- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Electroporation of Small Interfering RNAs into Tibialis Anterior Muscles of Mice
Published: Vol 12, Iss 11, Jun 5, 2022 DOI: 10.21769/BioProtoc.4428 Views: 3808
Reviewed by: Salma MerchantDoyel SenSHAY YOSEF GEULAAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Aging and wasting of skeletal muscle reduce organismal fitness. Regrettably, only limited interventions are currently available to address this unmet medical need. Many methods have been developed to study this condition, including the intramuscular electroporation of DNA plasmids. However, this technique requires surgery and high electrical fields, which cause tissue damage. Here, we report an optimized protocol for the electroporation of small interfering RNAs (siRNAs) into the tibialis anterior muscle of mice. This protocol does not require surgery and, because of the small siRNA size, mild electroporation conditions are utilized. By inducing target mRNA knockdown, this method can be used to interrogate gene function in muscles of mice from different strains, genotypes, and ages. Moreover, a complementary method for siRNA transfection into differentiated myotubes can be used for testing siRNA efficacy before in vivo use. Altogether, this streamlined protocol is instrumental for basic science and translational studies in muscles of mice and other animal models.
Keywords: ElectroporationBackground
Skeletal muscle is an important tissue with many fundamental functions (Wolfe, 2006; Nair, 2005). Consistently, diseases that affect skeletal muscle profoundly impact the organism’s fitness and survival (Demontis and Perrimon, 2009, 2010; Demontis et al., 2013a, 2014; Piccirillo et al., 2014; Rai and Demontis, 2016; Robles-Murguia et al., 2020; Rai et al., 2021a). Among the many muscle diseases, muscle wasting is a debilitating condition associated with aging and with diseases such as cancer, infections, kidney failure, sepsis, and neuromuscular disorders (Demontis et al., 2013b; Bonaldo and Sandri, 2013; Tsoli and Robertson, 2013; Piccirillo et al., 2014). Several studies have demonstrated that muscle wasting worsens disease outcome and decreases patient survival, whereas preserving skeletal muscle mass and function is protective (Zhou et al., 2010, Johnston et al., 2015). Skeletal muscles are composed of multinucleated syncytial cells known as fibers or myofibers. Myofiber types with distinct metabolic and contractile properties are differently abundant in muscles, and this varies in accordance with anatomical location and function (Schiaffino and Reggiani, 2011; Schiaffino et al., 2013). During muscle wasting, these myofiber types are differently susceptible and impacted by atrophic stimuli (Demontis et al., 2013b; Piccirillo et al., 2014; Bonaldo and Sandri, 2013; Ciciliot et al., 2013). Catabolic stimuli induce muscle wasting primarily via the induction of myofiber atrophy, whereas changes in the number of myofibers are not common. Mechanistically, a decrease in the size of myofibers occurs as a result of decreased synthesis and increased degradation of muscle protein, which is mediated by the autophagy/lysosome and ubiquitin-proteasome systems (Demontis et al., 2013b; Piccirillo et al., 2014; Bonaldo and Sandri, 2013). As for other muscle diseases, there are currently no therapies available in the clinic to prevent or cure age- and disease-associated muscle wasting. To address this unmet medical need, many experimental disease models and techniques for probing gene function in skeletal muscles have been developed over the years.
Electroporation has been used as a general method for gene delivery that is potentially applicable to all organisms and cell types (Ugen and Heller, 2003; Young and Dean, 2015). By using electrical fields, electroporation transiently destabilizes the plasma membrane and facilitates the electrophoretic movement and intracellular delivery of DNA plasmids (Ugen and Heller, 2003; Young and Dean, 2015). In skeletal muscle, this technique has been extensively used for the expression of plasmid-encoded transgenes and the subsequent assessment of gene function in skeletal muscle homeostasis (Aihara and Miyazaki, 1998; Rizzuto et al., 1999; Hoover and Magne Kalhovde, 2000; Sandri et al., 2003; Bertrand et al., 2003; Peng et al., 2005; Tevz et al., 2008). Moreover, this experimental approach has found translational application in improving the delivery of DNA plasmids, for mounting immune responses (Widera et al., 2000; Zucchelli et al., 2000; Khan et al., 2014; Vandermeulen et al., 2014; Haidari et al., 2019; Mpendo et al., 2020; Edupuganti et al., 2020), and for gene therapy in humans (Aihara and Miyazaki, 1998; Rizzuto et al., 1999; Mizui et al., 2004;Wong et al., 2005; Brolin et al., 2015).
Here, we report an optimized protocol for the electroporation of small interfering RNAs (siRNAs) into the tibialis anterior (TA) skeletal muscle of mice (protocol #1). We propose that this method can help to interrogate gene function in skeletal muscle, by inducing the acute knockdown of the intended target mRNAs in animal models. Moreover, we also report a protocol for siRNA-mediated knockdown in cultured myotubes (protocol #2), to complement in vivo studies with siRNA-mediated electroporation.
Knowledge gained with this siRNA electroporation technique could be similarly applied for improving the intramuscular delivery of other RNA species and RNA-based vaccines, as well as for other translational and basic science applications.
Experimental design and controls
In this protocol, the TA muscle in one hind leg is electroporated with siRNAs for targeting the intended mRNA, whereas the contralateral hind leg serves as control, and is electroporated with non-targeting (NT) siRNAs. Therefore, this experimental design provides a robust internal control unhindered by differences between individual animals. Nonetheless, to ensure that the results obtained from different animals in the same cohort can be cross-compared, it is important to utilize animals of the same age and sex, and reared in a consistent manner (siblings from the same litter should be utilized whenever possible).
A major advantage of the electroporation technique described here is that it allows probing gene function in animals of different ages. Previous studies have used 6-month-old mice as “young” controls (Sheard and Anderson, 2012) because, while postnatal muscle development mostly occurs in the first 3 months of age, growth still occurs in subsequent months (Ebert et al., 2015). However, whenever comparing electroporation experiments done at different ages, especially if these include ages at which postnatal development is not concluded, it is important to normalize the TA mass to the length of the tibia bone. This accounts for differences in whole body size when comparing muscles from different mice, because the tibia is typically static in fully-grown mice, and is not influenced by muscle atrophic and hypertrophic stimuli (Rowland, 2007; Shavlakadze et al., 2010; Puppa et al., 2014; Winbanks et al., 2016).
In addition to testing gene function in wild-type mice, siRNA electroporation can also be used to test the impact of mRNA knockdown in disease settings (e.g., models of cancer cachexia). In this scenario, the electroporation technique provides a suitable intervention for testing the requirement of a certain gene in disease progression. For example, the electroporation of siRNAs can be used to probe whether impeding the upregulation of a cancer-induced gene can preserve myofiber size and prevent cancer-induced TA mass loss. However, in this case, appropriate controls will consist of the electroporation of gene-targeting and control NT siRNAs, in contralateral legs of animals, with and without cancer (or another disease model).
Similarly, siRNA electroporation can be performed not only in wild-type animals but also concomitantly to another genetic intervention [e.g., Cre-Lox–mediated ablation of another gene in muscle (McCarthy et al., 2012)]. In this scenario, the electroporation probes the genetic interaction between the siRNA–targeted and the Cre–targeted genes. However, appropriate controls in this case will consist of the electroporation of gene–targeting and NT siRNAs in contralateral legs of animals with and without Cre–mediated ablation of the second gene tested.
Similar experimental design and controls are also used with the siRNA–mediated knockdown of target genes in cultured mouse C2C12 myotubes. Specifically, qRT-PCR and other cellular/molecular assays are used to test the outcome of siRNAs targeting the intended gene compared to control NT siRNAs. Although the percentage of mRNA knockdown needed to uncover a phenotype varies depending on the mRNA targeted, a previous estimate based on large-scale RNAi testing in Drosophila melanogaster suggests that phenotypes are typically uncovered when the reduction in mRNA levels is above 50% (Sopko et al., 2014; Graca et al., 2021).
We propose that testing siRNA efficacy of target gene knockdown in cultured myotubes may constitute an important step towards validation of siRNA reagents, before their use in vivo for TA muscle electroporation.
Limitations and comparison to other models
Because this protocol focuses on the electroporation of siRNAs, it is impacted by the known limitations associated with RNA interference, including the possibility that siRNAs target unintended mRNAs (RNAi off-target effects), and that the mRNA knockdown achieved via siRNAs is partial and insufficient to uncover a phenotype. These initial impediments of RNAi are largely surpassed by late-generation reagents (e.g., siRNA SMARTpools used here) (Setten et al., 2019; Neumeier and Meister, 2020).
Another limitation of siRNA delivery via electroporation is that it induces some, albeit minimal, muscle damage (McMahon and Wells, 2004; Skuk et al., 2013). In fact, the conditions reported in this protocol have been optimized to avoid extensive muscle damage, which has been possible because delivery of siRNAs requires remarkably lower electric fields and plasma membrane perturbation, when compared to the delivery of bulkier DNA plasmids (Aihara and Miyazaki, 1998; Rizzuto et al., 1999; Hoover and Magne Kalhovde, 2000; Sandri et al., 2003; Bertrand et al., 2003; Peng et al., 2005; Tevz et al., 2008), that have nonetheless lead to many landmark discoveries in muscle biology (Murgia et al., 2000; Sandri et al., 2004; Menzies et al., 2010). Specifically, the electrotransfer voltages used here have previously been demonstrated to be optimal (McMahon et al., 2001; Schertzer et al., 2006). Although the higher voltages may induce muscle damage when the skin is removed, and electrodes are placed directly on the muscle, the electroporation efficacy and damage are lower when the electrodes are placed on the skin without surgical incision, as reported here. Moreover, the method used here, by which the electrical field orientation is alternated (from lateral-medial to anterior-posterior), has been demonstrated to transduce more efficiently than the single orientation of the electrical field (Golzio et al., 2012). Therefore, in our opinion, the limitations deriving from the RNAi technology and electroporation are minimal due to the extensive optimization of siRNA reagents for this established technology, and because of the mild and non-surgical electroporation conditions that are used for siRNA delivery.
However, a major limitation of electroporation is that it allows for only a transient knockdown of gene function, which has been reported to be significant for up to 2–3 weeks (Tevz et al., 2008). Therefore, it is not possible to probe the long-term consequences of gene perturbation, as the electroporation only transiently affects mRNA levels. For the same reason, because some proteins are extremely long-lived (Savas et al., 2012; Toyama et al., 2013; Krishna et al., 2021), it may not be possible to impact their levels in the few-weeks timeframe of electroporation.
A further limitation of this protocol is that it allows for the analysis of gene function only in TA muscles. Therefore, it does not provide a means for testing gene function in muscles with different anatomical locations and physiological functions (Schiaffino and Reggiani, 2011; Schiaffino et al., 2013). Therefore, rather than surgery-mediated direct apposition to muscles, electroporation with electrodes apposed to the skin does not allow probing gene function in internal muscles, such as the soleus. Alternative methods for gene delivery to skeletal muscle are available, including adeno–associated viruses and sonodelivery (Decker et al., 2020; Manini et al., 2021), and they may overcome limitations of electroporation–mediated delivery if needed.
Advantages
Nonetheless, compared to other interventions, the siRNA electroporation protocol reported here offers several advantages. Some of the previously noted limitations (see paragraph above) can turn out to be advantageous under certain conditions. For example, the partial knockdown of the intended mRNA might be an issue and impede the uncovering of a phenotype. However, this might be advantageous when targeting a gene with fundamental functions, which would result in the death of the animal if completely ablated and/or impacted across all skeletal muscles.
It has been previously noted that, over generations, knockout animals accumulate compensatory background mutations that can reduce the phenotypic manifestation, and even lead to the complete disappearance of the phenotype (Rossi et al., 2015). In this respect, the electroporation of siRNAs provides an acute intervention for perturbing gene function, and it is therefore unencumbered by the compensatory adjustments that occur in classical genetic mutants over time. Similarly, the genetic background (i.e., the genetic makeup of the animal beyond the mutation in the intended gene) (Ungerer et al., 2003; Burnett et al., 2011; Lucanic et al., 2017; Hou et al., 2019; Koh et al., 2020), the cytoplasmic background (derived from the oocyte, with its set of mitochondria and cytoplasmic pathogens) (Toivonen et al., 2007; Joseph et al., 2013), and the microbiota (Ulgherait et al., 2016; Kim et al., 2017; Mamantopoulos et al., 2017; Poussin et al., 2018; Vujkovic-Cvijin et al., 2020) are examples of biological variables that are normally controlled only with a careful experimental design. For example, to ensure that all experimental animals do not differ in their cytoplasmic background, all experimental animals should be derived from the same mother, or at least from related mothers. To ensure that the animals are isogenic and do not differ in their genetic background, they should be back-crossed several times (typically 10×) against the same genetic reference strain.
The protocol reported here is based on the electroporation of siRNAs for the intended gene into the TA muscle of one hind leg, and the electroporation of control non-targeting (NT) siRNAs into the contralateral hind leg of the same animal. On this basis, any observed phenotype obtained with siRNAs for the intended gene versus NT siRNAs is not due to inter-individual differences in the genetic background, cytoplasmic background, or microbiota. Therefore, the protocol described here provides ideal settings for testing gene function in the TA muscle, without confounding biological variables.
Although skeletal muscle primarily consists of syncytial muscle cells (known as fibers or myofibers), there are many infiltrating and associated cells that are important for muscle homeostasis. These include immune cells, endothelial cells, fibroadipogenic progenitors, and satellite muscle stem cells. The electroporation likely targets all these cell types (Wong et al., 2005; Dean, 2013), and hence allows for the investigation of the function of a certain gene, not only in myofibers, but also in other muscle-associated cells.
Potential applications of this protocol and future directions
Due to its simplicity, this protocol can be applied to many different mouse strains and disease models. Moreover, it is potentially applicable to other rodent species that are generally used in research (such as rats), and other emerging rodent disease models, such as the long-lived naked-mole rat, and Octodon degus, a rodent that has been reported to develop spontaneous Alzheimer’s-like disease (Buffenstein, 2005; van Groen et al., 2011; Valenzano et al., 2017). Although these rodents radically differ from each other in regards to many features, including their life trajectories and disease predisposition, they all have TA muscles that are easily accessible for electroporation, without the need of any surgery. However, the TA muscle is bigger in rats; thus, the procedure reported here for mice would need to be scaled up. Alternatively, only a localized area of the TA could be electroporated and eventually sampled with a localized biopsy. Indeed, with appropriate adjustments, the siRNA electroporation described here could prove useful in investigating skeletal muscle biology also in larger model organisms, such as marmosets and monkeys (upon appropriate ethical approvals). In this case, the impact of siRNA electroporation into TA muscles could be tested by obtaining a biopsy of the electroporated area of the TA, without requiring sacrificing the animal (Joyce et al., 2012; Cotta et al., 2021). Beyond the TA, it may also be possible to adapt this method to test the impact of gene knockdown in other skeletal muscles. However, such additional testing would be limited to skeletal muscles located beneath the skin, and hence susceptible to electroporation without surgical incision.
In addition to RNAi–mediated knockdown, this electroporation method can also be used in conjunction with gene editing technologies such as CRISPR, to obtain target gene knockout and overexpression (Gaj et al., 2013; Doudna and Charpentier, 2014; Jiang and Doudna, 2017). For example, electroporation of short-guide RNAs (sgRNAs) into TA muscles of mouse models that express Cas9 or Cas9 fused with a transcriptional activator could lead to target gene deletion or overexpression, respectively, whereas electroporation of the contralateral leg with mock sgRNAs would provide a matched control.
Beyond its use in experimental animal models, it has been proposed that TA electroporation in humans may constitute a means for gene therapy, by improving the intramyofiber delivery of DNA vaccines (Widera et al., 2000; Zucchelli et al., 2000; Khan et al., 2014; Vandermeulen et al., 2014; Haidari et al., 2019; Mpendo et al., 2020; Edupuganti et al., 2020), and potentially also of recently-developed RNA-based vaccines, and for the production of cytokines, growth factors, and other therapeutic factors by the skeletal muscle of patients with a number of diseases (Aihara and Miyazaki, 1998; Rizzuto et al., 1999; Mizui et al., 2004; Wong et al., 2005;Brolin et al., 2015).
Materials and Reagents
Biological materials (protocol #1)
Laboratory animals:
C57BL/6J mice (The Jackson Laboratory, catalog number: 000664). However, this protocol can be used with any mouse strain, and it can be similarly applied also to other experimental rodent models.
CRITICAL: For electroporation, the animals should be 6 months of age minimum, to ensure postnatal muscle growth has been completed. However, younger animals can be used, if the intent is to probe the role of a gene in postnatal muscle growth.
Biological materials (protocol #2)
C2C12 murine myoblast cells:
The cells were obtained from the ATCC (ATCC, catalog number: CRL-1772), and were cultured in DMEM + GlutaMax with 10% (v/v) FBS media and 1% (v/v) penicillin/streptomycin at 37°C, with an atmosphere of 5% (v/v) CO2. For differentiation into myotubes, cells were grown up in DMEM + GlutaMax with 2% (v/v) HS media and 1% (v/v) penicillin/streptomycin, under the same conditions.
CRITICAL: All cells were tested every 6 months for Mycoplasma sp. contamination. The maximum passage for optimal results is 10.
Reagents (protocol #1 and #2)
Electroporation (protocol #1)
Hyaluronidase – Type IV-S from Bovine Testes (Sigma-Aldrich, catalog number: 4272)
5× siRNA Buffer (Horizon, catalog number: B-002000-UB-100)
1× PBS (Gibco, catalog number: 10010-023)
20 nM ON-TARGETplus siRNAs (delivered as individual siRNAs or as a SMARTpool of 4 siRNAs), such as the Non-targeting Control (Horizon, catalog number: D-001810-10-20) or siRNAs to target a gene of interest (Note: Other types of RNAi reagents could also be used in this protocol in place of the ON-TARGETplus siRNAs). Fluorophore-labeled siRNAs (siGLO reagents, such as the siGLO Red Transfection Indicator, D-001630-02-05) can also be used to monitor siRNA delivery when testing this protocol.
Isoflurane (Piramel Critical Care, catalog number: 66794-013-25)
Medical Oxygen [100% (v/v) O2]
Nair Depilatory Cream (Church & Dwight, or equivalent)
Cryopreservation (protocol #1)
Tragacanth Gum (Sigma-Aldrich, catalog number: G1128)
2-Methylbutane, i.e., isopentane (Sigma-Aldrich, catalog number: 277258)
Ethyl Alcohol, 140 Proof (Pharmco by Greenfield Global, catalog number: 111000140)
Cryo-sectioning (protocol #1)
Tissue-Tek O.C.T. Compound (Sakura Finetek USA, Inc., catalog number: 4583)
Hematoxylin Stain (Cancer Diagnostic, catalog number: SH5777)
Mount Quick Aqueous Mounting, 30 mL (Research Products International, catalog number: 195705)
Immunohistochemistry (protocol #1)
Bovine Serum Albumin (BSA; GoldBio, catalog number: A-420-1)
Triton X-100 (Sigma-Aldrich, catalog number: 9002-93-1)
SC-71-s Primary Antibody (Developmental Studies Hybridoma Bank, catalog number: SC-71)
BF-F3-s Primary Antibody (Developmental Studies Hybridoma Bank, catalog number: BF-F3)
Laminin α2 Antibody (Santa Cruz Biotechnology, catalog number: SC-59854)
Alexa Fluor 488 goat anti-mouse IgG1 (Thermo Fisher, Invitrogen, catalog number: A21121)
Alexa Fluor 555 goat anti-mouse IgM (Thermo Fisher, Invitrogen, catalog number: A21426)
Alexa Fluor 647 goat anti-rat IgG (Thermo Fisher, Invitrogen, catalog number: A21247)
DAPI (Sigma-Aldrich, catalog number: 10-236-276-001)
Slow Fade Gold Antifade (Thermo Fisher, Invitrogen, catalog number: S36937)
Rapid Dry Topcoat Polish (Electron Microscopy Sciences, catalog number: 72180)
Cell Culture (protocol #2)
DMEM, high glucose, GlutaMax Supplement (Gibco, catalog number: 10566106)
Fetal Bovine Serum (FBS; Gibco, catalog number: 10438-026)
1× Penicillin/Streptomycin 10,000 U/mL (P/S; Gibco, catalog number: 15140122)
Horse Serum (HS; Gibco, catalog number: 26050070)
Opti-MEM Reduced Serum Media (Gibco, catalog number: 31985062)
Lipofectamine 2000 (Invitrogen, catalog number: 11668019)
Cytosine β-D-arabinofuranoside (Sigma-Aldrich, catalog number: C1768)
0.25% Trypsin-EDTA (w/vol), phenol red (Gibco, catalog number: 25200056)
Myotube Immunostaining (protocol #2)
16% Paraformaldehyde (PFA) Aqueous Solution, EM Grade (Fisher Scientific, catalog number: 50-980-487)
Myosin 4 Monoclonal Antibody MF20 (Thermo Fisher Scientific, catalog number: 14-6503-82)
Alexa Fluor 555 goat anti-mouse IgG2b (Thermo Fisher, Invitrogen, catalog number: A21147)
Reagent setup (protocol #1 and #2) (see Recipes)
10% Tragacanth
Hyaluronidase stock solution
Hyaluronidase working solution
50 μM siRNA stock
2% BSA blocking buffer
Primary antibody staining solution (frozen slides)
Secondary antibody staining solution (frozen slides)
10% FBS
2% HS
1% P/S
4% PFA
Primary antibody staining solution (cell culture myotubes)
Secondary antibody staining solution (cell culture myotubes)
Equipment
For protocols #1 and #2
Induction chamber with nose cone (VetEquip V-10 Mobile Unit)
Isoflurane vaporizer (VetEquip, catalog number: 911103)
29 ½-gauge needles (Fisher Scientific, catalog number: 14-841-32) and U-100 insulin syringes (0.5 mL 0.33 × 12.7 mm; Exel INT, catalog number: 26028)
Electro Square Porator (ECM830 BTX Harvard Apparatus) and electrodes (Genetrodes, Straight, 10mm Gold Tip, catalog number: 45-0114, BTX Harvard Apparatus)
Petri dish, stackable lid 100 mm × 15 mm Sterile (Fisher Scientific, catalog number: FB0875712)
Exel International stainless steel disposable scalpel #11 (Fisher Scientific, catalog number: 14-840-01)
Carbon fiber digital caliper (Fisher Scientific, catalog number: 15-077-957)
Moloney forceps (Roboz Surgical Store, catalog number: RS-8254)
Graefe forceps (Roboz Surgical Store, catalog number: RS-5139)
Dissecting scissors, 4.5” Straight (Roboz Surgical Store, catalog number: RS-5912)
Operating scissors, 4.5” Straight (Roboz Surgical Store, catalog number: RS-6802)
Dressing forceps (World Precision Instruments, catalog number: 500365)
Cork sheets (Fisher Scientific, catalog number: 07-840-10)
Benchtop liquid nitrogen container, 2 L (Thermo Fisher Scientific, catalog number: 2123)
Versi-Dry Dispenser Roll, 20” by 100’ (Thermo Fisher Scientific, catalog number: 62070)
Insulated foam cooler
Precision balance (such as Sartorius Secura Analytical Balance 0.1mg)
250 mL beaker (DWK Life Sciences, Kimble)
Leica CM3050S Cryostat (Leica Biosystems, catalog number: 14903050S)
Superfrost plus microscope slides (Fisher Scientific, catalog number: 22-037-246)
Specimen disc, 30 mm (Leica Biosystems, catalog number: 14037008587)
Leica disposable blades low-profile 819 (Leica Biosystems, catalog number: 14035838925)
3 mL Transfer pipet (Falcon, catalog number: 357524)
Coverglass 22 × 30 mm, 1.5 thickness (Fisher Scientific, catalog number: NC1272771)
Single edge industrial razor blade (VWR, catalog number: 55411-050)
Microscope slide box 100p cork (Fisher Scientific, catalog number: 22-267294)
Kimwipes (Fisher Scientific, catalog number: 06-666A)
Wax pen Dako (Agilent, catalog number: S2002)
Laser-scanning confocal microscope (e.g., Nikon C2)
Nikon Elements software (Advanced Research version)
CO2 incubator (5% (vol/vol) CO2, 37°C)
Class II, Type A2 Biological Safety Cabinet
High capacity, benchtop centrifuge (Sorvall T6000D)
T150 cell culture flask (MidSci, catalog number: TP90151)
6-well Corning Costar Flat Bottom cell culture plates (Corning, catalog number: 3516)
Falcon 50-mL conical tubes (Fisher Scientific, catalog number: 352070)
Corning Costar 10-mL serological pipettes (Corning, catalog number: 4488)
Corning Costar 25-mL serological pipettes (Corning, catalog number: 4489)
Disposable 9-in Pasteur pipet (Fisher Scientific, catalog number: NC9496627)
Portable Pipette Controller (Drummond Scientific Company, catalog number: 4-000-101)
Hausser Scientific Hemocytometer (Fisher Scientific, catalog number: S17036)
Hand Tally counter (Fisher Scientific, catalog number: 07-905-6)
Inverted phase contrast microscope
Biohazardous waste container
Conical tube rack
Fluorescence microscope (Keyence BZ-X700)
Equipment setup
Surgical room (protocol #1)
All animal experiments and dissections should be performed in a designated procedure room with access to an isoflurane anesthesia chamber and oxygen. In the procedure room, sterilize the workstation with 70% alcohol and place down a surgical absorbent pad. Dress in proper personal protective equipment, including full gown, hair cover, shoe covers, face mask, and sterile gloves.
Preparation for dissecting tissues (protocol #1)
Prior to entering the procedure room, prepare for each sample: a 1.5-mL RNAse-free microcentrifuge tube, and a 2-cm × 2-cm cork pad for muscle mounting and cryopreservation. Gather all reagents necessary, including liquid nitrogen, isopentane, and tragacanth. Prepare a plastic beaker with 100 mL of isopentane, insert it into a Styrofoam pad, and float in liquid nitrogen inside an appropriate insulated container with a tight-closing lid. Allow isopentane to reach optimal temperature (-160°C) before freezing any tissue. Sterilize all surgical tools with 70% alcohol.
Cell culture preparation (protocol #2)
All cell culture experiments should be performed in a sterile Class II Type A2 Biological Safety cabinet, in a designated procedure room. All personnel should be trained according to BSL-2 protocols. Experiments should be performed using aseptic technique, and 70% ethanol used to sterilize the cell culture hood, all equipment, and reagents. Biohazard waste should be discarded in the appropriate manner according to institutional and state laws. The appropriate PPE, including gloves and a clean laboratory coat, must be worn. Prior to all experiments, the necessary reagents should be warmed in a 37°C water bath for a minimum of 30 min, unless otherwise stated in the protocol.
Software
GraphPad Prism (version 7 or higher)
ImageJ
Procedure
Electroporation of muscle • Timing ~2.5 h per mouse
Place the mouse in the induction chamber and deliver 3.0% (v/v) isoflurane at a 2.5-L/min flow rate, until the animal is fully anesthetized. Monitor the mouse’s breathing rate and test the efficiency of the anesthesia via the toe-pinch reflex.
CAUTION: Isoflurane is hazardous if vapor is inhaled. Ensure proper ventilation.
Remove the mouse from the induction chamber and transfer it to a surgical pad. Place the animal in the nose cone apparatus and maintain isoflurane levels.
Add a dime-sized amount of depilatory lotion to the leg for 10 s and wipe it with 70% ethanol.
Inject 30 μL of hyaluronidase working solution directly into the TA muscle. To do so, position the needle with a ~10° tilt onto the desired hind leg, oriented so that you can see the opening of the needle. Insert the needle starting from the lower attachment of the TA until reaching the upper part of the muscle. Slowly release all the solution while pulling the needle out from the muscle (Figure 1a–b). Ensure that all content is delivered. As a result, the muscle will inflate, but you should not see any subcutaneous liquid.
CRITICAL: Hyaluronidase degrades hyaluronic acid, a component of the extracellular matrix, and is crucial for helping the siRNAs spread through the TA.
Once injected, return the mouse to its cage, and allow it to recover for 2 h before anesthetizing it again.
CRITICAL: Ensure the mouse has access to food and water during recovery.
While the mouse is recovering, set up the electroporation machine and adjust the settings to read 20 ms, 1 Hz, and 80V. Test the electrodes by pulsing the tips into water. The pulse will produce bubbles if working properly.
CAUTION: The electrodes can cause damage to the skin if touched directly.
After 2 h from the hyaluronidase injection, anesthetize the mouse again and place it back in the nose cone, maintaining the same levels of isoflurane as before. Inject 30 μL of 50 μM siRNA directly into the TA muscle, in a similar manner as done for the hyaluronidase solution (step 4; Figure 1a–b).
Electroporate the muscle injected with siRNA by placing the Genetrodes electrodes on the previously-shaved skin parallel to the TA muscle, and pulsing four times (each pulse consisting of 1 Hz, 80 V, for 20 ms) with 1 s interval in-between the pulses (Figure 1c). Repeat this process with the electrodes placed perpendicular to the TA muscle (Figure 1d–e). Positioning the electrodes onto any area of the shaved skin on top of the TA will work, as long as firmly pressed on it.
CAUTION: Make sure the electrodes are not touching each other or your hands. Keep the electrodes 0.2 cm apart from each other.
Return the mouse to the cage for recovery. Plan to dissect the muscle for subsequent analyses 7 days post electroporation.

Figure 1. Electroporation of siRNAs into the myofibers of tibialis anterior muscles. A. Location of the tibialis anterior (TA) muscle and associated tibia bone in the hind leg. b. Injection of hyaluronidase and siRNAs into the TA: these solutions are slowly released while pulling out the needle. c, Set up of the electroporator apparatus. d–e. Parallel (d) and perpendicular (e) placement of electrodes on the skin on top of the TA for the electroporation of siRNAs.Muscle dissection and preservation • Timing ~7 min per mouse (protocol #1)
Place the mouse into the euthanasia chamber, and deliver 100% (v/v) CO2 at a 3-L/min flow rate for 2 min. Observe the animal for lack of breathing, and maintain the CO2 level for an additional minute. Remove the animal from the chamber, and perform cervical dislocation to confirm the animal’s death. Record its weight (relevant if comparing results obtained from animals of different ages, as explained in the background section), and immediately transfer to the surgical absorbent pad.
Position the animal on its back and spray the hind leg with 70% ethanol. Remove the skin surrounding the leg by making a small cut above the ankle with the operating scissors. Pull the skin back towards the thigh with the Moloney forceps to expose the TA.
CRITICAL: Carefully remove the fascia (i.e., the connective sheet that covers the muscle) by using the tips of the Moloney forceps.
CAUTION: All surgical tools are extremely sharp and could cause injury if not used properly.
Sever the distal tendon with a scalpel to release the muscle from the ankle. Using the Moloney forceps, pull the muscle toward the thigh, and hold the tissue in place. Detach the muscle from the proximal attachment and associated bone (i.e., the tibia) using the micro-dissecting scissors. Weigh the muscle and record the measurement.
To preserve the tissue for analysis, cut the TA in half at the mid-belly (the widest section of the muscle) with a scalpel. Prepare the sample for histology by spreading tragacanth on a prepared cork square. Using the Graefe forceps, drag the distal half of the TA muscle, tendon side down, into the tragacanth. Place the cork into the isopentane suspended in liquid nitrogen for a minimum of 30–45 s, to cryopreserve the tissue.
CAUTION: Isopentane is hazardous if inhaled. Avoid skin contact and wear protective equipment when handling.
Cut the remaining piece of the TA muscle in half again. Collect all the tissue in the prepared 1.5-mL microcentrifuge tube, and place in liquid nitrogen to snap-freeze for further analyses (such as western blot, RNA-seq, proteomics, etc.), following similar procedures as previously described (Meng et al., 2014).
CRITICAL: Perform an RNA extraction using one of the tissue pieces, to test the knockdown efficiency of the siRNA via qRT-PCR.
Using the operating scissors, cut at the middle of the knee to detach the leg from the animal. Once detached, locate the junction of the knee and tibia bone, and cut again. To expose the bone, press the foot of the animal towards the surgical pad, and pull the skin down. Measure the length of the tibia (in mm) using the digital caliper, and record. The measurement of the tibia length is used to normalize the weight of the TA muscle, in order to account for variability in body and muscle size among experimental animals.
Repeat steps 11–15 on the opposite leg.
Note: In order to provide an app0ropriate robust control, one TA is electroporated with siRNAs targeting a gene of interest, whereas the other TA in the contralateral leg is electroporated with control non-targeting siRNAs (Figure 2).
Temporarily store the cork and tubes on dry ice during dissections. Store all samples at -80°C for future analysis.
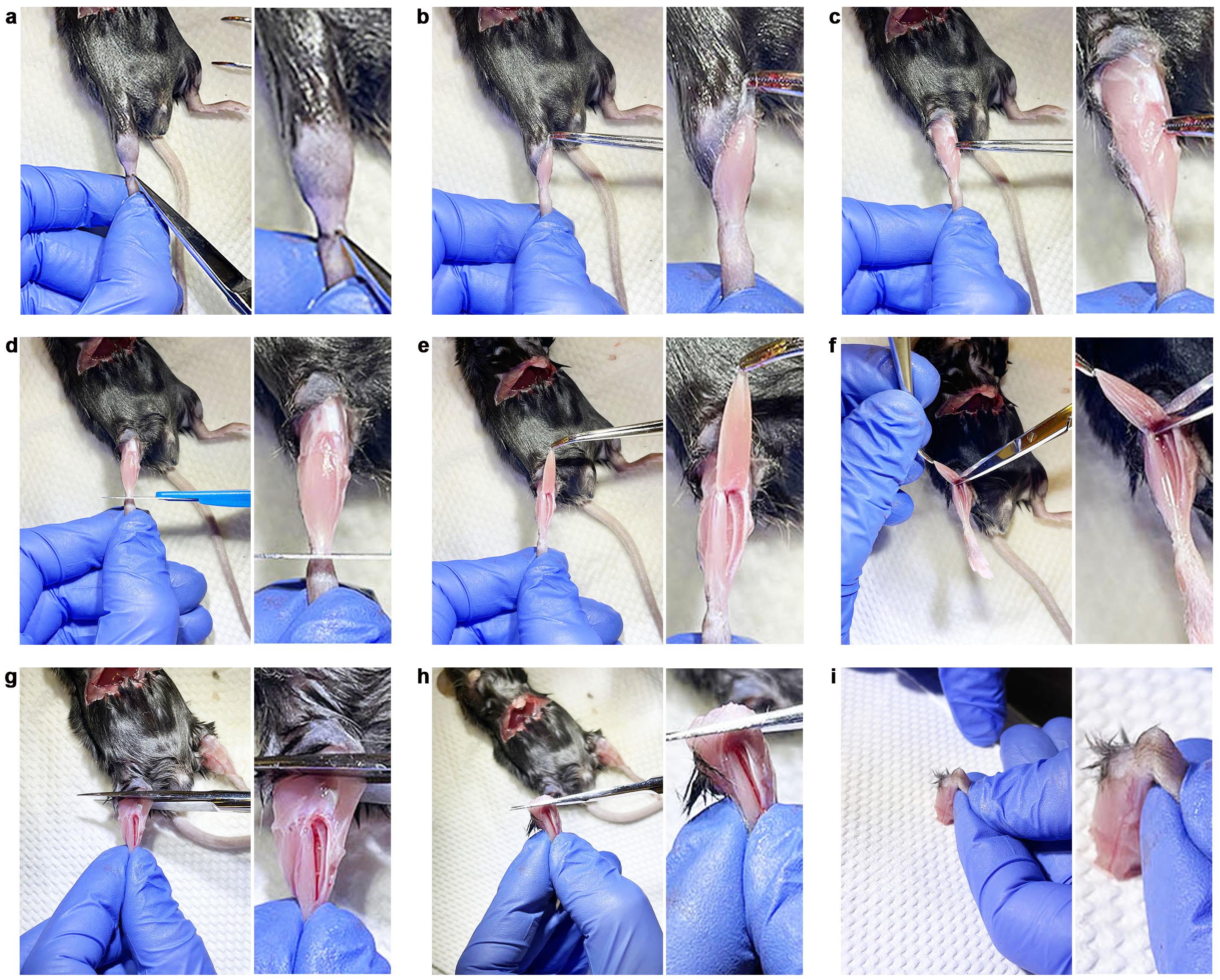
Figure 2. Dissection of the tibialis anterior muscle and tibia. a–i. Step-by-step visual representation of the dissection of the tibialis anterior (TA) muscle from the front of the hind leg. a. A small incision is made at the ankle using operating scissors. b. The skin is removed to expose the underlying TA muscle. c. The fascia is removed. d. The proximal tendon is severed, releasing the TA muscle. e. The TA muscle is pulled towards the thigh. f. The muscle is detached from the tibia bone. g. A cut is made at the level of the knee to detach the lower part of the hind leg. h. The junction between the knee and tibia bone is cut. i. The tibia is placed on the surgical pad, and the foot of the animal is pulled down to expose the bone, so that the full length of the tibia bone can be correctly measured.Cryo-sectioning of tissue • Timing ~15 min per sample (protocol #1)
Remove the tissue from the -80°C freezer, and place it in the cryostat chamber 30 min prior to sectioning. Adjust the specimen and chamber temperatures to -20°C.
CRITICAL: Specimen temperature varies depending on specimen type. For skeletal muscle, it is recommended a temperature range between -25°C and -15°C.
Place the specimen disc and brushes into the chamber, and align the disposable blade into the holder.
CAUTION: The disposable blade is sharp; use caution when handling to avoid injury. The cryostat handwheel should always be in the locked position when not in use.
Apply a sufficient amount of O.C.T. onto the 30-mm specimen disc, and mount the sample. Prepare four microscope slides per sample while the tissue freezes.
Once the tissue is frozen, insert the disc into the specimen head. Using the attached levers, orient the tissue into the desired position, and clamp down.
Adjust the trimming thickness to 10 µm, and bring the specimen towards the disposable blade, by unlocking the handwheel and using the motorized coarse feed. Begin trimming the muscle manually by rotating the handwheel clockwise until the tissue is smooth.
Trim the tissue again with the anti-roll plate down. Using a brush, smooth out the tissue and collect the sample onto a microscope slide.
TROUBLESHOOTING: If the sections curl or shred, adjust the knob attached to the anti-roll plate, to change its distance from the blade.
To test if the section is trimmed correctly, apply hematoxylin stain to the slide with a dropper, and wash with water after 10 s. Mount the slide and observe under a microscope. If correct, trim and collect the tissue until each additional microscope slide has up to three sections of tissue for subsequent immunostaining.
When finished, lock the handwheel and remove the specimen from the specimen head. Insert a razor blade between the disc and the tissue cork, to detach the sample from the freezing medium. Temporarily store the tissue on dry ice.
CAUTION: The razor blade is sharp, be careful when using to avoid injury.
Return the specimen head to the home position, and remove the disposable blade. Sweep all sectioning waste into the waste tray, and discard it in the biohazard waste.
CRITICAL: Do not leave the specimen in the cryostat chamber, as the chamber defrosts every 24 h.
Disinfect the chamber using 70% ethanol, close the chamber sliding window, and turn off the illumination.
Preserve the microscope slides in a slide-box and keep them at -20°C until needed. The slides can be stored at -20°C indefinitely.
Immunostaining slides • Timing ~1.5 h for day one and ~3 h for day two (protocol #1)
Remove the slides from -20°C storage and thaw them in a dry slide box for 10 min at room temperature.
Once thawed, outline a square around the desired tissue on the slide using a hydrophobic wax marker. Hydrate the slides with 1× PBS for 10 min, being careful not to touch the tissue.
Aspirate the PBS from the slides and add 2% BSA-0.1% Triton X-100 blocking solution for 1 h at room temperature.
Aspirate the blocking solution and add the primary antibody staining solution to the slides. Incubate overnight at 4°C. To identify TA myofiber types, the primary antibody solution should include anti-laminin α2, to detect myofiber boundaries, SC-71 antibodies, to immunostain for type IIA myofibers, and BF-F3 antibodies, to immunostain for type IIB myofibers. To minimize evaporation of the antibody solution, add water or soaked paper to the box containing the slides.
The next day, remove the slides from 4°C and aspirate the antibody solution. Wash the slides three times, using 1× PBS at room temperature. Incubate the slides with secondary antibodies at room temperature for 2 h. For the immunostaining of TA myofiber types, the secondary antibody solution should contain appropriate Alexa Fluor-conjugated antibodies, including AF647 goat anti-rat IgG to detect laminin, AF488 goat anti-mouse IgG1 to detect type IIA fibers, and AF555 goat anti-rabbit IgM to detect type IIB fibers. DAPI can also be included to stain the nuclei of myofibers.
Wash the slides twice with 1× PBS. After the final wash, prepare each slide individually by aspirating the 1× PBS from the slides, and removing the wax coating with a kimwipe.
To preserve for imaging, add two drops of antifade mounting medium to the slide, and place a cover glass over the tissue. Allow the slides to air dry for 24 h at room temperature.
CRITICAL: Fluorescent dyes are light-sensitive, store the slides in a dark place.
After 24 h, seal the slides by coating the edges with clear nail polish, and store at 4°C.
PAUSE POINT: Samples can be stored at 4°C for weeks or even months, but the signal intensity will decline over time.
Analyze the samples using a laser-scanning confocal microscope (Figure 3).
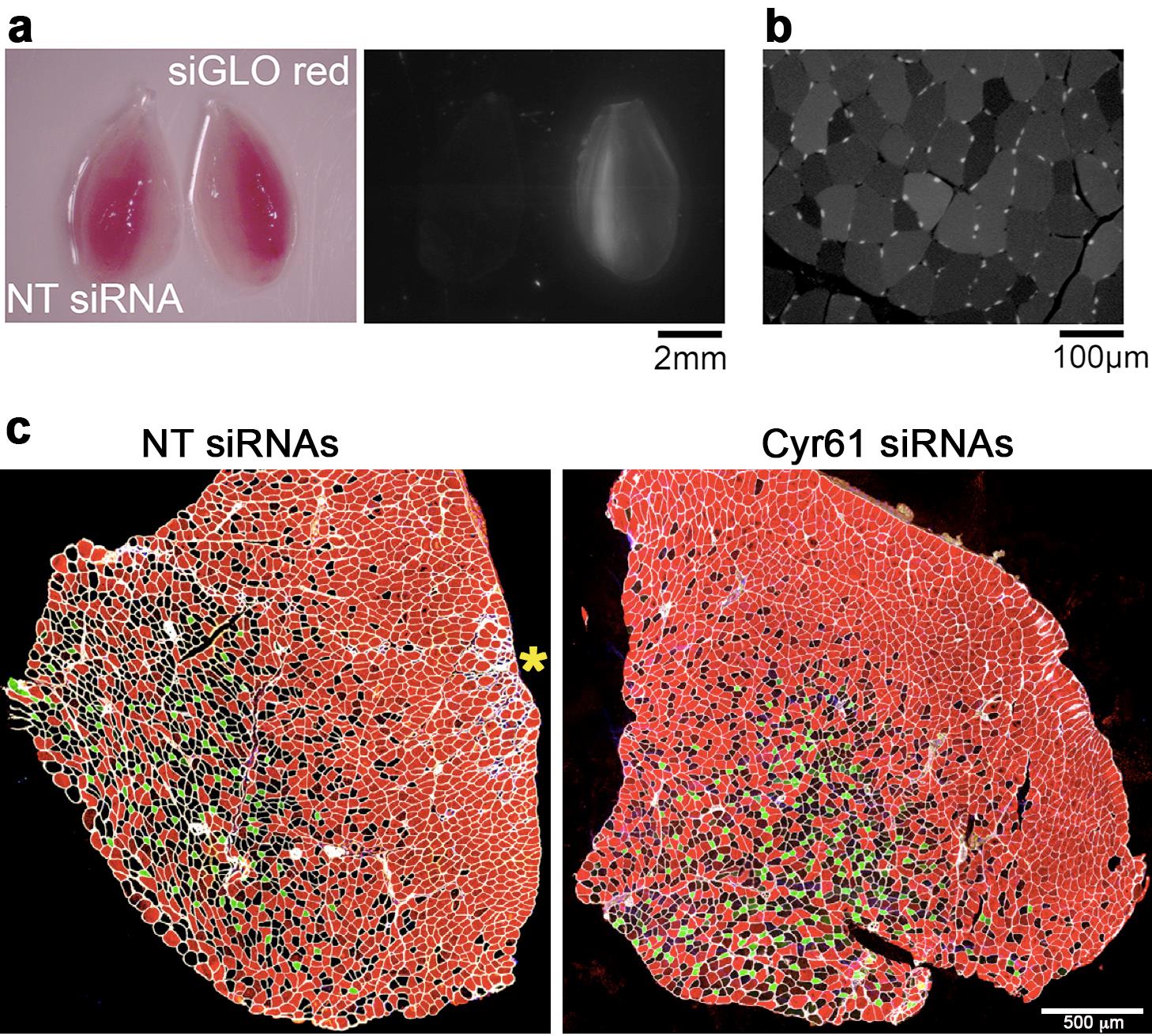
Figure 3. Outcome of siRNA electroporation into tibialis anterior muscles. a. Representative images of tibialis anterior (TA) skeletal muscles following electroporation of siGLO red and non-fluorescent control non-targeting (NT) siRNAs. Red fluorescence is detected in TA muscles electroporated with siGLO red, whereas no fluorescence is detected with NT siRNA electroporation. b. Red fluorescence is also detected in the myofibers of the TA muscle electroporated with siGLO red. c. Cross-sections of TA muscles 7 days after electroporation with control NT siRNAs and with siRNAs targeting the myokine Cyr61. Immunostaining for different myosin heavy chain isoforms identifies distinct myofiber types: type IIA (green), presumed IIX (black, i.e., lack of staining), and IIB (red) myofibers. Note the lack of damage/regeneration in the muscle electroporated with Cyr61 siRNAs, whereas only minor inflammatory infiltration and small regenerated fibers are marginally seen on the periphery to the right side (yellow asterisk) in the TA muscle electroporated with NT siRNAs. Cyr61 siRNAs induce a shift from type IIX to type IIB myofibers. Data in a–b is reproduced from Hunt LC et al. Cell Reports (2019), and data in c is from Hunt LC et al. Cell Reports (2021a).Confocal Imaging • Timing ~7 min per slide (protocol #1)
Turn on the Nikon C2 microscope. Open the imaging software program “NIS-Elements-Confocal”. For each laser channel, open the pinhole, and change the pixel size to 1024 × 1024 pixels.
NOTE: Equivalent laser scanning confocal microscopes and associated image acquisition software can be used for these analyses.
Mount the sample slide on the stage, with the coverslip down. Turn on the “Eye Port”, and use the 4× objective to observe the slide through the eyepiece. Adjust the stage until the lens is centered on the muscle section.
Turn off the eye port, and switch to the 10× objective. Select the Cy5 channel (405 nm), and scan the image live. Adjust the focus until the laminin-stained cell boundaries are clear. For each individual laser channel, optimize the signal amplification by adjusting the gain.
CRITICAL: Set the gain of the Cy5 channel using a control sample, and keep consistent for all samples.
Under the ND Acquisition tab, select lambda to individually check the confocal single channels: Cy5, FITC, TRITC, and DAPI. Then, select Larger Image, and scan the area in 4 × 4 fields, with an optimal path overlap of 10% stitching.
Select “start run” and save the image as both a ND2 and JPEG file for image analysis.
Image Analysis – Nikon Elements • Timing ~10 min per image (protocol #1)
Open the Nikon Elements – Advanced Research software. Select the analysis controls from the View pulldown menu, and dock the binary layers, thresholding, and object count functions.
Open the image as an ND2 file, and use the polygonal region of interest (ROI) option to draw an outline around the muscle section, to carefully establish a boundary for analysis. Under the result table, record the total area of the muscle, which will be later used to determine the total cross-sectional area (CSA).
CRITICAL: Outline as close to the border of the muscle as possible, to avoid detection of background by the analysis software.
Under the object count control, observe the restrictions box. Select both Area and MinFeret, and set the desired restrictions. For skeletal muscle, the restriction range is 100–10,000 μm2 and 10–150 μm, for area and Feret’s minimal diameter, respectively. Once set, select keep updating count.
CRITICAL: Restrictions and ranges can be adjusted according to the experimental model. Parameters can also be added under the results table.
Observe three working layers on the binary layers control: Cy5, FITC, and TRITC, representing laminin, type IIA fibers, and type IIB fibers, respectively. Set the threshold by selecting each layer individually, and adjusting the low/high option. Specifically, the software will display the detected fibers in a different color from the rest of the muscle based on the threshold limit. Using the reference layers, adjust the threshold to accurately represent the fibers by ensuring the threshold is identifying individual myofibers, based on the corresponding immunostaining (i.e., anti-IIA immunostaining only identifies IIA fibers). Once set, keep the thresholds the same for all images.
CRITICAL: For skeletal muscle, the low threshold for the Cy5 (Alexa 635) channel is always 0, and the high threshold for the FITC/TRITC (Alexa 488/555) channels is always 4095.
To determine type IIA myofibers, open the binary operations dialog box and create an intersection (AND) between the Cy5 and FITC layers. The software will recognize the fibers using the inverse threshold of laminin α2, to determine myofiber boundaries.
After the intersection is created, review the area of each fiber under the results table on the object count control. Use the reference layer for FITC as a guide, and delete any intersection that does not represent a fiber. Export the data into Excel, and label the sheet as the sample name and fiber type.
To determine type IIB myofibers, repeat steps 47–48 for the TRITC channel.
To find non-stained, presumed type IIX fibers, open the binary operations dialog box and create a subtraction (second layer from first layer) between the Cy5 and FITC layers. Repeat this step by using the newly created subtraction layer, and the TRITC layer. Review the area as before, deleting any non-fiber, and export the data to Excel.
De-select keep updating count, and close the image. Do not save changes to the image.
Repeat steps 47–51 for each sample (Figure 4).
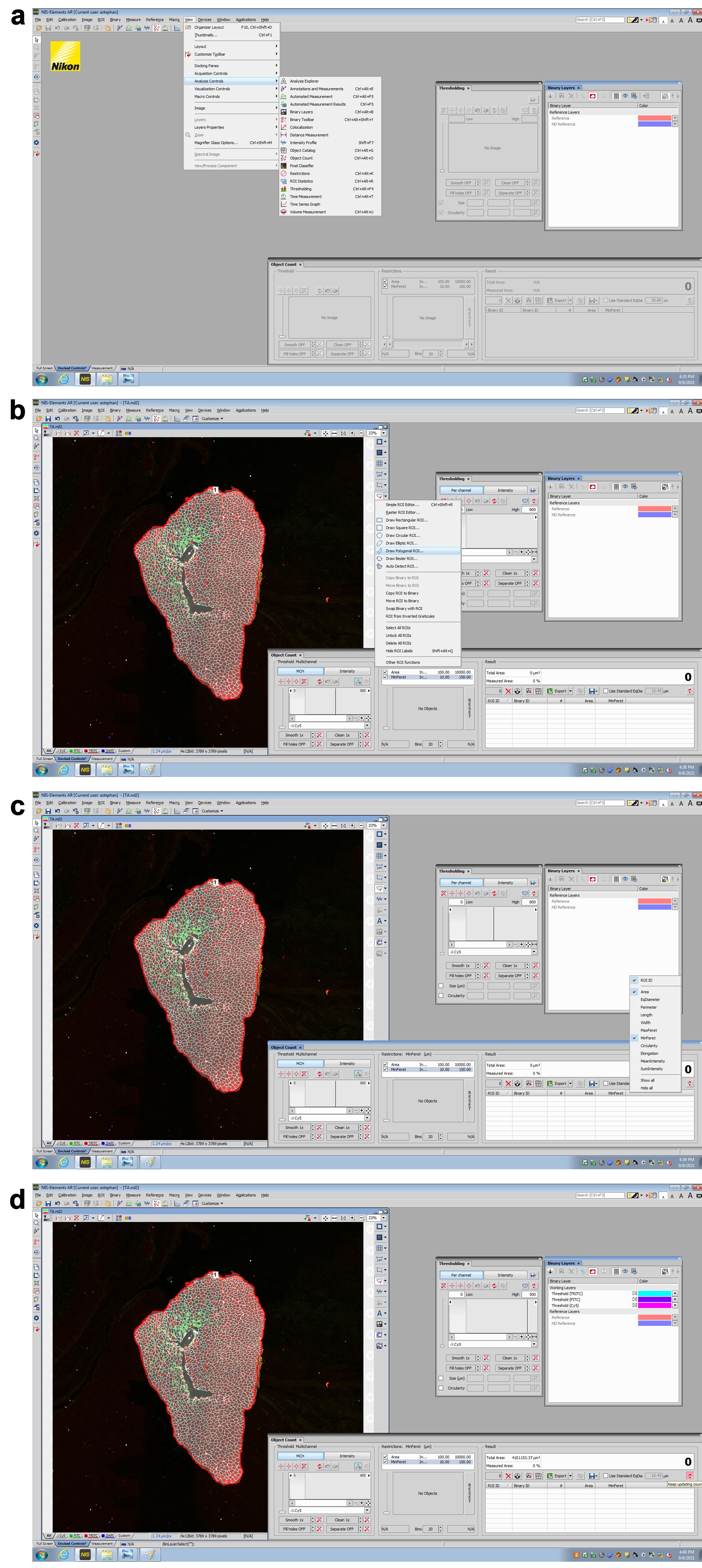

Figure 4. Analysis of TA myofiber size with the Nikon Elements software. a. The Nikon Elements software is opened, and the thresholding, binary layers, and object count functions are docked. b. The image is opened, and a polygonal region of interest (ROI) is drawn around the muscle border. c. On the object count function, the Area and MinFeret options are selected, and the restrictions are set to the desired range. The “keep updating count” function is selected. d. Three working layers (Cy5, FITC, and TRITC) are observed, and the thresholds are adjusted to accurately represent the fiber types (IIA, IIB, and IIX), and the myofiber boundary (indicated by anti-Laminin immunostaining). e. An intersection is created between the Cy5 and FITC channels, by using the binary operations dialog box to identify IIA fibers. f. An intersection is created between the Cy5 and TRITC channels, using the binary operations dialog box to identify IIB fibers. g. A subtraction is created between the Cy5 and FITC channels, using the binary operations dialog box. The step is repeated, using the newly created subtraction and the TRITC channel. Non-stained, presumed IIX fibers are detected. After steps e–g, the resulting data is exported to Excel for analysis.Average Feret’s Minimal Diameter and Percentages of Fibers Analysis using Excel and GraphPad Prism software • Timing ~15 min per sample group (protocol #1)
Add a new sheet to the Excel file with the Nikon Elements analysis data. Create two tables, one for the average Feret’s minimal diameter (FMD), and one for the percentage of fibers. Label the columns with the fiber types (IIA, IIB, and IIX), and the rows with the sample name.
To determine the FMD for each fiber type per sample, take the average of the Feret’s minimal diameter from the exported data. This analysis can also be performed for the cross-sectional area (CSA) of each fiber, by averaging the total area number recorded previously.
To obtain the percentage of fibers, count the total number of fibers for each fiber type per sample from the exported data. Sum the fiber counts (IIA, IIB, and IIX) together for the sample, and divide the individual fiber type count by the sum. Multiply that number by 100 to display the data in percentage.
Graph and analyze the results using GraphPad Prism, or an equivalent graphing software, by creating a new Grouped table, and selecting to “enter number of replicates in side-by-side sub columns”. Title each column as the name of the sample group, and label each row with the fiber type. The number of replicates corresponds to the number of samples per experiment group.
Copy the average FMD or percentage of fibers data calculated in the Excel file, and paste into the corresponding data table.
Graph the data as an individual interleaved scatter plot, and measure the variability by using the mean with standard deviation (SD).
Analyze the data using the 2-way ANOVA test under grouped analyses, to observe significant changes in fiber size or percentage of fibers. Change the parameters by selecting “within each row, compare columns” under the multiple comparisons tab.
Histogram Analysis using GraphPad Prism of the Feret’s Minimal Diameter data • Timing ~20 min (protocol #1)
Open the GraphPad Prism software and create a new Column table, choosing to “enter replicate values, stacked into columns”. Title each column as the name of the sample group, and duplicate the table for each additional fiber type.
For each fiber and sample, copy all the FMD data from the Nikon Elements Excel file and paste into the corresponding data tables.
Using the analyze function, select the frequency distribution option under the Column analyses. Change the parameters to “relative frequency (percentages)” and adjust the bin width and range accordingly. Graph the results as an interleaved bar graph.
Analyze the frequency distribution results, and select non-linear regression (curve fit) under the XY analyses. Under the parameters, use the Gaussian equation, keeping the default settings. This analysis can also be performed for the CSA of the fibers, using the “Area” data from the Nikon Elements Excel file.
C2C12 Cell Culture • Timing ~several days for growth and ~1 h for cell splitting (protocol #2)
Grow the cells in 10% (v/v) FBS DMEM + GlutaMax media at 37°C with 5% (v/v) CO2. When the cells reach 80% confluence, split them, and prepare plates for transfection.
To split the cells, remove the media from the flask, and wash twice with 1× PBS.
CRITICAL: Avoid adding PBS directly to the cells, as they could detach and be lost.
Detach the cells using 0.25% Trypsin-0.53mM EDTA (w/v) for 3 min at 37°C. To ensure detachment, tap the sides of the flask, and visualize under the microscope.
Neutralize the trypsinization by adding 10 mL of DMEM+10% (v/v) FBS back to the flask. Collect the cells in a 50-mL conical tube and centrifuge the cells at 2,000 × g for 10 min. To keep the cells growing, add 20 mL of 10% FBS media back to the flask, and place in the incubator.
Observe the pellet after centrifugation and aspirate the remaining media. Resuspend the pellet with 3 mL of growth media, adding 1 mL at a time, to ensure proper resuspension.
Count the cells by adding 10 µL of resuspended cells to both chambers of a hemocytometer. Visualize the central area of the chamber (1 mm2) under the microscope, and count the cells with a cell counter. Repeat the step for both chambers of the hemocytometer and average the two counts.
CRITICAL: Multiply the average count by the conversion factor 10,000 or 104 cells.
Prepare the appropriate amount of 6-well cell culture plates, depending on the number of siRNAs to be tested. Using the total cell number, calculate the volume needed from the suspension to have a final concentration of 20,000 cells/mL per well, with a total well volume of 2 mL.
CRITICAL: A minimum of 3 wells per siRNA are needed. Also include a siRNA-treated plate (one siRNA per well) for staining, imaging, and analyzing myotube diameter size.
Prepare a mixture of DMEM growth media and resuspended cells, and add the appropriate volume to the cell culture plates. Incubate the plates in 5% (v/v) CO2 at 37°C. Replace the growth media every other day, being careful not to detach the cells.
When the myoblasts reach 80–100% confluence, differentiate the cells to myotubes, by replacing the 10% (v/v) FBS growth media with 2% (v/v) HS differentiation media.
After four days of differentiation, treat the cells with Ara-c for two days in 10% FBS growth media, to kill any remaining myoblasts. The concentration of Ara-c on the first day of treatment should be 4.0 µg/mL, and 0.4 µg/mL on the second day.
Myotube siRNA Transfection • Timing ~45 min (protocol #2)
Prior to transfecting the cells, pre-warm Opti-MEM to 22°C and thaw the appropriate siRNAs on ice. Calculate the amount of reagent needed to have a total volume of 200 µL Opti-MEM, 8 µL of Lipofectamine 2000, and 8 µL of 50 µM siRNA per well.
CRITICAL: Include a non-targeting (NT) siRNA to use as control. The mixture volumes for Opti-MEM, Lipofectamine 2000, and siRNA can be adjusted based on plate size.
Dilute 8 µL of 50 µM siRNAs into 100 µL of Opti-MEM, and mix gently by pipetting. Repeat this step by adding 8 µL of Lipofectamine 2000 into 100 µL of Opti-MEM. Combine the two solutions, pipetting to mix, and incubate for 10 min.
During the incubation period, remove the media on the prepared 6-well plates, and add 1.8 mL of fresh 10% (v/v) FBS growth media.
After 10 min, add 200 µL of the transfection mixture to each well, and mix gently by rocking the plate back and forth. Incubate the plates with 5% (v/v) CO2 at 37°C for 48 h.
After 24 h, change the media to no serum media (containing 1% P/S).
To check the knockdown induced by the siRNAs, perform an RNA extraction 48 h post transfection, by using the Invitrogen TRIzol reagent and accompanying protocol. Run a qRT-PCR with mRNA-specific oligonucleotides, to quantify the knockdown of the siRNA-targeted mRNA, using the NT siRNA-treated samples as control.
Myotube Immunostaining and Imaging • Timing ~4 h for staining and ~1 h for imaging (protocol #2)
Add 1 mL of 4% PFA to each mL of culture media, and incubate for 10 min at room temperature (the final PFA concentration is 2%).
CRITICAL: Add the solution to the side of the well to avoid lifting the cells. For the same reason, the fixing solution is added directly to the cell culture medium.
Remove the PFA solution and wash the wells three times with 1× PBS. Aspirate the remaining 1× PBS, and block the wells by adding 1 mL of 2% BSA blocking buffer (2% BSA Triton X-100). Leave the plate gently shaking for 1 h at room temperature.
After blocking the wells, add 300 µL of the primary antibody solution, and gently shake the plate overnight at 4°C.
The following day, remove the antibody, and wash the wells three times with 1× PBS. Add 300 µL of the secondary antibody solution for 2 h at room temperature, gently shaking.
Wash the wells once with 1× PBS, then replace with 1 mL of fresh 1× PBS and store at 4°C, protecting the plate from light, until ready for imaging.
PAUSE POINT: The cells can remain in PBS for ~1 month, but the signal intensity will decline.
Image the plate using a fluorescent microscope with access to a 532-nm laser channel (TRITC). Using the 10× objective, image each well individually for further analysis.
CRITICAL: Image a hemocytometer with the brightfield setting, using the same objective as a scale (Figure 5).

Figure 5. Outcome of siRNA electroporation into C2C12 myoblasts and myotubes. a. Representative images of C2C12 myoblasts transfected with siGLO red demonstrate cellular uptake of the fluorescent siRNAs, whereas no fluorescence is seen in myoblasts transfected with non-fluorescent NT siRNAs. b. Similar results are found with transfection of siGLO red into C2C12 myotubes. Data in a–b is reproduced from Hunt LC et al. Cell Reports (2019).Myotube Diameter Measurement •Timing ~30 min per image (protocol #2)
Open ImageJ. Prior to measuring the myotubes, open the hemocytometer image. Using the straight-line tool, draw a line inside one of the 25 smaller squares (0.25 mm) located in the four corners of the hemocytometer.
Select the set scale option from the Analyze pulldown menu, and enter the size of the square into the “known distance” box as “250” and the unit of length as “µm”. Check the “global” option, and press OK. The scale should be 0.648pixels/µm.
CRITICAL: All images need to be set to the same scale for accurate analysis.
Open the stitched myotube image and zoom in on individual myotubes. Using the straight-line tool, draw three lines across the diameter, to indicate the beginning, middle, and end of each myotube. After each line, select the measurement option from the Analyze pulldown menu.
Repeat this until a minimum of 50 myotubes per group have been measured. Export the measurement data to Excel.
To determine the myotube diameter, average the three measurements per myotube. Graph the average of each myotube, compared to control, in GraphPad Prism or an equivalent analysis software. Perform appropriate statistical tests to determine whether a significant effect in myotube diameter size is induced by siRNAs (Figure 6).
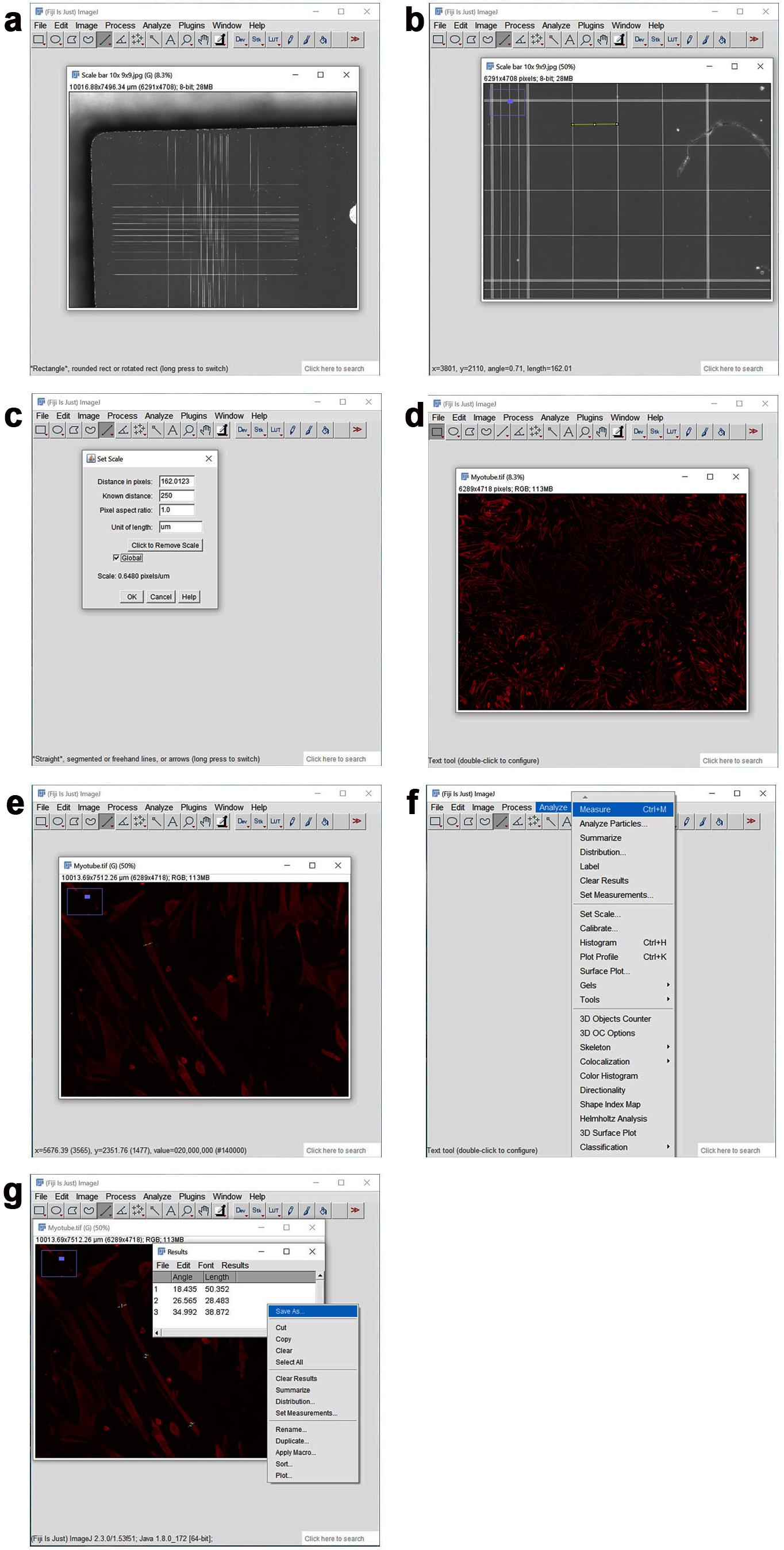
Figure 6. Determination of C2C12 myotube diameter with the ImageJ analysis software. a. The hemocytometer image is opened in ImageJ to establish a scale. b. By using the straight line tool, a line is drawn inside one of the 25 small squares located in one of the four corners of the hemocytometer. c. The set scale option is selected from the Analyze pulldown menu, and 250 μm is entered as the known pixel distance and unit, respectively. The global option is selected to set the scale as 0.648 pixels/μm. d. The stitched myotube image is opened. e. Individual myotubes are zoomed in. By using the straight line tool, three lines are drawn horizontally across the width of the myotube at the two ends, and in the middle of the myotube. f. After each straight line is drawn, the measurement option is selected from the Analyze pulldown menu. g. A minimum of 50 myotubes are measured, and the data is exported to Excel. The three measurements per myotube are averaged to determine the mean diameter size.Anticipated results. With the analyses reported above, TA muscles with a target gene knockdown can be analyzed to determine the resulting impact on myofiber size and myofiber type composition, but also for determining mRNA and protein changes, via follow-up applications such as RNA sequencing and western blot analysis of detergent-soluble and insoluble fractions (Rai et al., 2021b; Hunt et al., 2021a). To analyze myofiber size, muscle cryosections are stained with an anti-laminin antibody, to delineate the boundaries of all myofibers, and antibodies specific for myosin heavy chain isoforms are used to identify distinct myofiber types present in the TA muscle, i.e., fast glycolytic type 2B or IIB myofibers (MHC- 2B- positive) and smaller, more oxidative, type 2A or IIA myofibers (MHC-2A-positive), and 2X or IIX myofibers (MHC-2A-negative and 2B-negative), as previously done (Hunt et al., 2015, 2021a, 2021b). With these analyses, it is possible to determine whether siRNA-mediated knockdown of a target mRNA in TA muscles leads to changes in myofiber size and in the proportion of distinct myofiber types (Hunt et al., 2015, 2021a, 2021b). When measuring myofiber size in electroporated TA muscles, the Feret’s minimal diameter can be used, as this geometrical parameter enables reliable measurements of myofiber cross-sectional areas, unhindered by cellular distortion introduced by cryosectioning (Bloemberg and Quadrilatero, 2012). Similarly, estimation of the size of cultured C2C12 myotubes transfected with siRNAs provides insight into whether the siRNA-targeted mRNA has a general role in myotube size determination, compared to control NT siRNA transfection [however, myofiber type analysis is routinely not done in cell culture (Huang et al., 2021)]. Hunt et al. (2019) provides an example of similar results obtained with knockdown of the same protein in cultured C2C12 myotubes, and in mouse TA skeletal muscles in vivo: in both cases, knockdown of the E3 ubiquitin ligase UBR4 induces myofiber hypertrophy (Hunt et al., 2019).
Beyond these anticipated results, the protocol reported above can be coupled with the immunostaining of informative antigens and additional biochemical/molecular assays, to determine whether the acute knockdown of a target mRNA impacts any aspects of muscle homeostasis. This can consist in the estimation of myofiber size, when studying disease-associated muscle wasting, and in the determination of the abundance of organelles or markers associated with optimal muscle function or with muscle disease. In addition to determining cellular and molecular features, TA muscles that have been electroporated can also be analyzed functionally, to determine whether siRNAs targeting certain mRNAs lead to corresponding changes in the isometric and tetanic force (Hunt et al., 2019, 2021b).
Lastly, this method can overcome logistic limitations of other technical approaches. For example, the study of sarcopenia (i.e., the age-associated decline in skeletal muscle mass and strength) has been hindered by the relatively long time necessary to obtain old mice of a desired genotype. The protocol and optimized conditions reported here for siRNA electroporation into TA muscles may help overcome this limitation. Specifically, we propose that this approach may provide a rapid and robust method for achieving target gene knockdown in the TA muscles of old wild-type mice, and hence for rapidly assessing gene function in the context of sarcopenia.
Recipes
Reagent setup (protocol #1 and #2)
10% Tragacanth
Mix 10 mg of tragacanth gum in 100 mL of distilled water. Store at 4°C for two weeks.
Hyaluronidase stock solution
Mix 10 mg of hyaluronidase powder in 1 mL of 1× PBS. Store at -20°C for up to 1 year.
Hyaluronidase working solution
Dilute stock solution with 1× PBS to a final concentration of ~0.4 units/mL. Store at -20°C for up to 6 months.
50 μM siRNA stock
Resuspend 20 nmol siRNAs in 1× siRNA buffer for a final concentration of 50 μM. Store at -20°C for up to 6 months.
2% BSA blocking buffer
Mix 0.4 g of BSA powder (2% (wt/vol) final concentration) and 20 μL of Triton X-100 (0.1% (vol/vol) final concentration) in 20 mL of 1× PBS. Store at 4°C for up to 1 month.
Primary antibody staining solution (frozen slides)
Dilute primary antibody to a final concentration of 1:150 in 1× PBST (for 1.5 mL: 1.5 mL PBS, 1.5 μL Tween-20, 10 μL of primary antibody)
Secondary antibody staining solution (frozen slides)
Dilute secondary antibody to a final concentration of 1:200 in 1× PBST (for 2 mL: 2 mL PBS, 2 μL Tween-20, 10 μL of secondary antibody)
10% Fetal Bovine Serum (FBS)
Mix 50 mL of FBS with 500 mL DMEM + GlutaMax media.
2% Horse Serum (HS)
Mix 10 mL of HS with 500 mL DMEM + GlutaMax media.
1% Penicillin/Streptomycin
Add 5.55 mL of P/S to 550 mL DMEM + GlutaMax serum media.
4% PFA
Dilute 16% PFA with 1x PBS for a final concentration of 4% (4 mL of 16% PAF and 12 mL of PBS).
Primary antibody staining solution (cell culture myotubes)
Dilute primary antibody to a final concentration of 1:200 in 1× PBST (for 2 mL: 2 mL PBS, 2 μL Tween-20, 10 μL of primary antibody).
Secondary antibody staining solution (cell culture myotubes)
Dilute secondary antibody to a final concentration of 1:400 in 1× PBST (for 2 mL: 2 mL PBS, 2 μL Tween-20, 5 μL of secondary antibody).
Acknowledgments
Schemes were drawn with BioRender. The anti-MHC antibodies used to identify myofiber types were obtained from the Developmental Studies Hybridoma Bank. This work was supported by research grants to F.D. from the National Institute on Aging (R01AG055532 and R56AG63806). Research at St. Jude Children’s Research Hospital is supported by the American Lebanese Syrian Associated Charities (ALSAC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Author contributions: A.S. and F.D. wrote the manuscript. F.A.G. and L.C.H. provided feedback. All authors read and edited the manuscript.
Competing interests
The authors declare no competing interests.
Ethics
All experiments were performed in accordance with federal and local regulations. The St. Jude Children’s Research Hospital (SJCRH) animal care and use committee (IACUC) approved all protocols performed. Animals were housed in a ventilated, temperature-controlled facility in the Animal Resource Center at SJCRH in Memphis, Tennessee, USA.
References
- Aihara, H. and Miyazaki, J. (1998). Gene transfer into muscle by electroporation in vivo. Nat Biotechnol 16(9): 867-870.
- Bertrand, A., Ngo-Muller, V., Hentzen, D., Concordet, J. P., Daegelen, D. and Tuil, D. (2003). Muscle electrotransfer as a tool for studying muscle fiber-specific and nerve-dependent activity of promoters. Am J Physiol Cell Physiol 285(5): C1071-1081.
- Bloemberg, D. and Quadrilatero, J. (2012). Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PloS One 7(4): e35273.
- Bonaldo, P. and Sandri, M. (2013). Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6(1): 25-39.
- Brolin, C., Shiraishi, T., Hojman, P., Krag, T. O., Nielsen, P. E. and Gehl, J. (2015). Electroporation Enhanced Effect of Dystrophin Splice Switching PNA Oligomers in Normal and Dystrophic Muscle. Mol Ther Nucleic Acids 4: e267.
- Buffenstein, R. (2005). The naked mole-rat: a new long-living model for human aging research. J Gerontol A Biol Sci Med Sci 60(11): 1369-1377.
- Burnett, C., Valentini, S., Cabreiro, F., Goss, M., Somogyvari, M., Piper, M. D., Hoddinott, M., Sutphin, G. L., Leko, V., McElwee, J. J., et al. (2011). Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477(7365): 482-485.
- Ciciliot, S., Rossi, A. C., Dyar, K. A., Blaauw, B. and Schiaffino, S. (2013). Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 45(10): 2191-2199.
- Cotta, A., Carvalho, E., da-Cunha-Junior, A., Navarro, M. M., Paim, J. F., Valicek, J., Baptista-Junior, S., da Silveira, E. B., Lima, M. I., Carellos, E. V. M., et al. (2021). Muscle fat replacement and modified ragged red fibers in two patients with reversible infantile respiratory chain deficiency. Neuromuscul Disord 31(6): 551-557.
- Dean, D. A. (2013). Cell-specific targeting strategies for electroporation-mediated gene delivery in cells and animals. J Membr Biol 246(10): 737-744.
- Decker, R. E., Lamantia, Z. E., Emrick, T. S. and Figueiredo, M. L. (2020). Sonodelivery in Skeletal Muscle: Current Approaches and Future Potential. Bioengineering (Basel) 7(3): 107.
- Demontis, F., Patel, V. K., Swindell, W. R. and Perrimon, N. (2014). Intertissue control of the nucleolus via a myokine-dependent longevity pathway. Cell Rep 7(5): 1481-1494.
- Demontis, F. and Perrimon, N. (2009). Integration of Insulin receptor/Foxo signaling and dMyc activity during muscle growth regulates body size in Drosophila. Development 136(6): 983-993.
- Demontis, F. and Perrimon, N. (2010). FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell 143(5): 813-825.
- Demontis, F., Piccirillo, R., Goldberg, A. L. and Perrimon, N. (2013a). The influence of skeletal muscle on systemic aging and lifespan. Aging Cell 12(6): 943-949.
- Demontis, F., Piccirillo, R., Goldberg, A. L. and Perrimon, N. (2013b). Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis Model Mech 6(6): 1339-1352.
- Doudna, J. A. and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213): 1258096.
- Ebert, S. M., Dyle, M. C., Bullard, S. A., Dierdorff, J. M., Murry, D. J., Fox, D. K., Bongers, K. S., Lira, V. A., Meyerholz, D. K., Talley, J. J., et al. (2015). Identification and Small Molecule Inhibition of an Activating Transcription Factor 4 (ATF4)-dependent Pathway to Age-related Skeletal Muscle Weakness and Atrophy. J Biol Chem 290(42): 25497-25511.
- Edupuganti, S., S, C. D. R., Elizaga, M., Lu, Y., Han, X., Huang, Y., Swann, E., Polakowski, L., S, A. K., Keefer, M., Maenza, J., M, C. W., Yan, J., et al. (2020). Intramuscular and Intradermal Electroporation of HIV-1 PENNVAX-GP((R)) DNA Vaccine and IL-12 Is Safe, Tolerable, Acceptable in Healthy Adults. Vaccines (Basel) 8(4): 741.
- Gaj, T., Gersbach, C. A. and Barbas, C. F., 3rd (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering.Trends Biotechnol 31(7): 397-405.
- Golzio, M., Escoffre, J. M. and Teissie, J. (2012). shRNA-mediated gene knockdown in skeletal muscle. Methods Mol Biol 798: 491-501.
- Graca, F. A., Sheffield, N., Puppa, M., Finkelstein, D., Hunt, L. C. and Demontis, F. (2021). A large-scale transgenic RNAi screen identifies transcription factors that modulate myofiber size in Drosophila. PLoS Genet 17(11): e1009926.
- Haidari, G., Day, S., Wood, M., Ridgers, H., Cope, A. V., Fleck, S., Yan, C., Reijonen, K., Hannaman, D., Spentzou, A., et al. (2019). The Safety and Immunogenicity of GTU((R))MultiHIV DNA Vaccine Delivered by Transcutaneous and Intramuscular Injection With or Without Electroporation in HIV-1 Positive Subjects on Suppressive ART. Front Immunol 10: 2911.
- Hoover, F. and Magne Kalhovde, J. (2000). A double-injection DNA electroporation protocol to enhance in vivo gene delivery in skeletal muscle. Anal Biochem 285(1): 175-178.
- Hou, J., Tan, G., Fink, G. R., Andrews, B. J. and Boone, C. (2019). Complex modifier landscape underlying genetic background effects. Proc Natl Acad Sci U S A 116(11): 5045-5054.
- Huang, B., Jiao, Y., Zhu, Y., Ning, Z., Ye, Z., Li, Q. X., Hu, C. and Wang, C. (2021). Mdfi Promotes C2C12 Cell Differentiation and Positively Modulates Fast-to-Slow-Twitch Muscle Fiber Transformation. Front Cell Dev Biol 9: 605875.
- Hunt, L. C., Graca, F. A., Pagala, V., Wang, Y. D., Li, Y., Yuan, Z. F., Fan, Y., Labelle, M., Peng, J. and Demontis, F. (2021a). Integrated genomic and proteomic analyses identify stimulus-dependent molecular changes associated with distinct modes of skeletal muscle atrophy. Cell Rep 37(6): 109971.
- Hunt, L. C., Schadeberg, B., Stover, J., Haugen, B., Pagala, V., Wang, Y. D., Puglise, J., Barton, E. R., Peng, J. and Demontis, F. (2021b). Antagonistic control of myofiber size and muscle protein quality control by the ubiquitin ligase UBR4 during aging. Nat Commun 12(1): 1418.
- Hunt, L. C., Stover, J., Haugen, B., Shaw, T. I., Li, Y., Pagala, V. R., Finkelstein, D., Barton, E. R., Fan, Y., Labelle, M., et al. (2019). A Key Role for the Ubiquitin Ligase UBR4 in Myofiber Hypertrophy in Drosophila and Mice. Cell Rep 28(5): 1268-1281 e1266.
- Hunt, L. C., Xu, B., Finkelstein, D., Fan, Y., Carroll, P. A., Cheng, P. F., Eisenman, R. N. and Demontis, F. (2015). The glucose-sensing transcription factor MLX promotes myogenesis via myokine signaling. Genes Dev 29(23): 2475-2489.
- Jiang, F. and Doudna, J. A. (2017). CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys 46: 505-529.
- Johnston, A. J., Murphy, K. T., Jenkinson, L., Laine, D., Emmrich, K., Faou, P., Weston, R., Jayatilleke, K. M., Schloegel, J., Talbo, G., et al. (2015). Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 162(6): 1365-1378.
- Joseph, B., Corwin, J. A., Li, B., Atwell, S. and Kliebenstein, D. J. (2013). Cytoplasmic genetic variation and extensive cytonuclear interactions influence natural variation in the metabolome. Elife 2: e00776.
- Joyce, N. C., Oskarsson, B. and Jin, L. W. (2012). Muscle biopsy evaluation in neuromuscular disorders. Phys Med Rehabil Clin N Am 23(3): 609-631.
- Khan, A. S., Broderick, K. E. and Sardesai, N. Y. (2014). Clinical development of intramuscular electroporation: providing a "boost" for DNA vaccines. Methods Mol Biol 1121: 279-289.
- Kim, D., Hofstaedter, C. E., Zhao, C., Mattei, L., Tanes, C., Clarke, E., Lauder, A., Sherrill-Mix, S., Chehoud, C., Kelsen, J., Conrad, M., Collman, R. G., Baldassano, R., Bushman, F. D. and Bittinger, K. (2017). Optimizing methods and dodging pitfalls in microbiome research. Microbiome 5(1): 52.
- Koh, G., Zou, X. and Nik-Zainal, S. (2020). Mutational signatures: experimental design and analytical framework. Genome Biol 21(1): 37.
- Krishna, S., Arrojo, E. D. R., Capitanio, J. S., Ramachandra, R., Ellisman, M. and Hetzer, M. W. (2021). Identification of long-lived proteins in the mitochondria reveals increased stability of the electron transport chain. Dev Cell 56(21): 2952-2965 e2959.
- Lucanic, M., Plummer, W. T., Chen, E., Harke, J., Foulger, A. C., Onken, B., Coleman-Hulbert, A. L., Dumas, K. J., Guo, S., Johnson, E., et al. (2017). Impact of genetic background and experimental reproducibility on identifying chemical compounds with robust longevity effects. Nat Commun 8: 14256.
- Mamantopoulos, M., Ronchi, F., Van Hauwermeiren, F., Vieira-Silva, S., Yilmaz, B., Martens, L., Saeys, Y., Drexler, S. K., et al. (2017). Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 47(2): 339-348 e334.
- Manini, A., Abati, E., Nuredini, A., Corti, S. and Comi, G. P. (2021). Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence. Front Neurol 12: 814174.
- McCarthy, J. J., Srikuea, R., Kirby, T. J., Peterson, C. A. and Esser, K. A. (2012). Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skeletal muscle 2(1): 8.
- McMahon, J. M., Signori, E., Wells, K. E., Fazio, V. M. and Wells, D. J. (2001). Optimisation of electrotransfer of plasmid into skeletal muscle by pretreatment with hyaluronidase -- increased expression with reduced muscle damage. Gene therapy 8(16): 1264-1270.
- McMahon, J. M. and Wells, D. J. (2004). Electroporation for gene transfer to skeletal muscles: current status. BioDrugs 18(3): 155-165.
- Meng, H., Janssen, P. M., Grange, R. W., Yang, L., Beggs, A. H., Swanson, L. C., Cossette, S. A., Frase, A., Childers, M. K., Granzier, H., et al. (2014). Tissue triage and freezing for models of skeletal muscle disease. J Vis Exp (89): 51586.
- Menzies, F. M., Hourez, R., Imarisio, S., Raspe, M., Sadiq, O., Chandraratna, D., O'Kane, C., Rock, K. L., Reits, E., Goldberg, A. L., et al. (2010). Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Hum Mol Genet 19(23): 4573-4586.
- Mizui, M., Isaka, Y., Takabatake, Y., Mizuno, S., Nakamura, T., Ito, T., Imai, E. and Hori, M. (2004). Electroporation-mediated HGF gene transfer ameliorated cyclosporine nephrotoxicity. Kidney Int 65(6): 2041-2053.
- Mpendo, J., Mutua, G., Nanvubya, A., Anzala, O., Nyombayire, J., Karita, E., Dally, L., Hannaman, D., Price, M., Fast, P. E., et al. (2020). Acceptability and tolerability of repeated intramuscular electroporation of Multi-antigenic HIV (HIVMAG) DNA vaccine among healthy African participants in a phase 1 randomized controlled trial. PLoS One 15(5): e0233151.
- Murgia, M., Serrano, A. L., Calabria, E., Pallafacchina, G., Lomo, T. and Schiaffino, S. (2000). Ras is involved in nerve-activity-dependent regulation of muscle genes. Nat Cell Biol 2(3): 142-147.
- Nair, K. S. (2005). Aging muscle. Am J Clin Nutr 81(5): 953-963.
- Neumeier, J. and Meister, G. (2020). siRNA Specificity: RNAi Mechanisms and Strategies to Reduce Off-Target Effects. Front Plant Sci 11: 526455.
- Peng, B., Zhao, Y., Lu, H., Pang, W. and Xu, Y. (2005). In vivo plasmid DNA electroporation resulted in transfection of satellite cells and lasting transgene expression in regenerated muscle fibers.Biochem Biophys Res Commun 338(3): 1490-1498.
- Piccirillo, R., Demontis, F., Perrimon, N. and Goldberg, A. L. (2014). Mechanisms of muscle growth and atrophy in mammals and Drosophila.Dev Dyn (2) (243): 201-215.
- Poussin, C., Sierro, N., Boue, S., Battey, J., Scotti, E., Belcastro, V., Peitsch, M. C., Ivanov, N. V. and Hoeng, J. (2018). Interrogating the microbiome: experimental and computational considerations in support of study reproducibility. Drug Discov Today 23(9): 1644-1657.
- Puppa, M. J., Gao, S., Narsale, A. A. and Carson, J. A. (2014). Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma-induced cachexia. FASEB J 28(2): 998-1009.
- Rai, M., Coleman, Z., Curley, M., Nityanandam, A., Platt, A., Robles-Murguia, M., Jiao, J., Finkelstein, D., Wang, Y. D., Xu, B., Fan, Y. and Demontis, F. (2021a). Proteasome stress in skeletal muscle mounts a long-range protective response that delays retinal and brain aging. Cell Metab 33(6):1137-1154.e9.
- Rai, M., Curley, M., Coleman, Z., Nityanandam, A., Jiao, J., Graca, F. A., Hunt, L. C. and Demontis, F. (2021b). Analysis of proteostasis during aging with western blot of detergent-soluble and insoluble protein fractions. STAR Protoc 2(3): 100628.
- Rai, M. and Demontis, F. (2016). Systemic Nutrient and Stress Signaling via Myokines and Myometabolites. Annu Rev Physiol 78: 85-107.
- Rizzuto, G., Cappelletti, M., Maione, D., Savino, R., Lazzaro, D., Costa, P., Mathiesen, I., Cortese, R., Ciliberto, G., Laufer, R., La Monica, N. and Fattori, E. (1999). Efficient and regulated erythropoietin production by naked DNA injection and muscle electroporation. Proc Natl Acad Sci U S A 96(11): 6417-6422.
- Robles-Murguia, M., Rao, D., Finkelstein, D., Xu, B., Fan, Y. and Demontis, F. (2020). Muscle-derived Dpp regulates feeding initiation via endocrine modulation of brain dopamine biosynthesis. Genes Dev 34(1-2): 37-52.
- Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Holper, S., Kruger, M. and Stainier, D. Y. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524(7564): 230-233.
- Rowland, N. E. (2007). Food or fluid restriction in common laboratory animals: balancing welfare considerations with scientific inquiry. Comp Med 57(2): 149-160.
- Sandri, M., Bortoloso, E., Nori, A. and Volpe, P. (2003). Electrotransfer in differentiated myotubes: a novel, efficient procedure for functional gene transfer. Exp Cell Res 286(1): 87-95.
- Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., Walsh, K., Schiaffino, S., Lecker, S. H. and Goldberg, A. L. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117(3): 399-412.
- Savas, J. N., Toyama, B. H., Xu, T., Yates, J. R., 3rd and Hetzer, M. W. (2012). Extremely long-lived nuclear pore proteins in the rat brain. Science 335(6071): 942.
- Schertzer, J. D., Plant, D. R. and Lynch, G. S. (2006). Optimizing plasmid-based gene transfer for investigating skeletal muscle structure and function. Mol Ther 13(4): 795-803.
- Schiaffino, S., Dyar, K. A., Ciciliot, S., Blaauw, B. and Sandri, M. (2013). Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280(17): 4294-4314.
- Schiaffino, S. and Reggiani, C. (2011). Fiber types in mammalian skeletal muscles. Physiol Rev 91(4): 1447-1531.
- Setten, R. L., Rossi, J. J. and Han, S. P. (2019). The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov 18(6): 421-446.
- Shavlakadze, T., McGeachie, J. and Grounds, M. D. (2010). Delayed but excellent myogenic stem cell response of regenerating geriatric skeletal muscles in mice. Biogerontology 11(3): 363-376.
- Sheard, P. W. and Anderson, R. D. (2012). Age-related loss of muscle fibres is highly variable amongst mouse skeletal muscles. Biogerontology 13(2): 157-167.
- Skuk, D., Goulet, M. and Tremblay, J. P. (2013). Electroporation as a method to induce myofiber regeneration and increase the engraftment of myogenic cells in skeletal muscles of primates. J Neuropathol Exp Neurol 72(8): 723-734.
- Sopko, R., Foos, M., Vinayagam, A., Zhai, B., Binari, R., Hu, Y., Randklev, S., Perkins, L. A., Gygi, S. P. and Perrimon, N. (2014). Combining genetic perturbations and proteomics to examine kinase-phosphatase networks in Drosophila embryos. Dev Cell 31(1): 114-127.
- Tevz, G., Pavlin, D., Kamensek, U., Kranjc, S., Mesojednik, S., Coer, A., Sersa, G. and Cemazar, M. (2008). Gene electrotransfer into murine skeletal muscle: a systematic analysis of parameters for long-term gene expression. Technol Cancer Res Treat 7(2): 91-101.
- Toivonen, J. M., Walker, G. A., Martinez-Diaz, P., Bjedov, I., Driege, Y., Jacobs, H. T., Gems, D. and Partridge, L. (2007). No influence of Indy on lifespan in Drosophila after correction for genetic and cytoplasmic background effects. PLoS Genet 3(6): e95.
- Toyama, B. H., Savas, J. N., Park, S. K., Harris, M. S., Ingolia, N. T., Yates, J. R., 3rd and Hetzer, M. W. (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154(5): 971-982.
- Tsoli, M. and Robertson, G. (2013). Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab 24(4): 174-183.
- Ugen, K. E. and Heller, R. (2003). Electroporation as a method for the efficient in vivo delivery of therapeutic genes. DNA Cell Biol 22(12): 753.
- Ulgherait, M., Chen, A., Oliva, M. K., Kim, H. X., Canman, J. C., Ja, W. W. and Shirasu-Hiza, M. (2016). Dietary Restriction Extends the Lifespan of Circadian Mutants tim and per. Cell Metab 24(6): 763-764.
- Ungerer, M. C., Linder, C. R. and Rieseberg, L. H. (2003). Effects of genetic background on response to selection in experimental populations of Arabidopsis thaliana. Genetics 163(1): 277-286.
- Valenzano, D. R., Aboobaker, A., Seluanov, A. and Gorbunova, V. (2017). Non-canonical aging model systems and why we need them.EMBO J 36(8): 959-963.
- van Groen, T., Kadish, I., Popovic, N., Popovic, M., Caballero-Bleda, M., Bano-Otalora, B., Vivanco, P., Rol, M. A. and Madrid, J. A. (2011). Age-related brain pathology in Octodon degu: blood vessel, white matter and Alzheimer-like pathology. Neurobiol Aging 32(9): 1651-1661.
- Vandermeulen, G., Uyttenhove, C., De Plaen, E., Van den Eynde, B. J. and Preat, V. (2014). Intramuscular electroporation of a P1A-encoding plasmid vaccine delays P815 mastocytoma growth. Bioelectrochemistry 100: 112-118.
- Vujkovic-Cvijin, I., Sklar, J., Jiang, L., Natarajan, L., Knight, R. and Belkaid, Y. (2020). Host variables confound gut microbiota studies of human disease. Nature 587(7834): 448-454.
- Widera, G., Austin, M., Rabussay, D., Goldbeck, C., Barnett, S. W., Chen, M., Leung, L., Otten, G. R., Thudium, K., Selby, M. J. and Ulmer, J. B. (2000). Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J Immunol 164(9): 4635-4640.
- Winbanks, C. E., Murphy, K. T., Bernardo, B. C., Qian, H., Liu, Y., Sepulveda, P. V., Beyer, C., Hagg, A., Thomson, R. E., Chen, J. L., et al. (2016). Smad7 gene delivery prevents muscle wasting associated with cancer cachexia in mice. Sci Transl Med 8(348): 348ra398.
- Wolfe, R. R. (2006). The underappreciated role of muscle in health and disease. Am J Clin Nutr 84(3): 475-482.
- Wong, S. H., Lowes, K. N., Quigley, A. F., Marotta, R., Kita, M., Byrne, E., Kornberg, A. J., Cook, M. J. and Kapsa, R. M. (2005). DNA electroporation in vivo targets mature fibres in dystrophic mdx muscle. Neuromuscul Disord 15(9-10): 630-641.
- Young, J. L. and Dean, D. A. (2015). Electroporation-mediated gene delivery. Adv Genet 89: 49-88.
- Zhou, X., Wang, J. L., Lu, J., Song, Y., Kwak, K. S., Jiao, Q., Rosenfeld, R., Chen, Q., Boone, T., Simonet, W. S., et al. (2010). Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142(4): 531-543.
- Zucchelli, S., Capone, S., Fattori, E., Folgori, A., Di Marco, A., Casimiro, D., Simon, A. J., Laufer, R., La Monica, N., Cortese, R., et al. (2000). Enhancing B- and T-cell immune response to a hepatitis C virus E2 DNA vaccine by intramuscular electrical gene transfer. J Virol 74(24): 11598-11607.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Stephan, A., Graca, F. A., Hunt, L. C. and Demontis, F. (2022). Electroporation of Small Interfering RNAs into Tibialis Anterior Muscles of Mice. Bio-protocol 12(11): e4428. DOI: 10.21769/BioProtoc.4428.
Category
Cell Biology > Tissue analysis > Electroporation
Developmental Biology > Cell signaling
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.