- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CAMP-Membrane Interactions Using Fluorescence Spectroscopy
Published: Vol 3, Iss 15, Aug 5, 2013 DOI: 10.21769/BioProtoc.846 Views: 8860
Reviewed by: Fanglian He
Original research article
The authors used this protocol in:
Sep 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The molecular mechanism by which peptide antibiotics (also referred as cationic antimicrobial peptides-CAMPs) penetrate through the bacterial wall barrier, interact with, and disrupt their membrane is complex. It depends mainly on the peptide properties (structure, length, charge and hydrophobicity), on the characteristics of the cell wall matrix and the membrane itself.
Here, we present two fluorescence spectroscopic techniques, one for tracking the interaction of CAMPs with membranes, and the other for evaluating the ability of a peptide to cross the bacterial cell-wall and reach the membrane. The fluorescence approach is relatively simple, highly sensitive, non-invasive and allows time-scale investigation. It can be applied to lipid vesicles or intact bacteria. For membrane model systems such as liposomes, it allows to determine the binding kinetics of a peptide to vesicle and to assess the depth of penetration. By using bacterial strains carrying different mutations in their cell wall components, but not in their membrane, we can investigate how a specific element may affect the cell wall permeability to CAMPs (Saar-Dover et al., 2012).
In order to track the peptide-membrane interaction we conjugate a lipid environmentally sensitive NBD (7-nitrobenz-2-oxa-1, 3-diazole-4-yl) fluorophore to peptides. NBD fluorescence can increase up to approximately 10-fold upon interaction with membranes. Its high excitation wavelength (467 nm) and the high quantum yield reduce significantly the contribution of light scattering. NBD-labeled peptides exhibit fluorescence emission maxima around 540 nm in hydrophilic solution (Shai, 1999). However, upon interaction with lipid component such as the bacterial membrane, relocation of the NBD group into a more hydrophobic environment results in an increase in its fluorescence intensity and a blue shift of the emission maxima (Chattopadhyay and London, 1987). The first property is used to determine the binding constant of the peptide to the membrane. The second property is exploited to evaluate the depth of penetration (Merklinger et al., 2012; Zhao and Kinnunen, 2002). Here, we will focus on how to determine the binding constant. The advantage of the NBD moiety conjugation is that it allows the use of experimental conditions in which the lipid: peptide molar ratio range from < 100:1 up to > 15,000:1. The addition of NBD does not change the biological function of most of the peptide, as was found for different antimicrobial peptides such as paradaxin (Rapaport and Shai, 1992), dermaseptins (Pouny et al., 1992), cecropins (Gazit et al., 1994) and cathelicidin LL-37 (Oren et al., 1999). However, pre-examination must be done for each newly investigated peptide.
Materials and Reagents
- Peptides were synthesized by an Fmoc solid-phase method (Merrifield et al., 1982) on Rink amide-4-methylbenzhydrylamine hydrochloride salt (MBHA) resin. Fluorescent labeling with 4-chloro-7-nitrobenz-2-oxa-1, 3-diazole fluoride (NBD-F) or 5-(and-6)-carboxytetra-methylrhodamine succinimidyl ester (Rhodamine) was followed by peptide cleavage from the resin and purification by reverse phase high-performance liquid chromatography (RP-HPLC). See detailed methods in Oren et al. (1999) and Avrahami et al. (2001).
- Liposome suspension stock at total lipid concentration of 12.5 mM. For liposome preparation see Kliger et al. (1997)
- Bacterial suspension (OD600 nm adjusted to 4)
- Fluorescently labeled peptides solution (see Note 1)
- Double Distilled water (DDW) or Milli-Q reagent grade water
- Phosphate buffered saline (PBS) (pH 7.4)
- 70% (v/v) ethanol
- 5 x 5 mm quartz cuvette
Equipment
- Automatic peptide synthesizer ABI 433A (Applied Biosystems)
- Reverse phase high-performance liquid chromatography (RP-HPLC) Agilent HPLC 1100 (Hewlett Packard)
- SLM-Aminco Bowman series 2-luminescence spectrophotometer FA-355 (SLM-Aminco)
Procedure
- Binding of NBD-labeled peptide to membranes
The binding constant of a peptide is calculated from a titration of lipid vesicles, either small-unilamellar vesicles (SUVs, 10-50 μm size) or large-unilamellar vesicles (LUVs, 50-100 μm size) into NBD-peptide solution. To achieve an accurate result, at least 20 points should be recorded for each curve. An accepted dilution factor of the peptide solution is up to 10% and therefore no more than 40 μl of vesicle solution should be added. All assays were performed in room temperature (22-24 °C).- Dissolve NBD-peptide of the requested solution (such as DDW or PBS) to a final concentration of 0.1 μM (see Note 2). Each measurement uses 400 μl of peptide solution, therefore prepare 810 μl for two repetitions.
- Set the spectrophotometer to spectrum mode with excitation wavelength of 467 nm and emission wavelength of 500-600 nm. In our device, slits are usually set to 5-10 nm. Using wider slits will improve sensitivity but can increase background noise and therefore should be individually determined for each peptide.
- Add 400 μl peptide solution to a pre-cleaned cuvette (magnetic stirrer can be used) and read the signal output. Measure again every few minutes until no change is detected. This is the basal signal of the labeled peptide in the absence of membrane compounds.
- Add 1 μl from the LUVs suspension stock to the cuvette to reach an initial peptide/lipid ratio of 1:312 and read again. Calculation of ratio: 12.5 mM lipid is 12,500 μM that are being diluted 1:400 in a solution containing 0.1 μM peptide. Therefore, when 1 μl are added the ratio is 1:312 (12,500/401*10). When another 1 μl will be added the ratio will be 1: 622 and so on.
- Re-measure the signal intensity every 1 min until no change in the signal is detected. This will indicate that binding has reached equilibrium.
- Repeat step 1-d successively until no change in the peak maxima (around 530 nm) can be detected.
- Clean the cuvette by washing it three times with 70% (v/v) ethanol. Trace ethanol in the cuvette should be removed by rinsing with DDW.
- To account for background, the emissions of the vesicles alone at the same wavelength should be monitored and subtracted. Therefore, repeat steps 1-c~f using the same solvents but without dissolving peptide in it.
- Dissolve NBD-peptide of the requested solution (such as DDW or PBS) to a final concentration of 0.1 μM (see Note 2). Each measurement uses 400 μl of peptide solution, therefore prepare 810 μl for two repetitions.
- Cell-wall permeability assay
The assay is designed to compare the ability of a given peptide to penetrate the cell wall of a given bacterial strain and interact with its membrane (Saar-Dover et al., 2012). The relative elevation in NBD emission should be calculated for each strain and compared.- Grow you bacteria to an exponential stage, concentrate cells from the culture by centrifuging 5 ml at 1,300 x g), 3 min. Wash and re-centrifuge pallet twice with PBS. Adjust your bacterial suspension to OD600 nm = 4 in PBS.
- Dissolve NBD-peptide in PBS solution to a final concentration of 0.1-1 μM (use concentration that does not disrupt the cellular integrity, this can be determined separately using a SYTOX green assay (Saar-Dover et al., 2012).
- Set the spectrophotometer to kinetic mode with excitation wavelength of 467 nm and emission wavelength of 530 nm. In our device, SLM-Aminco Bowman series 2-luminescence spectrophotometer, slits are usually set to 5-10 nm. Using wider slits will improve sensitivity but can increase background noise and therefore should be individually determined for each peptide.
- Add 400 μl peptide solution to a pre-cleaned cuvette (magnetic stirrer can be used) and read the signal output until it stabilizes. This is the basal signal of the labeled peptide in the absence of membrane compounds.
- Add 10 μl from the bacterial suspension to the cuvette. Track the change in signal intensity with time until equilibrium is reached.
- Clean the cuvette by washing it three times with 70% (v/v) ethanol. Trace ethanol in the cuvette should be removed by rinsing with DDW.
- To account for background, the emissions of bacteria alone at the same wavelength should be monitored and subtracted. Therefore, repeat steps 2-c~e using the same solvents but without dissolving peptide in it.
- The signal intensity can be affected by oligomerization of the labeled peptide over the bacterial surface and self-quenching (reduced intensity). We therefore assess the level of peptide oligomerization by repeating the experiment using Rhodamine labeled peptides. Rhodamine is highly sensitive to quenching but unlike NBD, its emission is not affected strongly by the polarity of its environment.
- Grow you bacteria to an exponential stage, concentrate cells from the culture by centrifuging 5 ml at 1,300 x g), 3 min. Wash and re-centrifuge pallet twice with PBS. Adjust your bacterial suspension to OD600 nm = 4 in PBS.
Calculations
For binding of NBD-labeled peptide to membranes:
- Prepare a table of titration results- emission (Y) versus lipid concentration (X).
- Subtract the baseline value (solution only) from each Y value to correct for background.
- You should get a saturation curve, meaning a non-linear curve (Figure 1). Use a non-linear equation program solver (such as GraphPad Prism) to extract the best fitted equation.
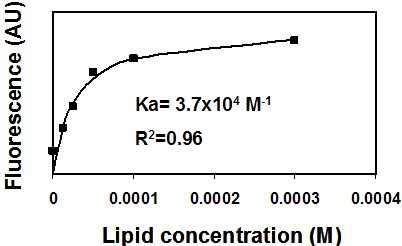
Figure 1. Binding of NBD-labeled peptide to membranes. A representative saturation curve describing an increase in NBD fluorescence upon titration of phosphatidylcholine: cholesterol (9:1) large unilamellar lipid vesicles (LUVs) into 0.2 μM NBD conjugated peptide (NBD-gp41 TMD). Nonlinear least-squares analysis was used to determine the affinity constant (Ka).
- Calculate the Ka (association constant). You can also determine the peptide: lipid ratio at saturation.
- Alternatively, a less preferable but still applicable way will be to calculate the slope from the linear part of you curve, as long as there are at least 10 points in that region.
- See more details in Rosenfeld et al. (2006).
For cell-wall permeability assay:
- Subtract the bacterial basal emission from the final emission value recorded after bacteria were added to peptide and the signal has stabilized.
- Calculate (in percentage) how much the addition of bacteria increased the emission relatively to the basal peptide emission.
- Compare the degree of change between any given bacterial strains (wild type versus mutants for example) to evaluate the role of a given component to cell-wall permeability to peptides.
- See more details in Saar-Dover et al. (2012).
Notes
- Peptide concentration (C) in molar units is determined spectroscopic using the Beer–Lambert equation:
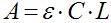
Where A is the actual absorbance at 467 nm (NBD) and at 530 nm (rhodamine). The molar absorption coefficient (ε) of NBD is 16,000 [cm/M], and that of rhodamine is 38,000 [cm/M]. L is the cuvette path length in centimeter. - Peptide concentration for the experiment should be at the low micromolar rang to reach very low peptide/lipid ratio and is also dependent on the labeling and purification quality. We generally use a concentration range of 0.1-1 μM of labeled peptides.
References
- Avrahami, D., Oren, Z. and Shai, Y. (2001). Effect of multiple aliphatic amino acids substitutions on the structure, function, and mode of action of diastereomeric membrane active peptides. Biochemistry 40(42): 12591-12603.
- Chattopadhyay, A. and London, E. (1987). Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26(1): 39-45.
- Gazit, E., Lee, W. J., Brey, P. T. and Shai, Y. (1994). Mode of action of the antibacterial cecropin B2: a spectrofluorometric study. Biochemistry 33(35): 10681-10692.
- Kliger, Y., Aharoni, A., Rapaport, D., Jones, P., Blumenthal, R. and Shai, Y. (1997). Fusion peptides derived from the HIV type 1 glycoprotein 41 associate within phospholipid membranes and inhibit cell-cell Fusion. Structure-function study. J Biol Chem 272(21): 13496-13505.
- Merklinger, E., Gofman, Y., Kedrov, A., Driessen, A. J., Ben-Tal, N., Shai, Y. and Rapaport, D. (2012). Membrane integration of a mitochondrial signal-anchored protein does not require additional proteinaceous factors. Biochem J 442(2): 381-389.
- Merrifield, R. B., Vizioli, L. D. and Boman, H. G. (1982). Synthesis of the antibacterial peptide cecropin A (1-33). Biochemistry 21(20): 5020-5031.
- Oren, Z., Lerman, J. C., Gudmundsson, G. H., Agerberth, B. and Shai, Y. (1999). Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem J 341 ( Pt 3): 501-513.
- Pouny, Y., Rapaport, D., Mor, A., Nicolas, P. and Shai, Y. (1992). Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 31(49): 12416-12423.
- Rapaport, D. and Shai, Y. (1992). Aggregation and organization of pardaxin in phospholipid membranes. A fluorescence energy transfer study. J Biol Chem 267(10): 6502-6509.
- Rosenfeld, Y., Papo, N. and Shai, Y. (2006). Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J Biol Chem 281(3): 1636-1643.
- Saar-Dover, R., Bitler, A., Nezer, R., Shmuel-Galia, L., Firon, A., Shimoni, E., Trieu-Cuot, P. and Shai, Y. (2012). D-alanylation of lipoteichoic acids confers resistance to cationic peptides in group B Streptococcus by increasing the cell wall density. PLoS Pathog 8(9): e1002891.
- Shai, Y. (1999). Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1462(1-2): 55-70.
- Zhao, H. and Kinnunen, P. K. (2002). Binding of the antimicrobial peptide temporin L to liposomes assessed by Trp fluorescence. J Biol Chem 277(28): 25170-25177.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Saar-Dover, R. and Shai, Y. (2013). CAMP-Membrane Interactions Using Fluorescence Spectroscopy. Bio-protocol 3(15): e846. DOI: 10.21769/BioProtoc.846.
- Saar-Dover, R., Bitler, A., Nezer, R., Shmuel-Galia, L., Firon, A., Shimoni, E., Trieu-Cuot, P. and Shai, Y. (2012). D-alanylation of lipoteichoic acids confers resistance to cationic peptides in group B Streptococcus by increasing the cell wall density. PLoS Pathog 8(9): e1002891.
Category
Microbiology > Microbe-host interactions > Bacterium
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.