- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Assay of the Glycosyltransferase Activity of a Heterologously Expressed Plant Protein
Published: Vol 4, Iss 21, Nov 5, 2014 DOI: 10.21769/BioProtoc.1285 Views: 12667
Reviewed by: Tie LiuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Glycosyltransferases are carbohydrate active enzymes containing catalytic modules involved in catalysing the biosynthesis of glycosidic bonds in oligo- and polysaccharides and glycoconjugates. One of the most comprehensive collections of Carbohydrate Active enZYmes is the CAZy database (http://www.cazy.org) comprising 120,000 glycosyltransferases allocated to 96 families based mainly on sequence homologies of their conserved and catalytically active domains (Cantarel et al., 2009). Interestingly, the glycosyltransferase activities of only about 1.6% of these proteins have been experimentally characterized (Lombard et al., 2014). In recent years, membrane-bound glycosyltransferases of a number of families have been shown to play a key role in the biosynthesis of plant cell-wall polysaccharides (Doblin et al., 2010; Scheller and Ulvskov, 2010; Driouich et al., 2012). They catalyze the transfer of glycosyl residues from donor nucleotide sugars to acceptors, forming the glycosidic bonds between adjacent glycosyl residues. Family 34 contains glycosyltransferases that have been shown to be involved in the biosynthesis of xyloglucans and transfer xylosyl residues to (1→4)-β-glucan chains (Keegstra and Cavalier, 2011). Our previous work suggests that Pinus radiata protein PrGT34B is a xyloglucan (1→6)-α-xylosyltransferase (Ade et al., 2014). Here, we describe a procedure for determining the xylosyltransferase activity of PrGT34B in vitro. We measured the transfer of xylose from the donor substrate UDP-xylose to different cello-oligosaccharide acceptor substrates under controlled reaction conditions. The assays include quantification of radioactively labeled reaction products and their identification by mass spectrometry. We also describe the purification, identification and quantification of the heterologously expressed recombinant protein PrGT34B in preparation for its use in the assays. This procedure may be applied to a wide range of glycosyltransferases in many different plant species.
Materials and Reagents
- Spodoptera frugiperda cells (Sf9) (Life Technologies, InvitrogenTM, catalog number: 11496-015 )
- Vector pFastBacHTaTM (Life Technologies, InvitrogenTM, catalog number: 10584-027 )
- Escherichia coli (E. coli) strains DH10Bac (Life Technologies, InvitrogenTM, catalog number: 10359-016 )
- 20% ethanol
- SDS
- β-mercaptoethanol
- Glycerol
- Bromophenol blue
- HEPES-NaOH (pH 7.0)
- MgCl2
- MnCl2
- Triton X-100
- Benzonase® nuclease (Novagen, catalog number: 70746 )
- Protease inhibitors (complete EDTA free, protease inhibitor cocktail tablet) (Roche Diagnostics, catalog number: 0 4693159001 )
- Imidazole
- Cellotetraose, cellopentaose and cellohexaose (Associates of Cape Cod, Northstar BioProducts®, catalog numbers: 400402-1 , 400404-1 , 400406-1 )
- Cellobiose (Sigma-Aldrich, catalog number: C7252 )
- UDP-D-xylose (CarboSource Services, Complex Carbohydrate Research Center, product code: UDP-Xylose )
- UDP-[14C-U]xylose (151.8 mCi/mmol) (PerkinElmer, catalog number: NEC543005UC )
- TALON metal affinity resin (Takara Bio Company, Clontech, catalog number: 635501 )
- BenchMark His-tagged protein standard (Life Technologies, InvitrogenTM, catalog number: LC5606 )
- Precision Plus protein unstained standard (Bio-Rad Laboratories, catalog number: 161-0363 )
- Mouse anti-His6 monoclonal antibody (GE Healthcare, product code: 27-4710-01 )
- Horseradish peroxidase (HRP) conjugated secondary sheep anti-mouse antibody (GE Healthcare, product code: NA931-100UL )
- SYPRO® Ruby protein gel stain (Life Technologies, Molecular Probes®, catalog number: S-21900 )
- Ion exchange resin Dowex 1x8-200 (chloride form) (Sigma-Aldrich, catalog number: 217425 )
- 2,5-Dihydroxybenzoic acid (Sigma-Aldrich, catalog number: 85707 )
- Starscint high flash-point LSC-cocktail (PerkinElmer)
- PAGE loading dye (see Recipes)
- Hypotonic lysis buffer (see Recipes)
- Equilibration/wash buffer for IMAC (see Recipes)
- Sample preparation buffer for IMAC (see Recipes)
- IMAC elution buffer (see Recipes)
- Reaction buffer for enzymatic assays (with radioactively labeled UDP-xylose) (see Recipes)
- Reaction buffer for enzymatic assays (without radioactively labeled UDP-xylose) (see Recipes)
Equipment
- Empty gravity-flow chromatography columns (20 ml volume) (Bio-Rad Laboratories, catalog number: 732-1010 )
- Micro Bio-Spin chromatography columns (Bio-Rad Laboratories, catalog number: 732-6204 )
- Amersham ECL detection system (GE Healthcare, product code: RPN2105 )
- PVDF transfer membrane (Amersham Hybond-P) (GE Healthcare, product code: 10600023 )
- NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific)
- LAS-3000 Lite imager with software LAS-3000 Image Reader (Fujifilm Corporation)
- FLA 2000 phosphorimager with software FLA 2000G (Fujifilm Corporation)
- Rackbeta LKB Wallac (PerkinElmer, model: 1209 )
- Voyager DETM PRO MALDI-TOF Workstation (Life Technologies, Applied Biosystems®)
Note: We believe that the reproducibility of the experiments does not depend on the precise equipment stated above but can be achieved using any other equivalent instrumentation.
Software
- Software Multi Gauge v2.2 (Fujifilm Corporation)
- Data Explorer (version 4.0.0.0) (Life Technologies, Applied Biosystems®)
- Software FLA 2000G (Fujifilm Corporation)
- Software LAS-3000 Image Reader (Fujifilm Corporation)
Procedure
- Heterologous expression of the glycosyltransferase
Note: There are a vast number of protocols describing procedures for the heterologous expression of proteins. Successful expression of plant glycosyltransferase proteins for enzymatic analyses has been achieved in bacteria, yeast and various insect cell systems. We do not give a detailed description of the heterologous expression of protein PrGT34B, but only outline the procedure we used and give some information that may be helpful.- In our study (Ade et al., 2014), we expressed the xylosyltransferase PrGT34B in Spodoptera frugiperda cells (Sf9) using an Autographa californica nuclear polyhydrosis virus based baculovirus expression vector system.
- The cell line Sf9 had previously been used to successfully express plant glycosyltransferases (Egelund et al., 2006). Furthermore, Sf9 cells are highly susceptible to infection with Autographa californica nuclear polyhydrosis virus and the corresponding baculovirus expression vector was shown to result in high expression levels of the desired gene product in insect cells (Condreay and Kost, 2007).
- The sequence of interest was cloned into vector pFastBacHTaTM, recombinant bacmid DNA was generated in E. coli strains DH10Bac or DH10MultiBac (Berger et al., 2004) and baculovirus was produced and amplified in Sf9 cells following the instructions in the manual “Bac-to-Bac Baculovirus Expression System”.
- Sf9 suspension cultures (200 ml), with a starting concentration of 2.0 x 106 cells ml-1, were inoculated with baculovirus (1:1,000 v/v of amplified virus stock to Sf9 cell suspension) and incubated with shaking (130 rpm, 72 h, 28 °C). Infected cells were harvested by centrifugation (500 x g, 10 min, 4 °C), and the cell pellets used for protein extraction (see below for details).
- The cell line Sf9 had previously been used to successfully express plant glycosyltransferases (Egelund et al., 2006). Furthermore, Sf9 cells are highly susceptible to infection with Autographa californica nuclear polyhydrosis virus and the corresponding baculovirus expression vector was shown to result in high expression levels of the desired gene product in insect cells (Condreay and Kost, 2007).
- For easy purification of the recombinant protein prior to its use in enzymatic assays, we strongly suggest that histidine tagged (6x histidine) fusion protein is expressed.
- In addition, many plant GTs, including xylosyltransferase PrGT34B, are type-II membrane bound proteins with a single transmembrane domain (TMD) close to the N-terminal end of the peptide, which acts as a signal anchor for the protein’s localization to the Golgi apparatus via the ER. For such proteins, we suggest expressing an N-terminus truncated version of the protein, lacking the N-terminal TMD but containing the complete functional domain. This may result in better solubility of the heterologously expressed protein and hence an easier extractability from the host cells (see below for details).
- If the His6-tag is to be fused to the protein’s N-terminus, the position of the N-terminal truncation of the protein has to be chosen carefully. We truncated the GT upstream of the most prominent stretch of charged amino acids between the TMD and the conserved domain to allow for an optimal presentation of the histidine-tag [please refer to Ade et al. (2014) for details on the exact positions of protein truncation and His6 tagging].
- In our study (Ade et al., 2014), we expressed the xylosyltransferase PrGT34B in Spodoptera frugiperda cells (Sf9) using an Autographa californica nuclear polyhydrosis virus based baculovirus expression vector system.
- Extraction of functional heterologous protein from Sf9 insect cells
- Extract native recombinant protein PrGT34B from 8 x 108 freshly pelleted Sf9 cells using ice-cold hypotonic lysis buffer (20 ml). Lyse the cells on ice (40 min) and then mix the lysate on an orbital shaker (50 rpm, 2 h, 4 °C).
- Clarify the lysate by centrifugation (14,000 x g, 5 min, 4 °C) and transfer the supernatant containing the Triton X-100 solubilized protein to a fresh tube.
- Proceed to the purification of the His6-tagged recombinant protein PrGT34B from the total extracted protein as soon as possible.
Note: We kept the protein extract on ice for no longer than 2 h before proceeding to protein purification to avoid loss of functional protein by protein degradation, protein precipitation or protein denaturation. - In addition, extract total protein from 8 x 108 freshly pelleted Sf9 cells infected with non-recombinant baculovirus or with baculovirus recombinant for empty vector pFastBacHTaTM for negative control reactions required to calculate background GT activities (see below).
- Extract native recombinant protein PrGT34B from 8 x 108 freshly pelleted Sf9 cells using ice-cold hypotonic lysis buffer (20 ml). Lyse the cells on ice (40 min) and then mix the lysate on an orbital shaker (50 rpm, 2 h, 4 °C).
- Purification of the His6-tagged recombinant GT using immobilized metal affinity chromatography (IMAC)
Note: The TALON resin consists of Sepharose beads with a tetradentate metal chelator that holds Co2+ ions in electronegative pockets. At neutral pH, the histidine side groups of the target protein’s polyhistidine tags are unprotonated and bind to the chelated cobalt ions. Under these conditions, increasing imidazole concentrations in the elution buffer during chromatography competitively elutes bound polyhistidine-tagged protein from the resin. Purification of the GT is required to precisely determine the quantity of specific protein to be used in the enzymatic assay. Only in this way can the specific enzymatic activity of the GT be calculated.- Load an empty gravity-flow chromatography column with 2 ml of a 50% (w/v) TALON metal affinity resin slurry in 20% ethanol at room temperature.
- After the resin has settled, apply a further frit and wash the column with double distilled water (ddH2O) (20 ml) at room temperature.
- Equilibrate the TALON resin with equilibration/wash buffer (10 ml) at room temperature.
- Mix the total protein lysate (20 ml) (see above) with ice-cold sample preparation buffer (5 ml).
- Load the ice-cold 25 ml of soluble protein lysate, adjusted to 50 mM HEPES-NaOH (pH 7.0), 100 mM NaCl, 5 mM imidazole, 10% glycerol, 1.6% Triton X-100, 1.6 mM MgCl2, on to the column.
Note: The increased ionic strength of the sample adjusted to 100 mM NaCl minimizes non-specific binding to the resin and stabilizes the soluble protein on the column during fractionation. The initial imidazole concentration of 5 mM increases the stringency for the affinity binding and decreases background protein binding. - Wash the TALON resin with equilibration/wash buffer (20 ml) and then elute His6-tagged protein from the column with IMAC elution buffer (15 ml) at room temperature.
- Capture successive fractions (1 ml) of the eluted protein and directly store on ice. Analyze aliquots (10 µl) by standard SDS-PAGE to determine the fraction with the highest concentration and purity of the recombinant protein to be used in subsequent enzymatic assays.
- Keep eluted soluble protein at 4 °C for only a short time before determining its enzymatic activity.
Note: We kept the purified protein on ice for no longer than 24 h before using it in GT assays to avoid loss of functional protein by protein degradation, protein precipitation or protein denaturation.
- Load an empty gravity-flow chromatography column with 2 ml of a 50% (w/v) TALON metal affinity resin slurry in 20% ethanol at room temperature.
- Identification of heterologously expressed recombinant protein PrGT34B by immunoblot analysis and peptide mass fingerprinting (PMF)
Note: Verification of the recombinant protein’s identity was achieved by a combination of immunoblot analysis of the His6-tagged protein and a mass spectrometric peptide analysis.- Denature an aliquot (10 µl) of the total protein extract (the total protein extracted from 5.0 x 105 Sf9 cells) and of the IMAC purified and eluted protein by heating in PAGE loading dye (5 min at 95 °C).
- Separate the denatured protein on a 12% polyacrylamide gel together with a suitable marker such as the BenchMark His-tagged protein standard.
- Transfer the protein to a PVDF transfer membrane using any kind of suitable electrophoretic protein transfer method.
- Detect the His6-tagged protein with a mouse anti-His6 monoclonal antibody (3,000-fold diluted) in combination with a horseradish peroxidase (HRP) conjugated secondary sheep anti-mouse antibody (7,000-fold diluted) using any kind of suitable hybridization protocol.
- Visualize chemiluminescent signals on the membrane using the Amersham ECL detection system and any kind of suitable analysis system such as the LAS-3000 Lite with the software LAS-3000 Image Reader.
- Optional: In addition to identification of the His6-tagged protein in the total protein extract and the IMAC eluted fraction by immunoblot analysis, separate a second aliquot of the IMAC purified heterologously expressed protein by standard SDS-PAGE. Stain the polyacrylamide gel with Coomassie blue, excise the relevant protein band and subject to PMF, using any kind of suitable commercial provider or academic proteomics facility.
Note: We recommend the analysis of the purified protein once by PMF to unambiguously verify its identity.
- Denature an aliquot (10 µl) of the total protein extract (the total protein extracted from 5.0 x 105 Sf9 cells) and of the IMAC purified and eluted protein by heating in PAGE loading dye (5 min at 95 °C).
- On-gel quantification of the purified protein PrGT34B
Note: The purity of the His6-tagged recombinant protein after IMAC purification is not 100%. Consequently, accurate quantification of one specific protein in a protein mixture is not possible by spectrophotometric methods such as the NanoDrop ND-1000 and requires alternative quantification methods.- Load equal volumes (10 µl) of different dilutions (e.g. undiluted, 2, 5, 10 and 20-fold diluted) of the IMAC purified heterologously expressed protein together with equal volumes (20 µl) of different dilutions of a quantitative protein mass ladder such as the Precision Plus protein unstained standard on a 12% polyacrylamide gel.
- Separate the protein by any kind of standard SDS-PAGE protocol.
- Stain the gel with the quantitative luminescent SYPRO Ruby protein gel stain following the manufacturer’s instructions.
- Visualize the fluorescence signals with any kind of suitable analysis system such as the FLA 2000 phosphorimager with FLA 2000G software.
Note: The emission maximum of the SYPRO Ruby protein stain is 610 nm. - Quantify the protein bands of the previously identified specific protein species on the gel based on a calibration curve obtained from the known quantities of protein in the reference bands of the various dilutions of the quantitative protein ladder using the software Multi Gauge v2.2.
Note: The 100, 50 and 20 kDa bands of undiluted Precision Plus protein standard (20 µl) correspond to 300, 1,500 and 300 ng of protein, respectively.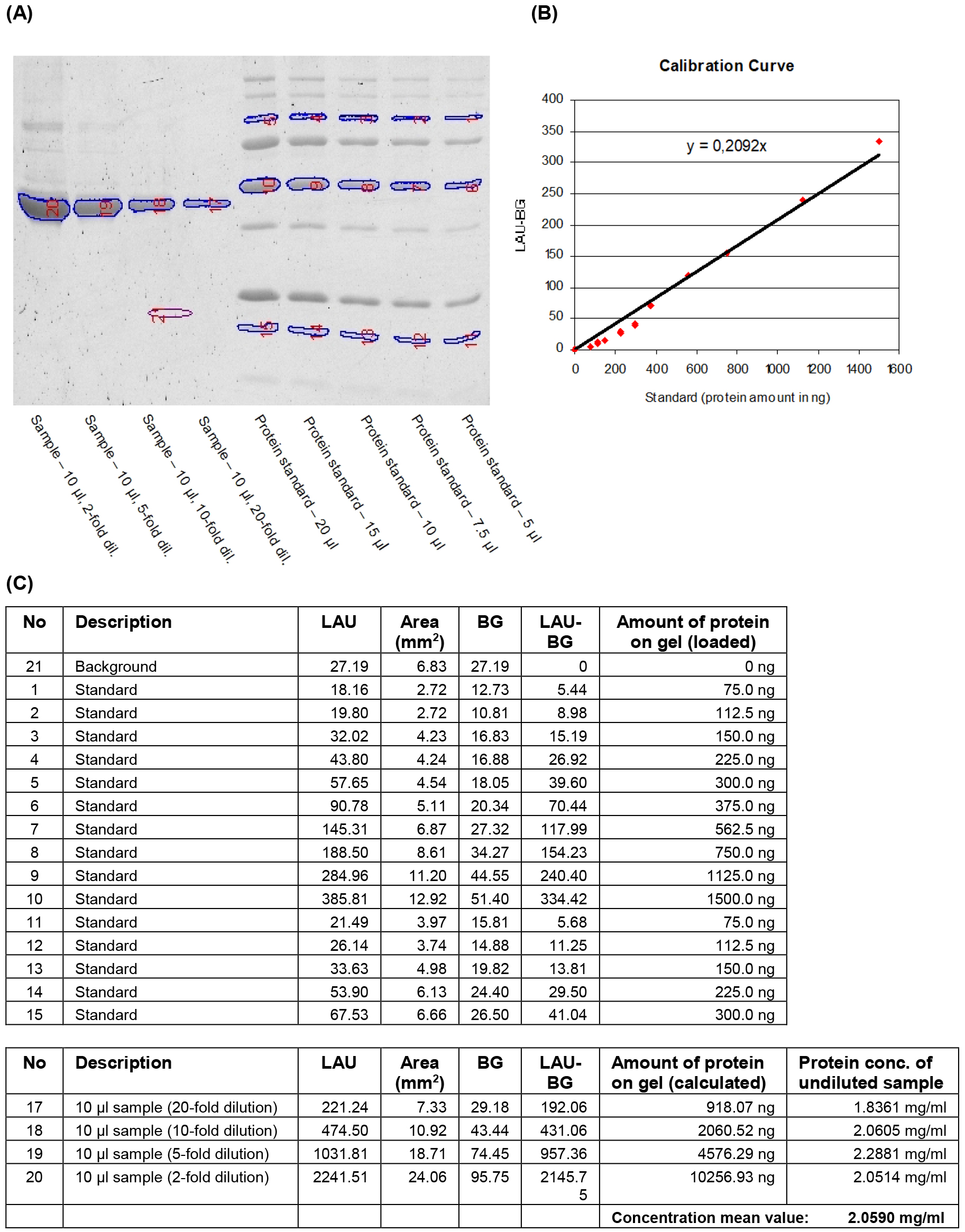
Figure 1. Quantification of IMAC purified heterologously expressed protein by fluorometric analysis of a SYPRO Ruby stained SDS-gel. Aliquots (10 μl) of four dilutions of the purified recombinant protein and five different volumes of the protein mass standard Precision Plus protein standard were separated on a 12% acrylamide gel. The gel was stained with SYPRO Ruby protein gel stain (Molecular Probes) and the fluorescent signals were visualised (A). Reference bands and bands to be quantified were manually selected and the signal intensities of the selected areas were detected as linear arbitrary units (LAU). The known protein quantities for each selected reference band and their background-corrected signal intensity values (LAU-BG) were used to generate a calibration curve (B). Protein amounts in the selected sample bands were calculated using the equation of the calibration curve. The final concentration of recombinant protein in the sample was determined by taking loaded volumes and dilution factors into account (C).
- Load equal volumes (10 µl) of different dilutions (e.g. undiluted, 2, 5, 10 and 20-fold diluted) of the IMAC purified heterologously expressed protein together with equal volumes (20 µl) of different dilutions of a quantitative protein mass ladder such as the Precision Plus protein unstained standard on a 12% polyacrylamide gel.
- Quantitative in vitro glycosyltransferase assay for the Pinus radiata xylosyltransferase PrGT34B
General information:- This protocol describes how to determine the in vitro GT activity of the xyloglucan xylosyltransferase PrGT34B by measuring, under constant reaction conditions, the transfer of the monosaccharide moiety from a nucleotide sugar such as UDP-xylose, used as the donor substrate, to an acceptor substrate such as a cello-oligosaccharide.
- An assay temperature of 25 °C and a pH of 6.5-7.0 were found to be optimal in a study of the activities of other GTs located in the Golgi apparatus in P. radiata (Mast et al., 2009). Furthermore, recent literature suggests that GT activities in the Golgi apparatus depend on a slightly acidic environment (Reyes and Orellana, 2008). However, successful enzymatic in vitro assays with enzymes closely related to the protein PrGT34B were done under neutral or slightly alkaline conditions in previous studies (Edwards et al., 1999; Faik et al., 2002; Cavalier and Keegstra, 2006). Consequently, we used a neutral pH for all assays described in the present protocol.
- For all GT activity assays described in this protocol, the protein PrGT34B was either in total Sf9 protein extract, adjusted to a total protein concentration of 10 mg/ml with hypotonic lysis buffer (see above), or in protein extract purified by IMAC and adjusted to a total protein concentration of 2.5 mg/ml with IMAC elution buffer (see above). In addition, dilutions of the extract purified by IMAC were prepared with IMAC elution buffer (see above) containing specific PrGT34B concentrations of 2,000, 100 and 40 µg/ml.
- Total protein extract contains HEPES-NaOH (pH 7.0) (50 mM), MgCl2 (2 mM), glycerol (10%) and Triton X-100 (2%) and the IMAC purified protein extract contains HEPES-NaOH (pH 7.0) (50 mM), NaCl (100 mM), imidazole (50 mM) and glycerol (10%).
Note: Xyloglucan xylosyltransferase activity of the heterologously expressed protein PrGT34B was assayed with radioactively labeled UDP-[14C-U]xylose and cellobiose, cellotetraose, cellopentaose or cellohexaose.
- Mix protein solution (total protein extract or IMAC-purified extract) (10 µl) with reaction buffer (40 µl), containing UDP-xylose, UDP-[14C]xylose and cello-oligosaccharide as acceptor substrate.
- Incubate (25 °C for e.g. 1, 17 or 44 h) the resulting reaction mixture (50 µl) containing the extracted protein in HEPES-NaOH (pH 7.0) (50 mM), MnCl2 (5 mM), MgCl2 (5 mM), UDP-xylose (1 or 4 mM), UDP-[14C]xylose (9 kBq, 54,300 dpm, 150 pmol) and cello-oligosaccharides (1 mM).
Notes:- These reaction conditions correspond to 50 nmol acceptor substrate and 50 or 200 nmol unlabelled UDP-sugar donor substrate in the reaction mixture, resulting in molar ratios of acceptor and donor of 1:1 or 1:4. The amount of 150 pmol UDP-[14C] xylose added to the reaction mixture corresponds to a concentration in the reaction mixture of only 3 µM, changing the total concentration of added UDP-xylose only insignificantly.
- The assay can be done with any other molar ratio between acceptor and donor substrate. Other GT activities can be assayed using appropriate nucleotide sugars and donor substrates in the assay mixture such as UDP-galactose and UDP-[14C]galactose as donor substrates and manno-oligosaccharides as acceptor substrates to assay the galactosyltransferase activity of galactomannan galactosyltransferases.
- These reaction conditions correspond to 50 nmol acceptor substrate and 50 or 200 nmol unlabelled UDP-sugar donor substrate in the reaction mixture, resulting in molar ratios of acceptor and donor of 1:1 or 1:4. The amount of 150 pmol UDP-[14C] xylose added to the reaction mixture corresponds to a concentration in the reaction mixture of only 3 µM, changing the total concentration of added UDP-xylose only insignificantly.
- Stop the reaction by heating (5 min, 95 °C), remove precipitated protein by centrifugation (10,000 x g, 2 min, 4 °C) and transfer the supernatant with the reaction products to a fresh tube and place on ice.
- Remove unincorporated nucleotide sugars (UDP-xylose and UDP-[14C]xylose) from the reaction mixture by anion exchange chromatography (AEC) using the ion exchange resin Dowex 1x8-200 (chloride form).
Note: The positively charged groups of the resin act as strong anion exchangers that stay charged over a wide pH range and bind negatively charged molecules such as nucleotide sugars in exchange for the counter ion chloride. - Mix Dowex 1 x 8-200 resin (1 g) with water (10 ml), allow the resin to swell over-night, wash the resin twice with fresh water (10 ml), allow the resin to settle (10 min), discard the supernatant and add fresh water to give a 50% (w/v) resin-water slurry.
- Directly mix the cleared supernatant from the assay reaction (50 µl) with the 50% (w/v) resin-water slurry (400 µl) and incubate on an orbital shaker (50 rpm, 5 min, at room temperature).
- Transfer the mixture to an empty Micro Bio-Spin chromatography column fitted with a frit to retain the resin and centrifuge (700 x g, 1 min, at room-temperature).
Note: The effluent may be collected in a fresh microcentrifuge tube. The Micro Bio-Spin column with the empty microcentrifuge tube may be placed into a 15 ml Falcon tube with lid, so the centrifugation can be done without the risk of contaminating the centrifuge. - Mix the complete effluent (250-300 µl), containing the uncharged reaction products with the incorporated radioactivity, with scintillation counting fluid (Starscint high flash-point LSC-cocktail) (1 ml).
- Measure the incorporated radioactivity with a liquid scintillation counter for 5 min.
Note: Conversion of measured “counts per minute” (cpm) into “disintegrations per minute” (dpm) is done automatically by the instrument using a calibration curve that is generated by plotting detected cpm values for external standards of known activity against their known spectral quench parameters [SQP(E)]. - Prepare control incubations for enzymatic assays identical to those described above but without protein and with protein extracts from Sf9 cells infected with non-recombinant virus to determine background radioactivity resulting from incomplete retention of unincorporated UDP-[14C]sugar during anion exchange chromatography, enzyme independent degradation of UDP-sugar and UDP-sugar hydrolyzing background activity. Treat these control incubations identically to the samples and measure the remaining unspecific background radioactivity in the AEC effluent.
- Calculate the xylosyltransferase activity as follows.
Incorporation of xylose into reaction products is calculated from the total radioactivity retained in the sample according to the formula “amount of incorporated xylose into acceptor substrate per assay (in pmol) = (A/B)*C”; with A representing the radioactivity retained in the sample following anion exchange chromatography (in dpm), B representing the total radioactivity added to the assay (in dpm) and C representing the total amount of UDP-sugar (labeled + unlabeled) added to the assay (in pmol).- GT activity can be shown as xylose incorporation per assay (in pmol/assay) or as xylose incorporation per time (in pmol/h) if the assays are incubated for a discrete time (e.g. 1, 17 and 44 h).
- For assays containing a known quantity of the heterologous protein PrGT34B, specific GT activity can be determined from the quantity (in pmol) of xylose incorporated into acceptor substrates per time and per quantity of specific enzyme (in pmol/h/µg).
- Background radioactivity detected in control assays (in dpm) may be used to calculate the unspecific activity in a sample or may be directly subtracted from the corresponding experimental sample to provide a background-corrected value for the calculation of enzymatic activity.
- GT activity can be shown as xylose incorporation per assay (in pmol/assay) or as xylose incorporation per time (in pmol/h) if the assays are incubated for a discrete time (e.g. 1, 17 and 44 h).
- This protocol describes how to determine the in vitro GT activity of the xyloglucan xylosyltransferase PrGT34B by measuring, under constant reaction conditions, the transfer of the monosaccharide moiety from a nucleotide sugar such as UDP-xylose, used as the donor substrate, to an acceptor substrate such as a cello-oligosaccharide.
- Identification and characterization of PrGT34B xylosyltransferase assay reaction products by MALDI-TOF MS
Notes:- Most mass spectrometric methods are not quantitative unless an internal standard is used that is in known amounts and is structurally identical to the reaction product. Consequently, the use of matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) described here can only be seen as a semi-quantitative method to determine GT activity. However, this procedure not only shows GT activity by detecting the expected reaction products but also allows the structural characterization of all reaction products generated by the enzyme PrGT34B.
- MALDI-TOF MS has been proven highly efficient for desorption and ionization of various biomolecules including oligosaccharides (Garozzo et al., 1995; Garozzo et al., 1997).
- Prepare the reaction mixture with extract containing the recombinant protein (or without recombinant protein for control reactions) exactly as described above (step 6F) but without adding radioactively labeled UDP-xylose.
- Incubate (25 °C for e.g. 17 h) the reaction mixture (50 µl), containing the extracted protein in HEPES-NaOH (pH 7.0) (50 mM), MnCl2 (5 mM), MgCl2 (5 mM), UDP-xylose (1 or 4 mM), and cello-oligosaccharides (1 mM).
Note: The incubation time was chosen sufficiently long to allow for maximum incorporation of xylose. - Stop the reaction by heating (5 min, 95 °C), remove precipitated protein by centrifugation (10,000 x g, 2 min, 4 °C) and transfer the supernatant with the reaction products to a fresh tube and place on ice.
- Mix an aliquot of the supernatant (1 µl) with the MALDI matrix 2,5-dihydroxybenzoic acid (10 mg/ml in ddH2O) (9 µl).
- Spot an aliquot of the sample-matrix mixture (1 µl) on a MALDI plate and air dry to allow crystallization of analyte and matrix molecules on the plate.
Note: It is important to minimize the glycerol concentration in the mixture to make sure the molecules crystallize uniformly on the plate. - Load the MALDI plate to a MALDI-TOF mass spectrometer such as the Voyager DETM PRO MALDI-TOF workstation.
Notes:- The spectrometer was calibrated using the three external standards des-ArgI-bradykinin (m/z = 904), angiotensin I (m/z = 1296) and [GluI]-fibrinopeptide B (m/z = 1570) diluted in 2,5-dihydroxybenzoic acid.
- Operate the spectrometer in the reflectron mode at an accelerating voltage of 20 kV with a delay time of 200 ns.
- The spectrometer was calibrated using the three external standards des-ArgI-bradykinin (m/z = 904), angiotensin I (m/z = 1296) and [GluI]-fibrinopeptide B (m/z = 1570) diluted in 2,5-dihydroxybenzoic acid.
- Compile mass spectra for each sample by plotting the mass (m) to charge (z) ratio (x-axis) of all detected ion species against their measured intensities (y-axis) using any suitable software such as the Data Explorer (version 4.0.0.0).
Note: Compilation of each mass spectrum should consist of the accumulated data from sufficient numbers of laser shots (e.g. 100 laser shots). - Identify substrates and GT reaction products in the mass spectrum based on the molecular weights of their singly charged (z = 1) sodium adduct ions [M+Na]+.
Notes:- Because of the high NaCl concentration in the sample-matrix mixture (> 1 mM), the dominant ion species in positive ionization mode are the sodium adducts of the protonated molecular ions of the analyte.
- Use the mass spectra generated for control reactions done without enzyme to identify peaks that correlate to reaction products of the recombinant protein PrGT34B.
- Choose a suitable base peak intensity threshold to identify peaks that are above the noise in the mass spectrum.
- Because of the high NaCl concentration in the sample-matrix mixture (> 1 mM), the dominant ion species in positive ionization mode are the sodium adducts of the protonated molecular ions of the analyte.
- Most mass spectrometric methods are not quantitative unless an internal standard is used that is in known amounts and is structurally identical to the reaction product. Consequently, the use of matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) described here can only be seen as a semi-quantitative method to determine GT activity. However, this procedure not only shows GT activity by detecting the expected reaction products but also allows the structural characterization of all reaction products generated by the enzyme PrGT34B.
Recipes
- PAGE loading dye
SDS (1%)
β-mercaptoethanol (1 mM)
Glycerol (10%)
Bromophenol blue - Hypotonic lysis buffer
50 mM HEPES-NaOH (pH 7.0)
2 mM MgCl2
10% glycerol (v/v)
2% Triton X-100 (v/v)
Benzonase® nuclease (10 U/ml)
Protease inhibitors
Prepare in double distilled water (ddH2O) - Equilibration/wash buffer for IMAC
50 mM HEPES-NaOH (pH 7.0)
100 mM NaCl
5 mM imidazole
10 % glycerol (v/v)
Prepare in ddH2O - Sample preparation buffer for IMAC
50 mM HEPES-NaOH (pH 7.0)
500 mM NaCl
25 mM imidazole
10% glycerol (v/v)
Prepare in ddH2O - IMAC elution buffer
50 mM HEPES-NaOH (pH 7.0)
100 mM NaCl
50 mM imidazole
10% glycerol (v/v)
Prepare in ddH2O - Reaction buffer for enzymatic assays (with radioactively labeled UDP-xylose)
50 mM HEPES-NaOH (pH 7.0)
6.25 mM MnCl2
6.25 mM MgCl2
1.25 or 5 mM UDP-xylose
1.25 mM oligosaccharide acceptor substrate (cellobiose, cellotetraose, cellopentaose or cellohexaose)
UDP-[14C]xylose (9 kBq, 54,300 dpm, 150 pmol)
Prepare in ddH2O - Reaction buffer for enzymatic assays (without radioactively labeled UDP-xylose)
50 mM HEPES-NaOH (pH 7.0)
6.25 mM MnCl2
6.25 mM MgCl2
1.25 or 5 mM UDP-xylose
1.25 mM oligosaccharide acceptor substrate (cellobiose, cellotetraose, cellopentaose or cellohexaose)
Prepare in ddH2O
Acknowledgments
This work was supported by funding from the New Zealand Foundation for Research, Science and Technology, and the University of Auckland. We thank CarboSource Services (Complex Carbohydrate Research Center, Athens, GA) for the UDP-xylose and Scion Research for a doctoral stipend for Carsten P. Ade. Parts of the procedure described here were modified from Cavalier and Keegstra (2006).
References
- Ade, C. P., Bemm, F., Dickson, J. M., Walter, C. and Harris, P. J. (2014). Family 34 glycosyltransferase (GT34) genes and proteins in Pinus radiata (radiata pine) and Pinus taeda (loblolly pine). Plant J 78(2): 305-318.
- Berger, I., Fitzgerald, D. J. and Richmond, T. J. (2004). Baculovirus expression system for heterologous multiprotein complexes. Nat Biotechnol 22(12): 1583-1587.
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. and Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37(Database issue): D233-238.
- Cavalier, D. M. and Keegstra, K. (2006). Two xyloglucan xylosyltransferases catalyze the addition of multiple xylosyl residues to cellohexaose. J Biol Chem 281(45): 34197-34207.
- Condreay, J. P. and Kost, T. A. (2007). Baculovirus expression vectors for insect and mammalian cells. Curr Drug Targets 8(10): 1126-1131.
- Doblin, M. S., Pettolino, F. and Bacic, A. (2010). Evans Review: Plant cell walls: the skeleton of the plant world. Functional Plant Biology 37(5): 357-381.
- Driouich, A., Follet-Gueye, M. L., Bernard, S., Kousar, S., Chevalier, L., Vicre-Gibouin, M. and Lerouxel, O. (2012). Golgi-mediated synthesis and secretion of matrix polysaccharides of the primary cell wall of higher plants. Front Plant Sci 3: 79.
- Edwards, M. E., Dickson, C. A., Chengappa, S., Sidebottom, C., Gidley, M. J. and Reid, J. S. (1999). Molecular characterisation of a membrane-bound galactosyltransferase of plant cell wall matrix polysaccharide biosynthesis. Plant J 19(6): 691-697.
- Egelund, J., Petersen, B. L., Motawia, M. S., Damager, I., Faik, A., Olsen, C. E., Ishii, T., Clausen, H., Ulvskov, P. and Geshi, N. (2006). Arabidopsis thaliana RGXT1 and RGXT2 encode Golgi-localized (1,3)-alpha-D-xylosyltransferases involved in the synthesis of pectic rhamnogalacturonan-II. Plant Cell 18(10): 2593-2607.
- Faik, A., Price, N. J., Raikhel, N. V. and Keegstra, K. (2002). An Arabidopsis gene encoding an alpha-xylosyltransferase involved in xyloglucan biosynthesis. Proc Natl Acad Sci U S A 99(11): 7797-7802.
- Garrozzo, D., Impallomeni, G., Spina, E., Sturiale, L. and Zanetti, F. (1995). Matrix‐assisted laser desorption/ionization mass spectrometry of polysaccharides. Rapid Commun Mass Spectrom 9(10): 937-941.
- Garozzo, D., Nasello, V., Spina, E. and Sturiale, L. (1997). Discrimination of isomeric oligosaccharides and sequencing of unknowns by post source decay matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 11(14): 1561-1566.
- Keegstra, K. and Cavalier, D. (2011). Glycosyltransferases of the GT34 and GT37 families. In Annual Plant Reviews, Volume 41. In: Ulvskov, P., (ed). Plant Polysaccharides, Biosynthesis and Bioengineering Oxford. Blackwell Publishing, pp. 235-249.
- Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42(Database issue): D490-495.
- Mast, S. W., Donaldson, L., Torr, K., Phillips, L., Flint, H., West, M., Strabala, T. J. and Wagner, A. (2009). Exploring the ultrastructural localization and biosynthesis of beta(1,4)-galactan in Pinus radiata compression wood. Plant Physiol 150(2): 573-583.
- Scheller, H. V. and Ulvskov, P. (2010). Hemicelluloses. Annu Rev Plant Biol 61: 263-289.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ade, C. P., Bemm, F., Dickson, J. M. J., Walter, C. and Harris, P. J. (2014). In vitro Assay of the Glycosyltransferase Activity of a Heterologously Expressed Plant Protein. Bio-protocol 4(21): e1285. DOI: 10.21769/BioProtoc.1285.
Category
Plant Science > Plant biochemistry > Carbohydrate
Plant Science > Plant biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.