- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Character-State Reconstruction to Infer Ancestral Protein-Protein Interaction Patterns

Published: Vol 5, Iss 16, Aug 20, 2015 DOI: 10.21769/BioProtoc.1566 Views: 13844
Reviewed by: Arsalan DaudiPrashanth SuravajhalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Protein-protein interactions are at the core of a plethora of developmental, physiological and biochemical processes. Consequently, insights into the origin and evolutionary dynamics of protein-protein interactions may provide information on the constraints and dynamics of specific biomolecular circuits and their impact on the organismal phenotype.
This protocol describes how ancestral protein-protein interaction patterns can be inferred using a set of known protein interactions from phylogenetically informative species. Although this protocol focuses on protein-protein interaction data, character-state reconstructions can in general be performed with other kinds of binary data in the same way.
Data
- Protein-protein interaction data
A comprehensive list of interactions for the protein family under study should be compiled. As interaction data are typically generated only for proteins whose sequences have been deposited in databases, a recently published comprehensive phylogeny of the protein family under study may yield an upper estimate of the number and phylogenetic breadth of interaction data to be expected. In many cases recently published phylogenetic relationships need to be extracted from the publications itself, however a growing number of phylogenies are being uploaded in online databases such as TreeBASE (http://treebase.org/treebase-web/home.html) or Dryad (http://datadryad.org/).- For obtaining data on protein-protein interactions, databases might be used. Prominent examples of such databases include BioGRID (http://thebiogrid.org/) (Chatr-Aryamontri et al., 2013), the Database of Interacting Proteins (http://dip.doe-mbi.ucla.edu/) (Salwinski et al., 2004), IntAct (http://www.ebi.ac.uk/intact/) (Orchard et al., 2014) and String (http://string-db.org/) (Franceschini et al., 2013), to mention but a few. Above all, UniProt (http://www.uniprot.org/) (UniProt 2015) provides cross-references to a number of these database, thus facilitating searches for potential interaction partners.
- Whereas database searches provide a good starting point, they very often do not capture all of the information available. It is therefore advisable to undertake a literature search. Special emphasize should be put on obtaining information from phylogenetically informative proteins, i.e. from proteins that occupy a position in the phylogeny that is critical for resolving the state of a particular trait (i.e. the character-state). Very often these are the early-diverging lineages, as their inclusion (together with more derived taxa) ensures that the whole phylogenetic breadth of a taxonomic group is captured. It might prove useful to obtain new experimental data for proteins that are phylogenetically especially informative. Indeed, generation of new protein-protein interaction data is often combined with character-state reconstruction to better understand the evolution of protein-protein interactions (Liu et al., 2010; Melzer et al., 2014; Li et al., 2015).
- For obtaining data on protein-protein interactions, databases might be used. Prominent examples of such databases include BioGRID (http://thebiogrid.org/) (Chatr-Aryamontri et al., 2013), the Database of Interacting Proteins (http://dip.doe-mbi.ucla.edu/) (Salwinski et al., 2004), IntAct (http://www.ebi.ac.uk/intact/) (Orchard et al., 2014) and String (http://string-db.org/) (Franceschini et al., 2013), to mention but a few. Above all, UniProt (http://www.uniprot.org/) (UniProt 2015) provides cross-references to a number of these database, thus facilitating searches for potential interaction partners.
- Sequence retrieval
Protein or nucleotide sequences for phylogenetic reconstructions can be retrieved from the NCBI nucleotide collection (http://www.ncbi.nlm.nih.gov/nuccore) or the NCBI protein collection (http://www.ncbi.nlm.nih.gov/protein).
Software
- For sequence alignment and subsequent phylogenetic reconstructions one or several of the following programs may be used:
Table 1. Programs for sequence alignments and phylogenetic reconstructionsProgram Purpose Reference ExPASy translate Translation of nucleotide sequences into amino acid sequences. (Artimo et al., 2012)
http://web.expasy.org/translate/Clustal 2 Sequence alignment. Suited especially for closely related sequences. (Larkin et al., 2007)
http://www.clustal.org/clustal2/MAFFT7 Sequence alignment. Suited for closely as well as more distantly related sequences. (Katoh and Standley, 2013)
http://mafft.cbrc.jp/alignment/software/RevTrans 1.4 Converting amino acid alignment into codon alignment. (Wernersson and Pedersen, 2003)
http://www.cbs.dtu.dk/services/RevTrans/MEGA 6 Sequence alignment and phylogenetic reconstruction. (Tamura et al., 2013) http://www.megasoftware.net/ MrBayes 3 Phylogenetic reconstruction. (Ronquist and Huelsenbeck, 2003)
http://mrbayes.sourceforge.net/index.php - To collate the character matrix
Microsoft Excel or a similar spreadsheet application - For character-state reconstruction:
Mesquite 3.02 (Maddison and Maddison, 2015) (http://mesquiteproject.org/)
Mesquite also provides extensive documentation: (http://mesquiteproject.wikispaces.com/)
Procedure
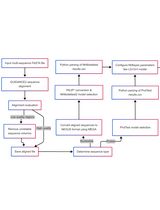
- Compilation of the character matrix
A character matrix is constructed that contains the names of the proteins and their interaction properties. This can be done using an Excel spreadsheet. Alternatively, data may be entered directly in Mesquite (Figure 1). It is possible to collate information for several interacting partners in separate columns. To conduct a likelihood character-state reconstruction with Mesquite (see below) data have to be coded categorically, i.e. ‘0’ for no interaction and ‘1’ for an interaction. Combinations for which the interactions status is unknown are left blank. Theoretically, one may also introduce three or more categories, e.g. ‘0’: no interaction; ‘1’: weak interaction; ‘2’ strong interaction. However, one needs to be aware of the fact that the categories are still discrete and do not follow a hierarchy (e.g. there is no constraint such that evolution has to proceed from ‘no’ to ‘weak’ to ‘strong’ interactions).
Coding of interactions can be complicated by the phylogenetic history of the interaction partner. Consider an example in which protein A interacts with protein B in a certain model organism. In another organism, one ortholog of A, termed A’ here, may exist, but two co-orthologs of B, (B’ and B’’) occur. If A’ interacts with B’ but not with B’’ it is difficult to assign an interaction status to A’ (Figure 2). One compromise is to designate A’ as interacting as long as an interaction with either B’ or B’’ is observed (Melzer et al., 2014). The situation gets more complicated if only incomplete data sets are available. Assume, for example, A’ is not interacting with B’’, but information on the interaction between A’ and B’ is not available. In this case, one may designate the interaction status of A’ as unknown to avoid the inclusion of false negatives in the dataset (Melzer et al., 2014). It is difficult to estimate how frequently these problems will appear in a particular dataset. It is therefore important to consider the phylogenetic history of the interaction partner in character-state reconstructions.
If the interaction data gathered rely on different methods it is helpful to also collect data for each method separately (Figure 1). This will later reveal whether the results of the character-state reconstruction depend on the method used to obtain the interaction data.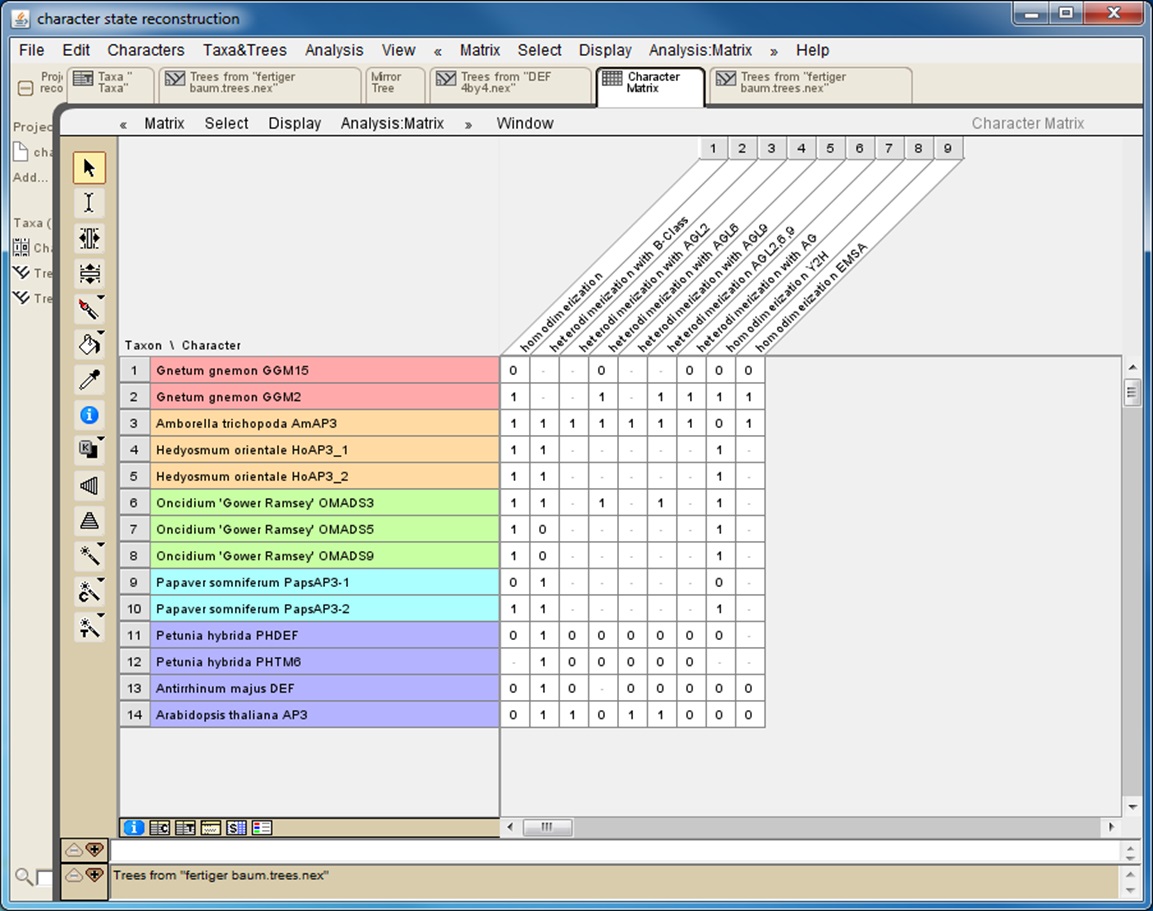
Figure 1. Screenshot from a character matrix in Mesquite. Protein names are listed in the second (coloured) column. Interaction characteristics are listed in subsequent columns. Data on homodimerization as well as on heterodimerization with other proteins and data obtained with different techniques (Y2H: yeast two-hybrid; EMSA: electrophoretic mobility shift assay) are collated in separate columns.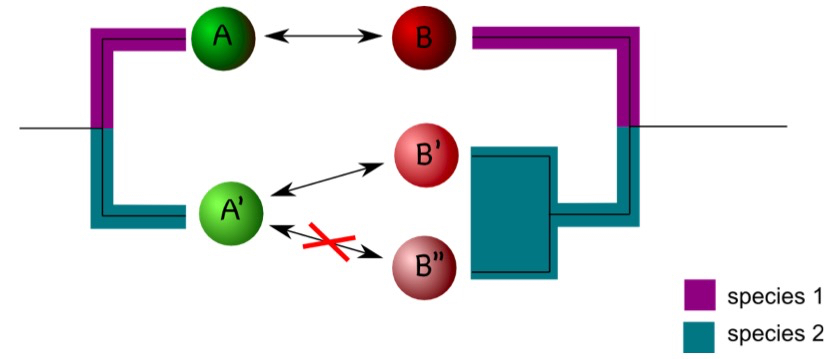
Figure 2. Duplications can complicate coding interactions. Proteins A and B interact in species 1 (as indicated by the double arrow). In species 2, A’ interacts with B’ but not with B’’. This raises the question as how to code the interaction status of A’. - Phylogenetic reconstruction
A phylogeny covering all of the proteins under study needs to be constructed using one of the many software tools available (e.g. MrBayes, MEGA 6, Bali-Phy, PhyML, see also Table 1). For an overview of basic concepts and methods in phylogeny reconstruction see De Bruyn et al. (2014). The phylogeny can be constructed using the sequences of the proteins under study. However, in principle every tree can be used as long as each protein is assigned to a specific position in the tree. Protein names in the character matrix described above and in the phylogenetic tree have to be identical to be later able to connect the two datasets. Mesquite also offers the possibility to manually draw trees; this may be used for cases in which a computational phylogenetic reconstruction is not feasible.
The phylogeny may contain proteins for which interaction data are not available. These will later be ignored by the character-state analysis. - Character-state reconstruction
The character-state reconstruction is done using Mesquite. For general instructions on how to handle Mesquite one may visit the ‘Mesquite ProjectTeam’ YouTube channel (https://www.youtube.com/channel/UCfSmgC0O_dWLI0PEoXZbS4Q).- Import/generate tree:
Mesquite allows to import trees from other files in several ways (http://mesquiteproject.wikispaces.com/Trees). If trees are read from NEXUS files note that Mesquite cannot handle some special characters (e.g. dash) if present in protein names. When importing a phylogenetic tree, the branch lengths will later be taken into consideration for the character-state reconstruction. If a manually drawn tree is used, all branch lengths will by default be set to 1. This may work well in a number of cases, but it should be kept in mind that proteins from early diverging taxa may possess artificially short branches under this setting (Figure 3). However, Mesquite also allows editing branch lengths (https://mesquiteproject.wikispaces.com/Trees).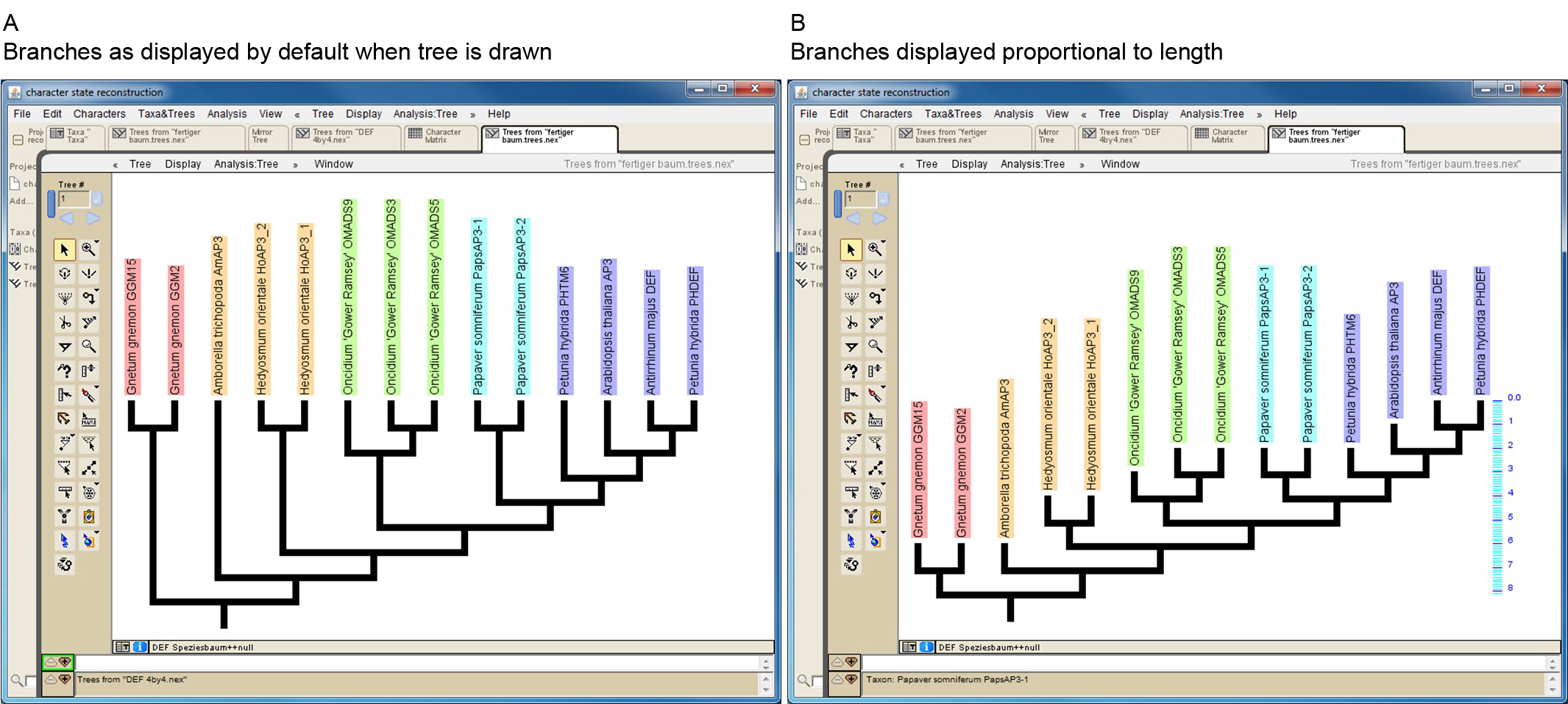
Figure 3. Screenshot of a manually drawn tree in Mesquite. A. Branches are by default displayed so that all tips reach the same level. However, branch lengths are by default set to one, as can be seen in B, where branches of the same tree are displayed proportional to length (see scaling on the right side of the tree). This reveals that some branches (e.g. that leading to Amborella trichopoda AmAP3) might be unreasonable short. - Import/generate data matrix:
There are several ways to generate or import a data matrix implemented in Mesquite (http://mesquiteproject.wikispaces.com/Characters+%26+Matrices). A straightforward approach is to generate a new blank data matrix with the required number of characters and copy/paste the interaction data from the original data source (i.e. from the Excel spreadsheet). The matrix needs to be specified as categorical to be used for the character state reconstruction. - Model specification and character reconstruction:
Mesquite provides an extensive documentation on the different settings for the character-state reconstruction: http://mesquiteproject.wikispaces.com/Ancestral+States. In our analyses we employed likelihood reconstruction methods (Melzer et al., 2014), but parsimony reconstructions are also available (Li et al., 2015). Two general models can be used for likelihood reconstructions: The ‘Markov k-state 1 parameter model’ (Mk1) and the ‘Asymmetrical Markov k-state 2 parameter model’ (AsymmMk). The principal difference between these two models is that the 2 parameter model allows ‘forward’ and ‘backward’ rates to be different, i.e. the probabilities for gaining and losing an interaction can be different. In the 1 parameter model, gaining and losing an interaction is equally probable. Biologically, it would in most cases make more sense to apply the 2 parameter model, as one may assume that it is more likely to lose an interaction than gaining it. However, several reports have shown that 2 parameter models can lead to implausible results if small to medium sized datasets (data on less than 100 protein-protein interactions) are being used (Mooers and Schluter, 1999; Pagel, 1999). A likelihood ratio test can be used to infer whether the 2 parameter model significantly improves the fit of the model to the data as compared to the 1 parameter model (Pagel, 1999; Ree and Donoghue, 1999). This test is performed by subtracting the - log probability values derived from the two models and multiplying the absolute value of the result by 2 (|(-logLMk1)-(-logLAsymmMk)|•2). The resulting number can be used as test statistic for a Chi-square test with one degree of freedom. The test is also integrated in Mesquite an can be conducted via Analysis: Tree > Values for Current Tree > Asymmetry Likelihood Ratio Test.
In the Mk1 and AsymmMk models, the rate of a character’s evolution is estimated by Mesquite (http://mesquiteproject.wikispaces.com/Processes+of+Character+Evolution#param). However, it is also possible to create own models with specific parameters (http://mesquiteproject.wikispaces.com/Ancestral+States#editingModels). This can be useful if, for example, the probability of gaining vs. losing an interaction is known from prior experimental evidence. - Evaluation of the results:
Results are best visualized using pie charts at the internal nodes of the tree (Figure 4). Mesquite offers the possibility to conduct the character-state reconstruction simultaneously over different phylogenetic trees. Also, several characters can be traced at once. This facilitates comparison of character-state reconstructions of one protein with different partners or comparison of character-state reconstructions based on different methods used to assay protein-protein interactions. - Export options:
Mesquite can export trees and character matrices in numerous ways (http://mesquiteproject.wikispaces.com/Interactions+with+Other+Programs). For a graphical representation of the character state reconstruction results we recommend to export the tree as PDF and use this file for further post-processing with graphics software such as Adobe Illustrator. For a direct comparison of the character state evolution of two different traits one may utilize the mirror tree function (Figure 4).
- Import/generate tree:
Representative data
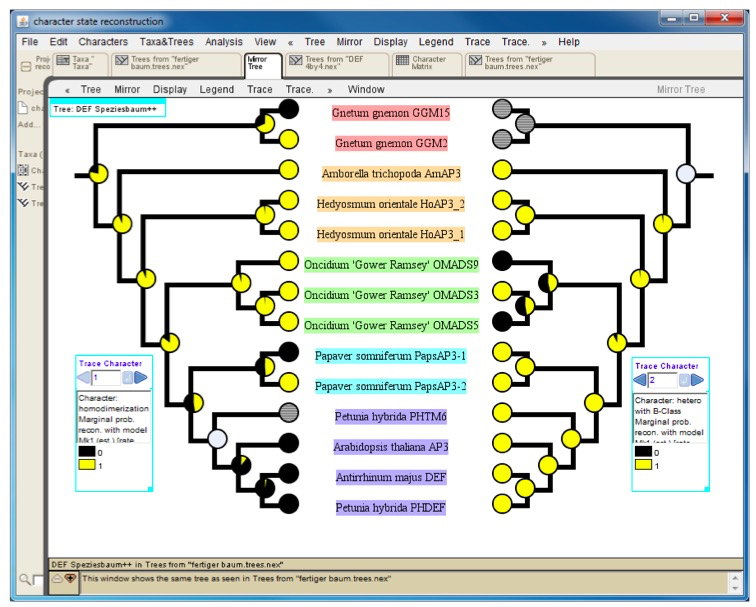
Figure 4. Mirror tree comparing results of character-state reconstruction for homodimerization (left) and heterodimerization (right) of a subfamily of plant transcription factors. Pie charts at internal nodes indicate the probability of the presence (yellow) or absence (black) of an interaction. Hatched circles at terminal positions (e.g. Petunia hybrida PHTM6 on the left tree) and grey circles at internal nodes designate an unknown interaction status.
Acknowledgments
This protocol was adapted from a previously published study (Melzer et al., 2014). This research was supported by a DFG grant to G. T. and R. M. (TH417/5–2). R. M. was supported by a post-doctoral fellowship of the Carl-Zeiss-Foundation.
References
- Artimo, P., Jonnalagedda, M., Arnold, K., Baratin, D., Csardi, G., de Castro, E., Duvaud, S., Flegel, V., Fortier, A., Gasteiger, E., Grosdidier, A., Hernandez, C., Ioannidis, V., Kuznetsov, D., Liechti, R., Moretti, S., Mostaguir, K., Redaschi, N., Rossier, G., Xenarios, I. and Stockinger, H. (2012). ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40(Web Server issue): W597-603.
- Chatr-Aryamontri, A., Breitkreutz, B. J., Heinicke, S., Boucher, L., Winter, A., Stark, C., Nixon, J., Ramage, L., Kolas, N., O'Donnell, L., Reguly, T., Breitkreutz, A., Sellam, A., Chen, D., Chang, C., Rust, J., Livstone, M., Oughtred, R., Dolinski, K. and Tyers, M. (2013). The BioGRID interaction database: 2013 update. Nucleic Acids Res 41(Database issue): D816-823.
- De Bruyn, A., Martin, D. P. and Lefeuvre, P. (2014). Phylogenetic reconstruction methods: an overview. Methods Mol Biol 1115: 257-277.
- Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., Roth, A., Lin, J., Minguez, P., Bork, P., von Mering, C. and Jensen, L. J. (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41(Database issue): D808-815.
- Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30(4): 772-780.
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. and Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23(21): 2947-2948.
- Li, L., Yu, X. X., Guo, C. C., Duan, X. S., Shan, H. Y., Zhang, R., Xu, G. X. and Kong, H. Z. (2015). Interactions among proteins of floral MADS-box genes in Nuphar pumila (Nymphaeaceae) and the most recent common ancestor of extant angiosperms help understand the underlying mechanisms of the origin of the flower. Journal of Systematics and Evolution: n/a-n/a.
- Liu, C., Zhang, J., Zhang, N., Shan, H., Su, K., Zhang, J., Meng, Z., Kong, H. and Chen, Z. (2010). Interactions among proteins of floral MADS-box genes in basal eudicots: implications for evolution of the regulatory network for flower development. Mol Biol Evol 27(7): 1598-1611.
- Maddison, W. P. and Maddison, D. R. (2015). Mesquite: a modular system for evolutionary analysis. Version 3.02.
- Melzer, R., Härter, A., Rumpler, F., Kim, S., Soltis, P. S., Soltis, D. E. and Theissen, G. (2014). DEF- and GLO-like proteins may have lost most of their interaction partners during angiosperm evolution. Ann Bot 114(7): 1431-1443.
- Mooers, A. O. and Schluter, D. (1999). Reconstructing ancestor states with maximum likelihood: Support for one- and two-rate models. Syst Biol 48: 623-633.
- Orchard, S., Ammari, M., Aranda, B., Breuza, L., Briganti, L., Broackes-Carter, F., Campbell, N. H., Chavali, G., Chen, C., del-Toro, N., Duesbury, M., Dumousseau, M., Galeota, E., Hinz, U., Iannuccelli, M., Jagannathan, S., Jimenez, R., Khadake, J., Lagreid, A., Licata, L., Lovering, R. C., Meldal, B., Melidoni, A. N., Milagros, M., Peluso, D., Perfetto, L., Porras, P., Raghunath, A., Ricard-Blum, S., Roechert, B., Stutz, A., Tognolli, M., van Roey, K., Cesareni, G. and Hermjakob, H. (2014). The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42(Database issue): D358-363.
- Pagel, M. (1999). The maximum likelihood approach to reconstructing ancestral character states of discrete characters on phylogenies. Syst Biol 48: 612-622.
- Ree, R. H. and Donoghue, M. J. (1999). Inferring rates of change in flower symmetry in asterid angiosperms. Syst Biol 48: 633-641.
- Ronquist, F. and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19(12): 1572-1574.
- Salwinski, L., Miller, C. S., Smith, A. J., Pettit, F. K., Bowie, J. U. and Eisenberg, D. (2004). The database of interacting proteins: 2004 update. Nucleic Acids Res 32(Database issue): D449-451.
- Tamura, K., Stecher, G., Peterson, D., Filipski, A. and Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12): 2725-2729.
- UniProt, C. (2015). UniProt: a hub for protein information. Nucleic Acids Res 43(Database issue): D204-212.
- Wernersson, R. and Pedersen, A. G. (2003). RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res 31(13): 3537-3539.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rümpler, F., Theißen, G. and Melzer, R. (2015). Character-State Reconstruction to Infer Ancestral Protein-Protein Interaction Patterns. Bio-protocol 5(16): e1566. DOI: 10.21769/BioProtoc.1566.
Category
Systems Biology > Interactome > Protein-protein interaction
Systems Biology > Proteomics > Whole organism
Systems Biology > Genomics > Phylogenetics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.