- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Transformation of the Cyanobacterium Leptolyngbya boryana by Electroporation
Published: Vol 5, Iss 24, Dec 20, 2015 DOI: 10.21769/BioProtoc.1690 Views: 12357
Reviewed by: Maria SinetovaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Leptolyngbya boryana (L. boryana) (formerly Plectonema boryanum) is a versatile, filamentous cyanobacterium that has the ability to fix nitrogen under microoxic conditions and to grow heterotrophically with glucose in the dark, providing an excellent system to investigate photosynthesis, nitrogen fixation, and their regulatory mechanisms. While L. boryana is not naturally transformable different from the unicellular cyanobacterium Synechocystis sp. PCC 6803, it can be transformed by electroporation. Here we describe the transformation of L. boryana by electroporation to isolate mutants in which a targeted gene is disrupted.
Keywords: CyanobacteriaMaterials and Reagents
- Bottle top filter system (0.22 µm) (Corning, catalog number: 430624 )
- Microcentrifuge tubes (1.5 ml) (Ina-optika corporation, BIO-BIK, catalog number: ST-0150F ) or its equivalent, sterilized by autoclave (121 ºC, 20 min)
- Micropipettes tips (121 ºC, 20 min)
- Sterile syringe filter (Millex-GV Syringe Filter Unit, 0.22 µm), used for filter sterilization of solutions of antibiotics and glucose (Thermo Fisher Scientific, Millipore, catalog number: SLGV033RV ) or its equivalent
- Petri dish [sterile Petri dishes (90 mm x 15 mm)] (ASONE Corporation, catalog number: 1-7484-01-30 ) or its equivalent
- Pulse cuvettes (Gene Pulser cuvette, 0.1 cm) (Bio-Rad Laboratories, AbD Serotec®, catalog number: 1652089 )
- Hybond N+ filter (disc 82 mm diameter) (GE Healthcare, Amersham, catalog number: RPN82B ), sterilized by autoclave (121 ºC, 20 min).
- Leptolyngbya boryana (wild type or dg5) (grown on a BG-11 agar plate supplemented with 20 mM HEPES-KOH, pH 7.5 and 30 mM glucose)
Note: The dg5 strain was isolated from the wild type as a natural mutant that grows much faster heterotrophically in the dark than wild type (Fujita et al., 1996). Recently we identified that the mutation responsible for the dg5 phenotype is one adenine insertion causing a frameshift in the cytM gene encoding cytochrome cM (Hiraide et al., 2015). - Sterilized water (150 ml)
Note: Water is purified by RiOs Essential Water Purification System (Merck Millipore Corporation) or WEX system (Yamato). Distilled water can be also used. Autoclave (121 ºC, 20 min) and chill on ice before use. - NaNO3 (Nacalai tesque, catalog number: 31617-35 )
- 1 M K2HPO4 solution (Wako Pure Chemical Industries, Siyaku, catalog number: 164-04295 )
- 7.5% (w/v) MgSO4.7H2O (Nacalai tesque, catalog number: 21003-75 )
- 3.6% (w/v) CaCl2.2H2O (Nacalai tesque, catalog number: 06731-05 )
- 2.0% (w/v) Na2CO3 (Nacalai tesque, catalog number: 31311-25 )
- Citric acid (Wako Pure Chemical Industries, Siyaku, catalog number: 038-05521 )
- Ammonium iron (III) citrate, brown (Wako Pure Chemical Industries, Siyaku, catalog number: 092-00802 )
- Ethylenediamine-N, N, N', N'-tetraacetic acid, disodium salt, dihydrate (EDTA-Na2) (Dojindo, catalog number: 345-01865 )
- H3BO4 (Wako Pure Chemical Industries, Siyaku, catalog number: 021-02195 )
- MnCl2.4H2O (Nacalai tesque, catalog number: 21211-45 )
- ZnSO4.7H2O (Wako Pure Chemical Industries, Siyaku, catalog number: 268-00405 )
- Na2MoO4.2H2O (Wako Pure Chemical Industries, Siyaku, catalog number: 197-02485 )
- CuSO4.5H2O (Wako Pure Chemical Industries, Siyaku, catalog number: 033-04415 )
- Co(NO3)2.6H2O (Wako Pure Chemical Industries, Siyaku, catalog number: 031-03752 )
- 1 M Glucose (filter sterilized) (Wako Pure Chemical Industries, Siyaku, catalog number: 049-31165 )
- 50 mg/ml kanamycin sulfate (filter sterilized) (Wako Pure Chemical Industries, Siyaku, catalog number: 111-00344 )
- 50 mg/ml chloramphenicol (dissolve in ethanol) (Nacalai tesque, catalog number: 08027-72 )
- 10 mg/ml streptomycin sulfate (filter sterilized) (Meiji Seika Pharma, catalog number: 4987222665643 )
- 50 mg/ml erythromycin (filter sterilized) (Wako Pure Chemical Industries, Siyaku, catalog number: 054-05101 )
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Nacalai tesque, catalog number: 17514-15 )
- Plasmid preparation kit (GenElute HP Plasmid Midiprep Kit) (Sigma-Aldrich, catalog number: NA0200 )
- Linearized plasmid solution (> 2 µg µl–1)
Note: The concentration of plasmid is determined by a spectrophotometer before the enzyme digestion. - Bacto Agar [1.5% (w/v)] (BD, catalog number: 214010 )
- Ferric citrate solution (see Recipes)
- Trace metal A5+Co solution (1 L) (see Recipes)
- 2x BG-11 solution (1 L) (see Recipes)
- BG-11 agar plates (supplemented with 20 mM HEPES-KOH, pH 7.5, with or without appropriate antibiotics, Table 1) solidified with Bacto Agar [1.5% (w/v)] (BD, catalog number: 214010) (see Recipes)
Equipment
- Forceps
- Micropipettes ( P-1000 , P-200 and P-20 , or their equivalents) (Gilson)
- Clean bench
- Autoclave (TOMY DIGITAL BIOLOGY CO., model: SX-500 or its equivalent)
- Gene pulser Xcell (Bio-Rad Laboratories, AbD Serotec®, catalog numbers: 1652666 , 1652668 and 1652669 )
- Spectrophotometer (Shimadzu, model: UV-1700 or GE Healthcare, model: NanoVue Plus or its equivalent)
- Aspirator (with a vacuum pump)
- Growth cabinet (with appropriate fluorescence light bulbs)
Procedure
- Plasmid for targeted mutagenesis is constructed (Figure 1). Plasmids carry a chimeric DNA fragment consisting of the upstream sequence of the target gene, drug resistance cartridge (Table 1), and downstream sequence of the target gene (Figure 1B). The upstream and downstream sequences should be more than 1 kb each, for stable homologous recombination. If the upstream and downstream fragments are obtained by PCR, their nucleotide sequences in the plasmid should be confirmed by Sanger sequencing to avoid introduction of unintended mutation(s) outside of the target gene via homologous recombination. The vectors are normally pUC18/19 derivatives.
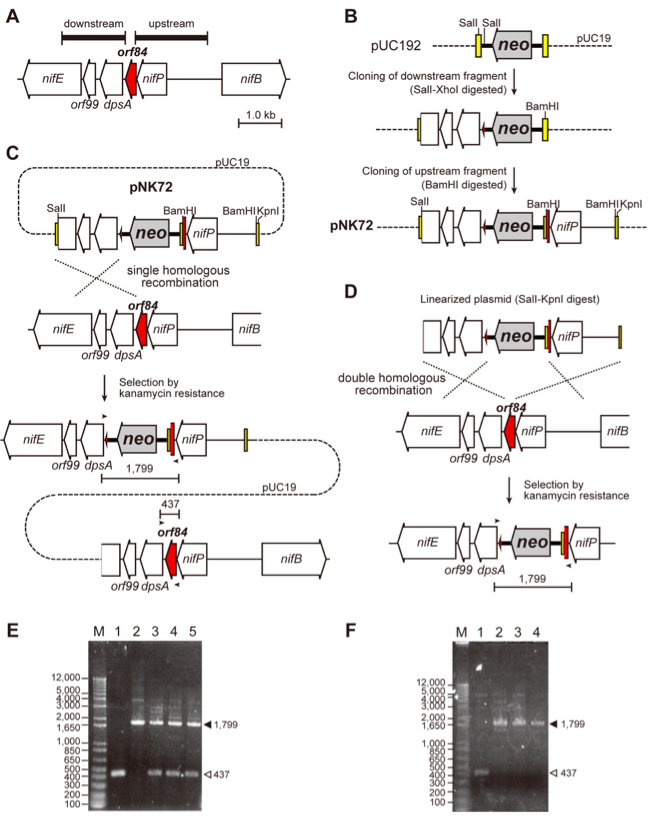
Figure 1. An example of plasmid construction, homologous recombination and PCR analysis of mutants in L. boryana. A. Gene arrangement of the target gene (the orf84 gene, LBWT_23230, shown in red) in L. boryana (Tsujimoto et al., 2014). The 1.7-kb upstream and 1.5-kb downstream fragments (thick horizontal bars) of the orf84 gene are amplified by PCR to construct plasmid. B. Construction of the plasmid pNK72 to isolate an orf84-disrupted mutant (∆orf84). The two amplified fragments were cloned into a vector. In this case, pUC192 was used as the vector, which was constructed by the insertion of the neo gene (kanamycin resistance gene from pMC19, Fujita et al., 1992) into the BamHI site of pUC19 (dashed line with its multi-cloning sites shown by yellow boxes). The 1.5-kb downstream fragment (digested with SalI-XhoI) was cloned into the SalI sites of pUC192, and subsequently the 1.7-kb upstream fragment (digested with BamHI) was cloned into the BamHI site, yielding the plasmid pNK72. C. Gene arrangement of a single recombinant, in which the plasmid pNK72 is incorporated into the chromosome by a single homologous recombination event. Both wild-type and knock-out copies are present in the chromosome. In this case the single recombination event occurs between the 1.5-kb downstream fragment of the plasmid and the corresponding chromosomal part. Small horizontal arrows indicate primers for PCR shown in panel E. D. Gene arrangement of the ∆orf84 mutant, in which the orf84 gene was replaced with the neo gene by a double recombination event. The plasmid was previously digested with KpnI and SalI for electroporation to avoid to generate single recombinants. Small horizontal arrows indicate primers for PCR shown in panel F. E. PCR analysis of the single recombinants of orf84. Two DNA fragments were detected. The longer (a filled triangle, 1,799 bp) and shorter (an open triangle, 437 bp) fragments are corresponding to those of knock-out and wild-type copies, respectively, in the three transformants (lanes 3-5). The longer fragment was detected in PCR with the plasmid pNK72 as the template (lane 2). The shorter fragment (the wild-type copy) was detected in the wild-type cells (lane 1). F. PCR analysis of ∆orf84. Only single DNA fragment corresponding to the knock-out copy of orf84 (a filled triangle, 1,799 bp) was detected in the two transformants (lanes 3 and 4). The shorter and longer fragments (lanes 1 and 2) were the same as E. (unpublished results, Kotani and Fujita). - The plasmid prepared by a plasmid preparation kit was linearized by restriction enzyme(s) to minimize the probability that the plasmid would integrate into the genome via a single recombination event resulting in a merodiploid that harbors both knock-out and wild-type copies in the genome (Figure 1C). The digestion site(s) can be selected from anywhere in the vector part. It should be careful not to digest the parts of homologous recombination and the drug resistance marker. The digested plasmid was precipitated by ethanol and dissolved in sterilized water to be >2 µg µl–1.
- L. boryana is cultivated on an agar plate of BG-11 containing 30 mM glucose under low light conditions (<10 µmol m-2 s-1) at 30°C for 2-7 days (Note 1).
- All procedures below should be performed under aseptic conditions. Cells on the agar plate are suspended in sterilized water (ca. 2 ml per one agar plate) and the suspension is transferred into a bottle top filter (Figure 2A-C. Notes 2 and 3). In this stage, Hybond N+ filters should be placed onto new BG-11 agar plates for cell recovery after electroporation (step 11).
- The cells are collected on the surface of the bottle top filter by aspiration (Figure 2D).

Figure 2. Preparation of L. boryana cells for pulse application - Approximately 50 ml of sterilized water, chilled on ice, is added to the filter to suspend the cells. The cells are again collected on the filter by aspiration. This washing procedure is repeated three times (Figure 3C. Notes 4-5). Centrifugation (1,200 x g, 10 min) is another option for washing cells with water. In this case, note that L. boryana cells form very loose pellets even by high-speed centrifugation (for example, 44,000 x g, 30 min) due to long trichomes.
- The cells on the filter are suspended in a small aliquot (500 µl) of sterilized water (Figure 2D) and the suspension is transferred into a microcentrifuge tube (1.5 ml) to store on ice until pulse application (Note 3).
- A small aliquot (50 µl) of the suspension used for pulse application is dispensed into another microcentrifuge tube (Note 3) and the concentrated plasmid solution (typically 10 µl of 2 µg µl-1) is added to the tube and mixed well.
- The mixture is transferred into a cuvette with a gap width of 0.1 cm and chilled on ice (Figure 2E-F, Note 3).
- A single exponential decay pulse (setting: voltage, 1.41 kV; capacitance, 50 µF; and resistance, 250 ohms) is applied. The time constant will be approximately 10 ms. The time constant and the actual voltage should be recorded on your note (Notes 5-6).
- An aliquot (350 µl) of BG-11 liquid medium is added to the cuvette and the cell suspension is recovered with the blue tip of a P-1000 type micropipette dispenser (Figure 3A) to spread onto a Hybond N+ filter overlaid on a BG-11 agar plate without antibiotics (Figure 3B-C).
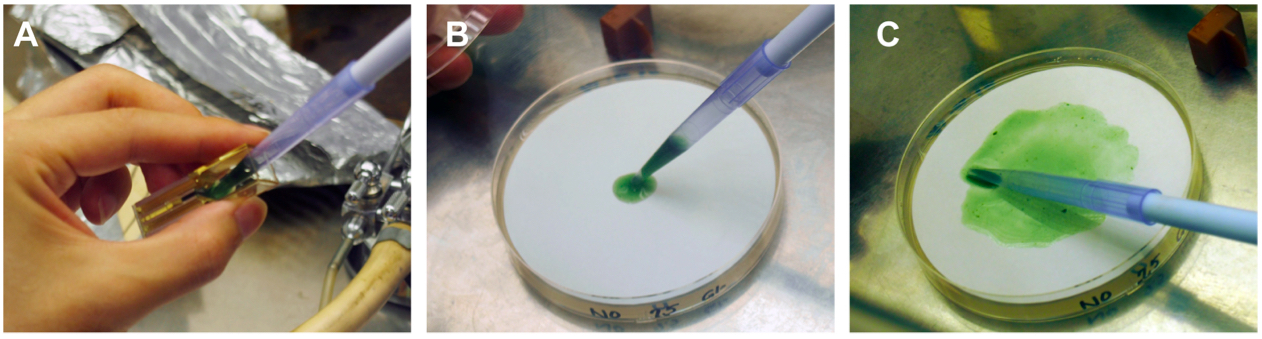
Figure 3. Recovery of pulse-applied cells from a cuvette and inoculation on a non-selective plate - The agar plate is incubated at 30 °C under low light conditions (<10 µmol m-2 s-1) for 2 days. During this period the antibiotic resistance gene is expressed to confer the cell’s resistance to the specific antibiotic.
- The Hybond N+ filer is transferred onto a new BG-11 agar plate containing appropriate antibiotics (final concentrations are shown in Table 1). The agar plate is incubated at 30 °C under high light conditions (ca. 100 µmol m-2 s-1).
- Colonies showing resistance against the antibiotics appear after approximately 10 to 14 days (Figure 4). You can pick them up with toothpicks or tips (for a P-200-type micropipette) that should be previously autoclaved (121 °C, 20 min).
- Some colonies are single recombinants and some are double recombinants. PCR analysis should be carried out to identify double recombinants (Figure 1E-F; Notes 7 -8).
Table 1. List of antibiotics that can be used for transformation of L. boryana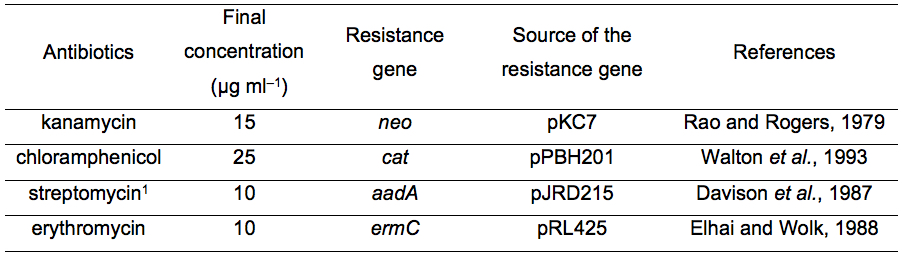
1This antibiotic has not yet been used for targeted mutagenesis. However, we confirmed that transformants harboring pJRD215 (with the streptomycin resistance gene) showed resistance to streptomycin (unpublished results).
Representative data
Representative results in targeted gene disruption are shown in Table 2. In most cases almost all transformants appeared on selective plates were double recombinants. A procedure of repeated inoculations of transformants to segregate cells in which all wild-type copies are completely replaced with the mutant copies is required in Synechocystis sp. PCC 6803, but this process is not needed in L. boryana (Note 8).
Table 2. Examples of electroporation of L. boryana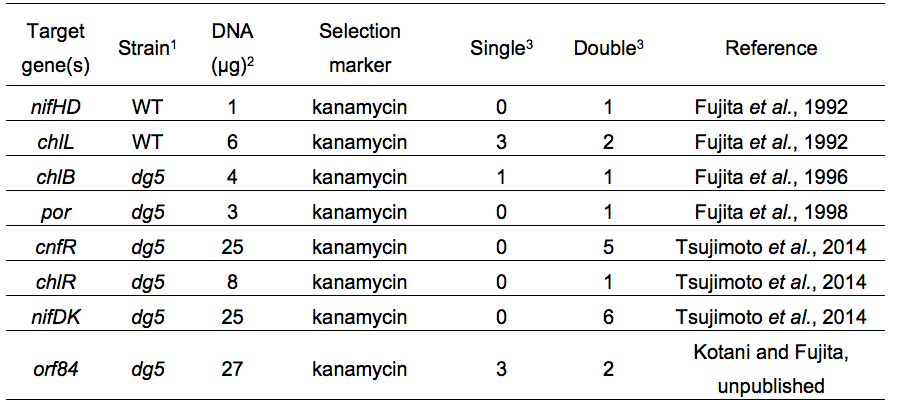
1WT, wild type
2Total DNA amount in the cell suspension for pulse application
3Number of colonies. Single or double recombinant was confirmed by Southern blot analysis or PCR
Figure 4. Typical appearance of transformants on a selective plate. Two tiny green colonies (red arrows) appeared 14 days after pulse application.
Notes
- Liquid culture of BG-11 (containing 20 mM HEPES-KOH, pH 7.5 and 30 mM glucose) can be used (Fujita et al., 1992; Fujita et al., 1996, Fujita et al., 1998; Kimata-Ariga et al., 2000).
- To suspend cells on an agar plate, we use a sterile bent glass rod that is used for spreading Escherichia coli cells in transformation.
- Concentrated cell suspension may be too viscous to dispense by a normal micropipette tip. Thus, we prepare special tips (both of P-1000 and P-200 tips) by cutting the ends of normal tips with scissors (to be a diameter of approximately 2 mm) and used them for handling dense cell suspensions. These tips are autoclaved (121 °C, 20 min) before use.
- In the original procedure (Fujita et al., 1992), 1 mM HEPES-KOH (pH 7.5) and 10% (w/v) glycerol were sequentially used for washing cells. We recently found that using water instead gives essentially the same results as described here.
- Insufficient washing of cells and/or impurity of plasmid preparation may cause arc discharge in pulse application, resulting in transformation failure.
- Original settings were; voltage, 1.25 kV; capacitance 25 µF; and resistance, 600 ohms (Fujita et al., 1992). Using another electroporator (BTX Electro Cell Manipulator 600), the settings are; voltage, 1.45 kV; capacitance 50 µF; and resistance, 256 ohms.
- While only single recombinants may be isolated in one experiment, double recombinants can be eventually isolated in further trials (Figure 1). However, if double recombinants are never isolated even after many times trials of transformation, it is probable that the target gene is essential for growth under the conditions.
- Differentiation from a single recombinant to a double recombinant has never been observed even though many times of subculturing.
- For transformation with a shuttle vector, derivatives of pPBH201 (Kimata-Ariga et al., 2000; Yamamoto et al., 2009; Yamamoto et al., 2011), a relatively low concentration of plasmid (<0.1 µg µl-1) is enough to isolate transformants.
Recipes
- Ferric citrate solution
60 mg citric acid
60 mg ammonium iron (III) citrate, brown
10 mg EDTA.Na2 - Trace metal A5+Co solution (1 L)
2.86 g H3BO4
1.81 g MnCl2.4H2O
0.22 g ZnSO4.7H2O
0.39 g Na2MoO4.2H2O
79 mg CuSO4.5H2O
49 mg Co(NO3)2.6H2O - 2x BG-11 solution (1 L)
3.0 g NaNO3 dissolved in 932 ml distilled water
460 µl of 1 M K2HPO4 solution
2.0 ml of 7.5% (w/v) MgSO4.7H2O
2.0 ml of 3.6% (w/v) CaCl2.2H2O
2.0 ml of 2.0% (w/v) Na2CO3
20.0 ml of Ferric citrate solution
Serially add 2.0 ml of Trace metal A5+Co
40 ml of 1 M HEPES-KOH, pH 7.5
Stored at 4 ºC
Note: BG-11 liquid medium (Rippka et al., 1979) supplemented with 20 mM HEPES-KOH (pH 7.5). For BG-11 liquid medium, the equal amounts of 2x BG-11 and distilled water are mixed, and autoclaved (121 ºC, 20 min). - BG-11 agar plates
The equal amounts of 2x BG-11 and distilled water containing:
Bacto Agar [3.0% (w/v)] is separately autoclaved (121 ºC, 20 min). Allow the media to cool about 50 ºC (Be careful not to be solidified at this stage). The solutions of 2x BG-11 and agar are mixed in a clean bench. If needed, appropriate antibiotic solution and glucose solution are added. Then, pour about 30 ml of medium into each Petri dish. When the BG-11 agar medium solidifies, invert the agar plates and store at 4 ºC.
Acknowledgments
This protocol was adapted from the previously published studies, Hiraide et al. (2015), Tsujimoto et al. (2014) and Fujita et al. (1992). The original protocol was described in Fujita et al. (1992), and we modified it as described here. We thank Yasuhiro Takahashi, Tomohiro Matsumura, Toshiharu Hase, and Hiroshi Matsubara for initial works on establishment of this transformation system. We thank Douglas K. Walton, Carl E. Bauer and Peter Wolk for donating plasmids pPBH201, pJRD215 and pRL425, respectively. We thank Chie Tomatsu for providing technical help. This work was supported by the Japan Society for the Promotion of Science (JSPS) (Grants-in-Aid for Scientific Research Nos. 05740481, 06740601, 07740617, 08836006, 11740445, 23370020, 23000007, 26660084, 15H04387 and 15H01397), Precursory Research for Embryonic Science and Technology (PRESTO) and the Advanced Low Carbon Technology Research and Development Program (ALCA).
References
- Davison, J., Heusterspreute, M., Chevalier, N., Ha-Thi, V. and Brunel, F. (1987). Vectors with restriction site banks. V. pJRD215, a wide-host-range cosmid vector with multiple cloning sites. Gene 51(2-3): 275-280.
- Elhai, J. and Wolk, C. P. (1988). Conjugal transfer of DNA to cyanobacteria. Methods Enzymol 167: 747-754.
- Fujita, Y., Takagi, H. and Hase, T. (1996). Identification of the chlB gene and the gene product essential for the light-independent chlorophyll biosynthesis in the cyanobacterium Plectonema boryanum. Plant Cell Physiol 37(3): 313-323.
- Fujita, Y., Takagi, H. and Hase, T. (1998). Cloning of the gene encoding a protochlorophyllide reductase: the physiological significance of the co-existence of light-dependent and -independent protochlorophyllide reduction systems in the cyanobacterium Plectonema boryanum. Plant Cell Physiol 39(2): 177-185.
- Fujita, Y., Takahashi, Y., Chuganji, M. and Fujita, Y. (1992). The nifH-like (frxC) gene is involved in the biosynthesis of chlorophyll in the filamentous cyanobacterium Plectonema boryanum. Plant Cell Physiol 33(1): 81-92.
- Hiraide, Y., Oshima, K., Fujisawa, T., Uesaka, K., Hirose, Y., Tsujimoto, R., Yamamoto, H., Okamoto, S., Nakamura, Y., Terauchi, K., Omata, T., Ihara, K., Hattori, M. and Fujita, Y. (2015). Loss of cytochrome cM stimulates cyanobacterial heterotrophic growth in the dark. Plant Cell Physiol 56(2): 334-345.
- Kimata-Ariga, Y., Matsumura, T., Kada, S., Fujimoto, H., Fujita, Y., Endo, T., Mano, J., Sato, F. and Hase, T. (2000). Differential electron flow around photosystem I by two C(4)-photosynthetic-cell-specific ferredoxins. EMBO J 19(19): 5041-5050.
- Rao, R. N. and Rogers, S. G. (1979). Plasmid pKC7: a vector containing ten restriction endonuclease sites suitable for cloning DNA segments. Gene 7(1): 79-82.
- Rippka, Deruelles, J., Waterbury, J. B., Herdman, M. and Stanier, R. Y. (1979). Genetic assignments, strain histories and properties of pure cultures of cyanobacteria. J Gen Microbiol 111, 1-61.
- Tsujimoto, R., Kamiya, N. and Fujita, Y. (2014). Transcriptional regulators ChlR and CnfR are essential for diazotrophic growth in nonheterocystous cyanobacteria. Proc Natl Acad Sci U S A 111(18): 6762-6767.
- Walton, D. K., Gendel, S. M. and Atherly, A. G. (1993). DNA sequence and shuttle vector construction of plasmid pGL3 from Plectonema boryanum PCC 6306. Nucleic Acids Res 21(3): 746.
- Yamamoto, H., Kurumiya, S., Ohashi, R. and Fujita, Y. (2009). Oxygen sensitivity of a nitrogenase-like protochlorophyllide reductase from the cyanobacterium Leptolyngbya boryana. Plant Cell Physiol 50(9): 1663-1673.
- Yamamoto, H., Kurumiya, S., Ohashi, R. and Fujita, Y. (2011). Functional evaluation of a nitrogenase-like protochlorophyllide reductase encoded by the chloroplast DNA of Physcomitrella patens in the cyanobacterium Leptolyngbya boryana. Plant Cell Physiol 52(11): 1983-1993.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tsujimoto, R., Kotani, H., Nonaka, A., Miyahara, Y., Hiraide, Y. and Fujita, Y. (2015). Transformation of the Cyanobacterium Leptolyngbya boryana by Electroporation. Bio-protocol 5(24): e1690. DOI: 10.21769/BioProtoc.1690.
Category
Microbiology > Microbial genetics > Transformation
Molecular Biology > DNA > Transformation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.