Surface plasmon resonance analysis of dynamic biological interactions with biomaterials. Biomaterials 21(18): 1823-1835.
- Lee, T. H., Heng, C., Swann, M. J., Gehman, J. D., Separovic, F. and Aguilar, M. I. (2010). Real-time quantitative analysis of lipid disordering by aurein 1.2 during membrane adsorption, destabilisation and lysis. Biochim Biophys Acta 1798(10): 1977-1986.
- Mader, J. S. and Hoskin, D. W. (2006). Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin Investig Drugs 15(8): 933-946.
- Mozsolits, H. and Aguilar, M. I. (2002). Surface plasmon resonance spectroscopy: an emerging tool for the study of peptide-membrane interactions. Biopolymers 66(1): 3-18.
- Payne, J. A., Bleackley, M. R., Lee, T. H., Shafee, T. M., Poon, I. K., Hulett, M. D., Aguilar, M. I., van der Weerden, N. L. and Anderson, M. A. (2016). The plant defensin NaD1 introduces membrane disorder through a specific interaction with the lipid, phosphatidylinositol 4,5 bisphosphate. Biochim Biophys Acta 1858(6): 1099-1109.
- Ramsden, J. J. (1999). Molecular orientation in lipid bilayers. Philosophical Magazine B 79(3): 381-386.
- Salamon, Z. and Tollin, G. (2001). Optical anisotropy in lipid bilayer membranes: coupled plasmon-waveguide resonance measurements of molecular orientation, polarizability, and shape. Biophys J 80(3): 1557-1567.
- van der Weerden, N. L., Bleackley, M. R. and Anderson, M. A. (2013). Properties and mechanisms of action of naturally occurring antifungal peptides. Cell Mol Life Sci 70(19): 3545-3570.
- Zasloff, M. (2002). Antimicrobial peptides of multicellular organisms. Nature 415(6870): 389-395.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Payne, J. A., Lee, T., Anderson, M. A. and Aguilar, M. (2017). Examination of the Interaction between a Membrane Active Peptide and Artificial Bilayers by Dual Polarisation Interferometry. Bio-protocol 7(1): e2087. DOI: 10.21769/BioProtoc.2087.
Category
Biochemistry > Lipid > Lipid-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.