- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Optimized CRISPR-Cas9-based Strategy for Complex Gene Targeting in Murine Embryonic Stem Cells for Germline Transmission

Published: Vol 12, Iss 10, May 20, 2022 DOI: 10.21769/BioProtoc.4423 Views: 3672
Reviewed by: Giusy TornilloNingfei AnVishal NehruAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Although CRISPR-Cas9 genome editing can be performed directly in single-cell mouse zygotes, the targeting efficiency for more complex modifications such as the insertion of two loxP sites, multiple mutations in cis, or the precise insertion or deletion of longer DNA sequences often remains low (Cohen, 2016). Thus, targeting and validation of correct genomic modification in murine embryonic stem cells (ESCs) with subsequent injection into early-stage mouse embryos may still be preferable, allowing for large-scale screening in vitro before transfer of thoroughly characterized and genetically defined ESC clones into the germline. This procedure can result in a reduction of animal numbers with cost effectiveness and compliance with the 3R principle of animal welfare regulations. Here, we demonstrate that after transfection of homology templates and PX458 CRISPR-Cas9 plasmids, EGFP-positive ESCs can be sorted with a flow cytometer for the enrichment of CRISPR-Cas9-expressing cells. Cell sorting obviates antibiotic selection and therefore allows for more gentle culture conditions and faster outgrowth of ESC clones, which are then screened by qPCR for correct genomic modifications. qPCR screening is more convenient and less time-consuming compared to analyzing PCR samples on agarose gels. Positive ESC clones are validated by PCR analysis and sequencing and can serve for injection into early-stage mouse embryos for the generation of chimeric mice with germline transmission. Therefore, we describe here a simple and straightforward protocol for CRISPR-Cas9-directed gene targeting in ESCs.
Graphical abstract:

Background
The advent of CRISPR-Cas9 technology has led to a surge in interest in gene editing, due to its simplicity, speed, and low cost. CRISPR-Cas9 relies upon RNA-DNA binding, providing a significant simplification over previous gene editing methods like zinc-finger nucleases (ZFN) and transcription activator-like effector nucleases (TALEN), which rely upon protein-DNA binding at the site of interest and are therefore more cumbersome to engineer and not ubiquitously useful (González Castro et al., 2021). Due to its efficacy in vivo, CRISPR-Cas9 has quickly become a standard method for the development of genetically engineered mouse mutants. CRISPR-Cas9 machinery, composed of single guide RNAs (sgRNA), homology templates, and Cas9 protein, can be injected into zygotes for creation of chimeric offspring (Qin et al., 2016; Hall et al., 2018; Muñoz-Santos et al., 2020). However, depending on the nature of the desired genetic modification, the efficacy of correct mutagenesis can vary considerably, with a concomitant increase in mouse numbers and associated burden when creating new mouse lines.
An alternative strategy for utilizing CRISPR-Cas9 technology to create mutant mouse lines is to enhance classical gene targeting in murine embryonic stem cells (ESCs) (Oji et al., 2016). CRISPR-Cas9-mediated DNA double-strand breaks induce homology directed repair (HDR) for more efficient introduction of complex or multiple genetic alterations into the germline. Correctly mutated ESCs can be injected into early-stage mouse embryos to yield chimeric mice (Qin and Wang, 2019). Thus, ex vivo gene editing in ESCs can reduce mouse numbers for generation of specific gene modified lines and is aimed at improving the workflow in accordance with the principles of the 3Rs (replace, reduce and refine).
Here, we report an optimized workflow for the induction of targeted mutations in murine ESCs for subsequent germline transmission in mice. We established this protocol to introduce specific point mutations at two positions in exon 7 and exon 17 in the murine Malt1 locus in one step (O'Neill et al., 2021). The two missense mutations yield glutamate to alanine exchanges at amino acid positions 325 and 814 within the two TRAF6 binding motifs of the MALT1A protein and specifically abolish the interaction with the ubiquitin ligase TRAF6. This simple and straightforward protocol should be generally useful for almost any type of targeted mutagenesis of the mouse germline. In short, murine ESCs are transfected with CRISPR-Cas9 machinery using a lipid-based transfection system. Successfully lipofected cells are enriched via an EGFP marker by cell sorting and screened by qPCR using primers specific to the targeted mutation site. The use of lipofection and cell sorting yields mutant ESC clones more conveniently and faster than with classical protocols utilizing electroporation and subsequent selection by antibiotic resistance. The resulting ESC clones are potentially totipotent and competent for germline transmission upon injection into C57BL/6 mouse embryos, yielding chimeric mice for generation of the desired mouse line. This protocol provides a framework for researchers who seek to develop mouse models with complex, difficult or multiple mutation loci, including deletion of large stretches of DNA, insertion of exon-flanking loxP sites for conditional models, and insertion of reporter genes at specific loci. This workflow allows for high-throughput screening of ESC clones and an overall reduction of mouse numbers and associated burden.
Materials and Reagents
Cell lines and oligonucleotides
R1/E (129S1/X1) murine embryonic stem cells (ESCs)
Murine embryonic feeder (MEF) cells (prepared from wildtype mice)
Top10 chemically competent E. coli (produced in-house (transformation efficiency: 5 × 106) or commercially available cells [e.g., XL10-Gold Ultracompetent cells (Agilent, catalog number: 200317)]
Homology templates - Ultramer DNA Oligos, 2 nanomole scale (IDT)
Single guide (sg) RNA oligonucleotides - forward and reverse (Eurofins, 100 µM)
qPCR primers (Eurofins, 100 µM)
PX458 vector (pSpCas9(BB)-2A-GFP) (Addgene, catalog number: 48138)
Materials
10 cm Petri dishes (Corning, catalog number: 353003)
Pipette tips - various
96-well flat-bottom plates (Corning, catalog number: 3997)
96-well round-bottomed plates (Greiner, catalog number: 6500101)
3 mL FACS tubes (Falcon, catalog number: 352063)
6-well plates (ThermoFisher Scientific, catalog number: 140685)
100 µm cell strainer (Neolab, catalog number: GF-0061)
Lipofectamine 3000 transfection kit (Invitrogen, catalog number: L3000-001)
BbsI restriction enzyme (NEB, catalog number: R0539L, 10 units/µL)
T4 DNA ligase (Thermo Fisher Scientific, catalog number: EL0011)
NEB buffer 2.1 (10×) (NEB, catalog number: B7202S)
Sterile, nuclease-free water (VWR, catalog number: PD092)
NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, catalog number: 7400609.50)
SOC medium (Life Technologies, catalog number: 15544034)
Ampicillin (Gibco, catalog number: 11593027)
LB agar (Carl Roth, catalog number: X9963)
LB (Luria/Miller) medium (Carl Roth, catalog number: X968.3)
Miniprep kit (DNA isolation kit) (Macherey-Nagel, catalog number: 740499.250)
Mitomycin C (MMC) (Sigma-Aldrich, catalog number: M4287-5x2mg)
DPBS (ThermoFisher Scientific, catalog number: 14190144)
OptiMEM medium (ThermoFisher Scientific, catalog number: 31985062)
Proteinase K (ThermoFisher Scientific, catalog number: 26160)
Trypsin-EDTA 0.05% (ThermoFisher Scientific, catalog number: 25300-054)
DMSO (Carl Roth, catalog number: A994.1)
DMEM medium (Gibco, catalog number: 41966-129)
KnockoutTM DMEM medium (Life Technologies, catalog number: 10829018)
Glutamax (Invitrogen, catalog number: 35050061)
NEAA (Gibco, catalog number: 11140-035)
Pen/Strep (Gibco, catalog number: 15140-122)
Fetal bovine serum (FBS) (Gibco, catalog number: 10270-106) – regular charge for MEF cell culture
ESC FBS (tested for ESC culture to maintain totipotency)
β-Mercaptoethanol (Thermo Fisher, catalog number: 31350010)
LIF/ESGRO (Millipore, catalog number: ESG1107)
EDTA (Carl Roth, catalog number: 8043.1)
Sodium chloride (NaCl) (Carl Roth, catalog number: 3957.2)
Tris (Carl Roth, catalog number: AE15.2)
1× SYBRTM Safe DNA Gel Stain (ThermoFisher Scientific, catalog number: S33102)
Ethidium Bromide solution 0.07% (PanReac AppliChem, catalog number: 1239-45-8)
Glycerol (Carl Roth, catalog number: 4043.3)
Sodium dodecyl sulfate (SDS) (Carl Roth, catalog number: 2326.2)
Bromophenol blue (Sigma-Aldrich, 115-39-9)
Xylene cyanol (Sigma-Aldrich, 2650-17-1)
10× annealing buffer (see Recipes)
MEF medium (see Recipes)
ESC medium (see Recipes)
2× MEF Freezing medium (see Recipes)
2× ESC Freezing medium (see Recipes)
10× MMC stock (see Recipes)
10× DNA loading buffer (see Recipes)
Equipment
Microscope (Carl ZeissTM, Axiovert 40 CFL equipped with a ZeissTM A-Plan 2.5×/0.06 objective and a ZeissTM E-PI 10×/20 ocular)
Fluorescence microscope (Life Technologies, EVOS FL)
Vortex (Merck, Vortex Genie 2, catalog number: Z258423-1EA)
Gel imaging system/UV transilluminator (INTAS)
NanodropTM 2000 (ThermoFisher Scientific, ND-2000)
Micropipettes (Eppendorf, variable volumes)
Multi-channel micropipettes (Eppendorf, variable volumes)
Tabletop cooling centrifuge (Eppendorf, 5417R)
Sterile bench (Thermofisher Scientific, HERASafe KS/Telstar, Telstar Bio II A)
LightCycler® Real-time PCR machine (Roche, LightCycler 480 II)
Autoclave (various)
Cell sorter (MoFlo, Cytomation, with Summit 4.3 software)
Electrophoresis chamber (Carl Roth, Midi)
Electrophoresis power supply (Sigma-Aldrich, Consort EV243)
Software
MoFlo® computer software (Summit 4.3 Software, Beckman Coulter); FlowJo (BD)
LightCycler® 480 software (https://lifescience.roche.com/en_de/products/lightcycler14301-480-software-version-15.html)
CRISPR-Cas9 guide design software such as CRISPick by BROAD Institute (numerous other options are available, see Cui et al., 2018)
Procedure
The general aim of this protocol is to manipulate the germline of murine embryonic stem cells (ESCs) by introducing one or more mutations simultaneously and possibly even within the same gene. For this, ESCs are transfected with one or more CRISPR plasmids encoding sgRNA(s) that introduce double-stranded DNA breaks at the site(s) of mutagenesis. Homology templates that contain the desired mutations are co-transfected with the guide(s) to induce homology directed repair (HDR). Afterwards, single cell-derived ESC clones are grown, isolated, and screened for correct genetic manipulation of the genomic sequence(s).
Selection of CRISPR single guide RNA (sgRNA) sequences (CRISPR guides)
Use a CRISPR-Cas9 sgRNA design program to find optimal spCas9 (S. pyogenes Cas9) cleavage sites and corresponding 20 nucleotide guide sequences near the target gene locus, for example, CRISPick from the Broad Institute based on algorithms developed by Doench and colleagues (Doench et al., 2016; Sanson et al., 2018). It is important that the Cas9 cleavage site (located 3 to 4 base pairs upstream of the S. pyogenes PAM sequence 5’-NGG) be as close as possible to the site of mutation since an inverse correlation between mutation efficiency and cut-to-mutation distance has been described (Paquet et al., 2016). For improved expression of the guide RNA by the U6 RNA polymerase III promoter of the PX458 plasmid, it is recommended to add an extra “G” at the 5‘ of the sgRNA if the 20-nt guide sequence does not already begin with a “G” (Ran et al., 2013). For cloning, a guide oligo (with a BbsI 5’-CACC sticky end) and the corresponding reverse complement guide oligo (with a BbsI 5’-AAAC sticky end) must be annealed to obtain a double-stranded oligonucleotide that can be cloned into the BbsI site of the PX458 vector (pSpCas9 backbone) (Figure 1). Note that the guide sequence does not include the 5’-NGG PAM sequence itself. For further details see Ran et al. (2013).

Figure 1. sGuide RNA oligonucleotide design example taken from O'Neill et al. (2021), Figure S1, Malt1 exon 17 targeting. A 20-bp guide sequence was identified by a CRISPR design algorithm (Doench et al., 2016) and subsequently used for designing the forward oligo. An “extra G” (blue) was added to the 5’ end of the 20-bp guide sequence, followed by the BbsI 5’-CACC sticky end (green). The reverse oligo is the complement of the 20-bp guide sequence plus a “C” complementary to the “extra G (blue)” and has a 5’-AAAC sticky end (green). The annealed oligo has sticky end overhangs (green) for ligation into the BbsI sites of the PX458 CRISPR plasmid.Design of ssODN Homology Templates
For the generation of point mutations or the introduction of small insertions or deletions, it is possible to use approximately 200 bp single-stranded oligodesoxyribonucleotides (ssODN) as homology templates for HDR. These can be ordered from companies specializing in high-precision DNA synthesis. The desired alteration(s) should be placed at the center of the ssODN. If possible, the PAM sequence, which directs Cas9 to the target site, should be silently mutated within the homology template sequence to prevent Cas9-directed cleavage of the homology template or the successfully modified target locus. Alternatively, or in addition to PAM mutation, silent mutations within the 20-nt guide sequence can be introduced, which at the same time facilitates the design of a mutant allele-specific PCR primer (there is no specific number of silent mutations that can be universally recommended; in our hands, four silent mutations proved to be appropriate, as shown in Figure 2). Moreover, an artificial restriction site may be generated within the homology template by silent mutagenesis, offering an additional option for screening ESC clones. An example HDR is shown in Figure 2. We want to mention that, for another project, we achieved successful targeting by using a linear double-stranded DNA homology template of approximately 1,400 bp total length with 700 bp homology arms flanking the site of mutagenesis (this might be useful for larger scale genetic manipulations such as deletion or insertion of longer stretches of DNA).
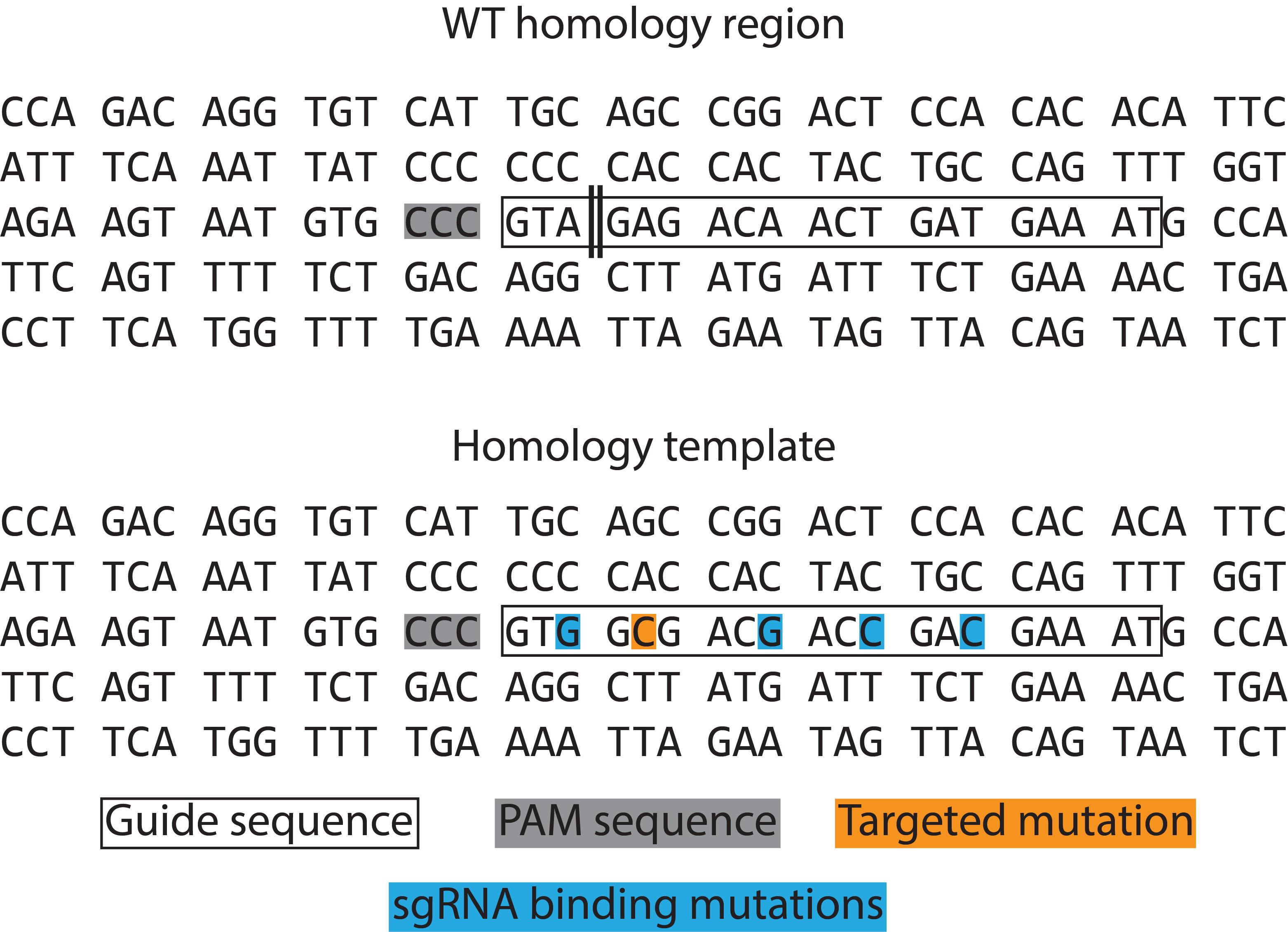
Figure 2. Homology template design example taken from O'Neill et al. (2021). Malt1 exon 17 targeting with the ‘antisense guide’ sequence as shown in Figure 1. The desired targeted mutation is shown in orange. The “CCC” triplet codon (representing the “GGG” PAM sequence since an antisense guide was utilized here) (grey) could not be silently mutated. Instead, silent mutations (blue) were inserted to prevent binding of the sgRNA to the homology template. For comparison, the wildtype sequence of the homology region is shown above with the “GGG” PAM in grey and the 20-nt guide sequence indicated by a box. The approximate expected cleavage site in the wildtype sequence is indicated with a double line.Cloning CRISPR guide oligos into the PX458 plasmid
This protocol uses the plasmid PX458, which contains a cassette for transient co-expression of SpCas9 and EGFP and a cassette into which a custom designed CRISPR guide oligo can be ligated to generate a single guide sequence that is expressed under the control of a U6 RNA polymerase III promoter (Ran et al., 2013). When transfected into ESCs, the PX458-guide construct leads to expression of the sgRNA, Cas9, and EGFP. EGFP-positive ESCs can be enriched by FACS for further culture on feeder cells and outgrowth of single ESC clones.
Anneal the forward and reverse sgRNA oligonucleotides. Set up the annealing reaction by mixing:
Sense guide oligonucleotide (100 µM) 2 µL Antisense guide oligonucleotide (100 µM) 2 µL Annealing buffer (10×) 2 µL Water (nuclease-free) 14 µL Heat the mixture to 95°C in a heating block for 5 min. Turn off the heating block and allow mixture to cool to room temperature (approximately 3 h). Dilute annealed oligonucleotides 1:400 in nuclease-free water.
Digest the PX458 vector with BbsI for guide insertion. Set up the following restriction digest reaction:
PX458 2 µg NEB 2.1 buffer (10×) 2 µL BbsI (10 units/µL) 1.5µL Water (nuclease-free) add to 20 µL total Incubate for 1 h at 37°C on a heating block. Mix restriction reaction with 2 µL of 10× DNA loading buffer and separate on a 1% agarose gel (containing 0.5 µg/mL ethidium bromide or 1× SYBRTM Safe DNA Gel Stain) at 120 V for 1 h. Cut out the linearized PX458 vector fragment (9,270 bp), purify with a gel purification kit, and measure concentration on a NanodropTM.
Ligate the annealed oligonucleotides (from step1; 1:400 diluted = 25 nM) into the PX458 vector (from step 2). Set up the following ligation mix:
PX458 (linearized) 100 ng Annealed guide oligos (25 nM) 2 µL T4 ligase (5 units/µL) 2 µL T4 ligase buffer (5×) 2 µL Water (nuclease-free) add to 10 µL total Incubate the ligation mix at room temperature for 15 min.
For transformation into E. coli, add 2 µL of ligated sgRNA guides (from step 3) to 25 µL chemically competent E. coli. Incubate mixture on ice for 5 min, heat-shock at 42°C for 40 s, and place on ice for 5 min. Mix E. coli with 150 µL of SOC medium without antibiotics and incubate for 1 h at 37°C. Spread the total cell volume on LB plates containing Ampicillin and incubate overnight at 37°C. Select colonies and inoculate into 5 mL of LB medium with Ampicillin and grow overnight at 37°C and 180 rpm. Isolate plasmid DNA using a DNA isolation kit and quantify DNA via NanodropTM. Following sequencing verification of correct guide insertion, the PX458 CRISPR guide plasmid can be used for transfection into murine ESCs for co-expression of the sgRNA and Cas9, with EGFP as a reporter for successful transfection and enrichment via flow cytometric cell sorting.
Preparation and Culture of Murine Embryonic Fibroblasts (MEFs)
ESCs need to be cultured on a layer of mouse embryonic fibroblasts (MEFs), which act as feeder cells. MEFs are generated from 12.5 dpc (days post coitum) wildtype mouse embryos and can be passaged four times before being growth arrested by treatment with Mitomycin C (MMC) so that they then can serve as feeder cells for ESC culture.
Harvesting MEFs
Prepare murine MEF cells from wildtype mouse embryos on day 12.5. Briefly, in a cell culture dish containing PBS, separate each embryo from the yolk sac and remove and discard the head and liver. Subsequently, each embryo is processed separately: mesh remaining tissue through a 100 µm mesh into MEF medium and transfer cells of one embryo to a 6-cm cell culture dish. Let MEF cells grow overnight. On the next day, wash away non-adherent, dead cells and allow cells to grow further until confluent (approximately 2 days).
Passaging MEFs
Detach adherent MEF cells from the surface of the 6 cm dish by washing cells with PBS, adding 2 mL trypsin, and incubating 5 min at 37°C. Stop trypsinization by adding MEF medium. Transfer harvested MEFs from the 6 cm dish to one 15 cm plate and let grow in the incubator. Upon confluency, perform a second passage from the 15 cm plate (use 8 mL trypsin) to four 15 cm dishes. MEF cells can now be frozen in liquid nitrogen (see step 3) or treated with Mitomycin C (MMC) for use in ESC culture (see step 4).
Freezing and thawing MEFs
Harvest MEFs from one confluent 15 cm dish (approximately 2 × 107 cells) by trypsinization, as described in step 2. Take up harvested cells in 2 mL of MEF medium and put on ice. Add 2 mL of ice-cold 2× Freezing medium (1:1) and transfer 1 mL aliquots (containing approx. 5 × 106 cells) to cryo-vials, which are placed in a styrofoam box for slow freezing in a -80°C freezer. After 24 h, transfer the frozen cells to liquid nitrogen for long-term storage. For thawing, float a cryo-vial in a 37°C water bath and transfer the thawed cell suspension into 10 mL of PBS. Centrifuge at 350 × g for 5 min, take up the cell pellet in 10 mL of MEF medium, distribute the cells to four 10 cm dishes and incubate at 37°C/5% CO2. Either let cells grow for 2 days for further passaging or use on the next day for treatment with MMC (see step 4).
Treatment of MEFs with Mitomycin (MMC)
MEF cell density for use as an MMC-treated feeder cell layer is 2.5 × 106 cells per 10 cm dish. Treat MEF cells with MMC (10 µg/mL in MEF medium) and incubate at 37°C for 2 to 3 h to inhibit further proliferation. Remove medium containing MMC, wash cells thoroughly three times with PBS, and add fresh MEF medium. MCC-treated MEFs are now ready for use as a base layer for ESC growth (and should be used within 2 days after MMC treatment).
ESC culture (R1/E cells)
ESCs must be cultured under conditions that allow for maintenance of totipotency. For this, ESCs are cultured on a layer of feeder cells and must be kept at appropriate cell density in ESC medium containing FBS that has been approved for ESC culture (can be purchased from commercial sources or obtained from labs specializing in ESC culture). Murine R1/E ESCs are always grown on a confluent (but not too dense) feeder layer of MMC-treated MEFs (approximately 40,000 feeder cells/cm2).
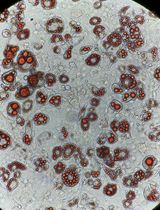
Seed approximately 30,000 R1/E ESCs per cm2 on feeder cells in ESC medium. Medium is exchanged daily, and ESCs are usually split after 2 days of culture at appropriate cell density and morphology: this condition is reached when the ESCs form numerous distinct colonies with sharp edges that together cover approximately one-third of the surface of the culture dish without touching each other (Figure 3). For splitting, ESCs are washed with PBS and treated for 5 min with trypsin, and trypsin is inactivated by addition of ESC medium. A single cell suspension is obtained by vigorous pipetting with an electric pipetting device: draw the cell suspension into a 10 mL pipet, press the tip of the pipette onto the bottom of the cell culture dish, and force the cell suspension out of the pipet. Repeat this 10 times. Count cells and seed 30,000 cells/cm2 on fresh feeder cells. With some experience, it may not be necessary to always count the ESCs, and after visual inspection of the cells under the microscope it is possible to estimate an acceptable split factor, usually 1:6 for ESCs at optimal growth conditions and cell density (a split factor of 1:6 means that the cells are harvested from a cell culture plate and 1/6 of the cells are transferred into a new cell culture plate of the same format).
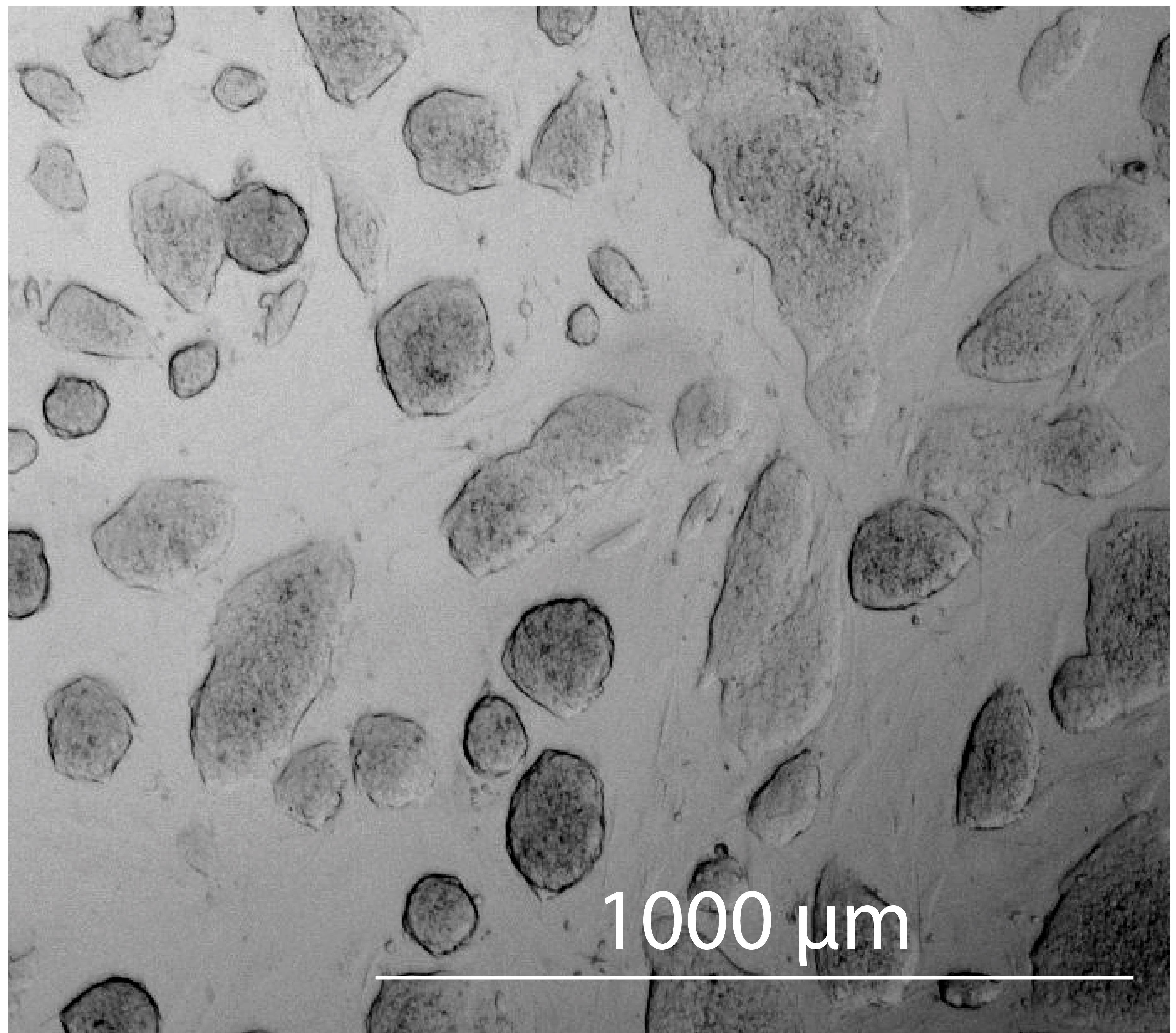
Figure 3. Morphology of R1/E ESCs. R1/E cells form distinct colonies and cover approximately one-third of the plate surface.ESCs can be frozen in liquid nitrogen: for this, ESCs are trypsinized as described above, cells are counted, spun down at 350 × g, adjusted to 5 × 106 cells/mL in ESC medium, and shortly pre-cooled on ice. An equal volume of ice-cold 2× ESC Freezing medium is added and, after mixing, 1 mL of cell suspension is distributed to cryo-vials for freezing at -80°C within a styrofoam box with subsequent transfer to liquid nitrogen. For later thawing, the cryo-vial is placed in a 37°C water bath and, when content is liquid, the cells are transferred into 10 mL PBS in a 15 mL tube, spun at 350 × g, and the cell pellet is taken up in ESC medium. Cells from each cryo-vial (2.5 × 106 cells) are seeded on feeder cells in a 10 cm dish.
Transfection of ESCs
Reported protocols typically use electroporation to transfect ESCs with plasmids and homology templates. Electroporation requires large amounts of DNA (20 µg) as well as many cells (commonly approximately 5 × 106 cells grown in a 10 cm dish), and many cells die due to the harsh electroporation conditions. Lipofection proves to be gentler, allowing the use of much lower cell numbers (200,000 cells grown in a 6-well plate) and smaller DNA amounts (2 µg or less). This results in approximately 5% transfection efficiency as measured by the percentage of EGFP-positive cells in flow cytometry, while only a small proportion of transfected cells with high EGFP expression can be seen under a fluorescence microscope (Figure 4). Transfection of ESCs is performed in a 6-well format using lipofection via the Lipofectamine 3000 kit based on the manufacturer’s protocol.
Approximately 16 h prior to transfection, seed 200,000 ESCs per well of a 6-well plate in 2 mL of ESC medium (on feeder cells).
On the day of transfection, prepare mixtures in Tube A and Tube B:
Tube A: 125 µL OptiMEM medium + 5 µL Lipofectamine 3000 reagent.
Tube B: 125 µL OptiMEM medium + 6 µL P3000 reagent + 6 µg DNA (PX458 plasmids and homology templates, evenly distributed to the maximum total DNA amount of 6 µg).
Mix the contents of tubes A and B together and incubate at room temperature for 15 min.
Transfer the entire transfection volume to the ESCs in the 6-well (step 1).
Incubate the ESCs with the DNA-liposome complexes for 6 h.
After incubation, replace the medium with fresh ESC medium and let cells grow for 24 h in the incubator before sorting EGFP-positive cells by FACS.
FACS-based cell sorting of ESCs and monoclonal ESC clone growth
Successfully transfected ESCs express EGFP encoded by the PX458 plasmid and can therefore be enriched to more than 95% by FACS (Figure 4B). Only 2000 sorted EGFP-positive ESCs are needed to be seeded on a feeder layer in a 10 cm dish for the outgrowth of up to 100 ESC clones. In contrast to classical protocols that use DNA constructs with antibiotic resistance cassettes for selection, FACS-sorted ESCs do not need to be treated with antibiotics, and therefore the ESC plates do not contain millions of dying cells in culture, which would have to be carefully washed away. Monoclonal ESC clones grow with ESC medium exchanged every second day and are ready for picking within 7 days, which is approximately 2 days faster than experienced with classical protocols using electroporation and antibiotic selection.
Twenty-four hours after lipofection, wash ESCs in the 6-well plates (from Procedure F, step 6) 1× with PBS.
Add 500 µL of trypsin, incubate 5 min, add 500 µL of ESC medium, and pipette vigorously up and down to generate a single cell suspension containing ESCs and feeder cells.
Transfer the cell suspension to a new 6-well plate. Within 1 h, most feeder cells but not the ESCs will re-adhere. Gently remove the ESCs and transfer to a 3 mL tube for cell sorting.
Using a cell sorter equipped with a 100 µm nozzle, sort EGFP-positive ESCs into ESC medium. Plate 2000 sorted cells onto a 10 cm dish pre-laid with 3 × 106 MMC-treated MEF cells and allow cells to grow for approximately 7 days with medium changes every second day (Figure 5A). Up to 100 ESC colonies can be expected to grow per 10 cm dish. Therefore, it is recommended to seed ten 10 cm dishes per condition to obtain sufficient numbers of colonies for screening (we recommend screening approximately 500 colonies).
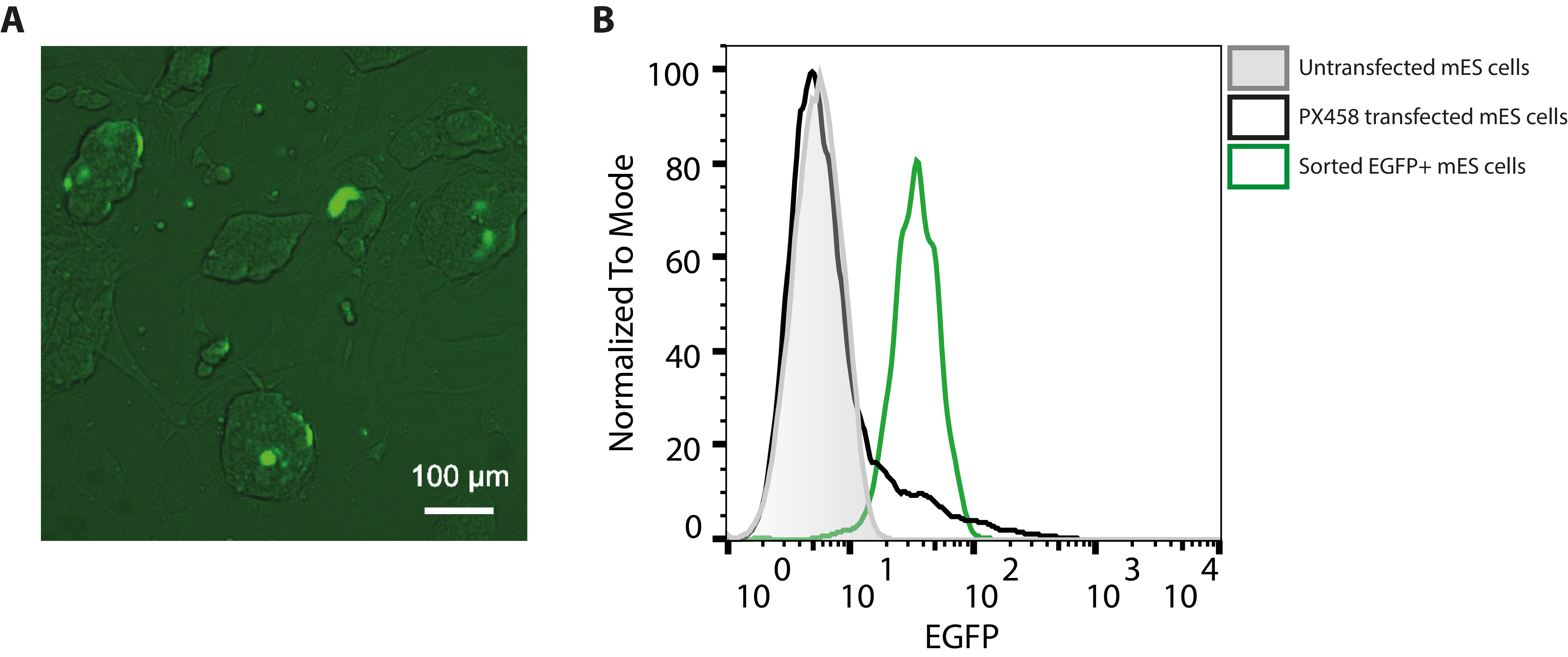
Figure 4. ESCs 24 h after lipofection with PX458. (A) Cells with high EGFP expression seen under a fluorescence microscope. (B) Flow cytometry allows for sensitive detection of ESCs with even low to moderate EGFP expression (black curve) when compared to untransfected cells (grey curve). Cell sorting leads to enrichment of EGFP-expressing cells to over 95% (green curve).Picking ESC clones
Monoclonal ESC clones can be picked approximately 7 days after cell sorting when distinct 3-dimensional colonies have grown on the 10-cm plates and can be seen under a microscope (Figure 5A).
Prior to picking colonies, seed 2 flat-bottomed 96-well plates with MMC-treated MEF cells in 100 µL of ESC medium (approximately 10,000 cells per well). Keep plates in the incubator until needed.
Wash the 10 cm plates containing ESC colonies with 10 mL of PBS and add 10 mL of fresh PBS to the plates.
Place a microscope under the sterile work bench to visualize the single ESC colonies (Figure 5B). Ensure that there is sufficient space to approach the plate from the side with a 100 µL pipette. Detach each colony from the feeder layer by gently dislodging it from the surface with the pipette tip and drawing it up in a volume of 50 µL. Care should be taken to extract the entire ESC colony to avoid distribution of ESCs to the plate and therefore cross-contamination of other monoclonal colonies.
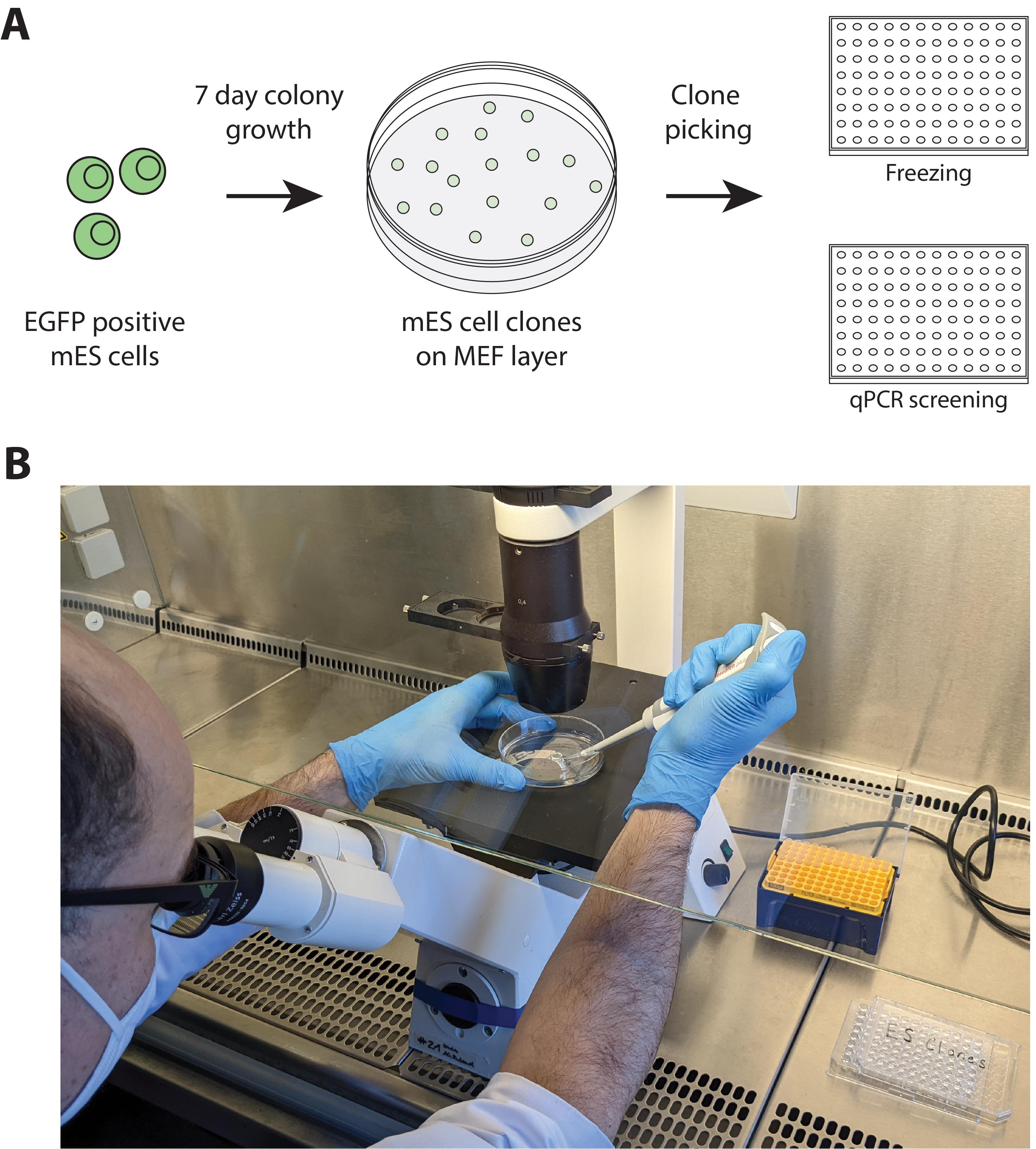
Figure 5. Cell picking workflow and set-up. (A) EGFP positive cells from cell sorting are plated at a density of 2000 cells per 10 cm dish on a layer of MMC-treated MEF cells. Following a 7-day growth period, 50–100 monoclonal ESC clones can be picked from each 10 cm dish into duplicate 96-well plates for freezing or qPCR screening, respectively. (B) Microscope set-up for clone picking under a sterile cell culture hood.Transfer the picked colonies to a round-bottomed 96-well plate. Pick 96 clones to fill the plate and as many plates as are needed for screening (we typically pick five 96-well plates). Proceed as quickly as possible to step 5 (it is recommended to work in tandem: one person picking and another processing the picked clones).
Following collection of 96 colonies, add 30 µL of trypsin to each round-bottomed 96-well containing the picked ESC clones and incubate the plate at 37°C for 10 min. Add 100 µL of ESC medium to each well to stop trypsinization.
Using a multichannel micropipette, vigorously pipette up and down in each well to dissociate the ESC clone aggregates into single cells and split the contents of each well into the two flat-bottomed plates containing MEF cells (prepared in step 1). Incubate both plates for 2 days. Plate #1 is used for screening, and plate #2 is frozen for later recovery of positive clones identified by qPCR screening and sequence analysis.
Before freezing ESCs in 96-well plates it can be of value to inspect every single 96-well for judging cell density and take notes about the cell culture plate format into which the cells should be seeded upon defrosting (this can be estimated by the 1:6 split factor as described in Procedure E, step 1). For freezing ESCs in 96-well plates, wash cells in Plate #2 (from step 6) 1× with PBS, trypsinize cells by adding 30 µL of trypsin per well, incubate for 5 min in the incubator, mix with 70 µL of ESC medium to stop the trypsinization reaction, and pipette vigorously to resuspend cells. Pre-cool the plate shortly on ice, then add 100 µL of ice-cold 2× ESC freezing medium, mix and freeze plate within a styrofoam box at -80°C, and later transfer to liquid nitrogen. For thawing cells from a 96-well plate, thaw the plate in a 37°C incubator, transfer desired clones to 15 mL Falcon tubes in 5 mL of medium, spin the cells down at 350 × g, aspirate the supernatant, wash the cells once with 1 mL of PBS, take up the cells in 100 µL of ESC medium, and transfer to an appropriate well size in sufficient medium (usually 48- or 24-well format).
qPCR screening for correctly targeted ESC clones
It is recommended to establish specific and sensitive qPCR protocols at the very beginning of the project as part of homology template design because it is vital that correctly manipulated ESC clones can be identified by well-established screening strategies. It may be valuable to get a DNA template synthesized that corresponds to the genomic locus after correct insertion of the homology template, which can serve as a positive control for specific and sensitive amplification of the mutated locus. Screening conditions can be even better simulated if genomic DNA from wildtype ESCs (prepared as described in steps 1 to 3 below) is spiked in when establishing PCR conditions. Test serial dilutions of the positive control templates down to a copy number of 1. Detectable amplification of 10 copies of the positive control template is usually sensitive enough to be able to later detect positive ESC clones by screening genomic DNA of ESC clones. To prevent false-positive signals during screening, be careful to avoid contamination of the work place and of equipment with “positive control” DNA fragments or positive screening amplicons; positive control DNA or amplicons should never be handled where later steps of the protocol take place (ESC culture, transfection, clone picking, and screening PCR).
To prepare genomic DNA of ESC clones from Plate #1 (from Procedure H step 6) for qPCR screening, remove medium from the 96-well plate, wash cells once with PBS, and then add 200 µL of nuclease-free water to each well. In a 96-well PCR cycler, incubate the plate at 95°C for 15 min.
Add Proteinase K to each well to a final concentration of 200 µg/mL and incubate at 55°C for 90 min for protein digestion. Heat plate to 95°C for 15 min to inactivate Proteinase K. Samples are now ready for screening.
Design PCR screening primers that are specific for the amplification of correctly targeted sequences for the identification of successfully manipulated ESC clones. In case of site directed mutagenesis, one PCR primer should be mutation-specific by covering the mutation site (according to the design of the homology template as described in Procedure B), and the other primer should be located outside the homology template sequence. See, for example, Figure S1B in O'Neill et al. (2021). Potential positive clones will amplify at lower cycle threshold (Ct) values during qPCR. In the case of inserting multiple mutations within one ESC clone, qPCR protocols and mutation specific primers will need to be designed for each mutation site, and each qPCR screening will need to be performed separately.
Perform qPCR according to your established protocol(s). Most likely, the majority of genomic ESC DNA samples will generate background signals at high Ct values (>35), depending on the specificity of the PCR protocol. However, a subset of samples will give rise to robust amplification at much lower Ct values (Figure 6). Those clones are candidates with genomic modifications which need to be further analyzed by sequencing (O’Neill et al., 2021, Figure S1C, D).
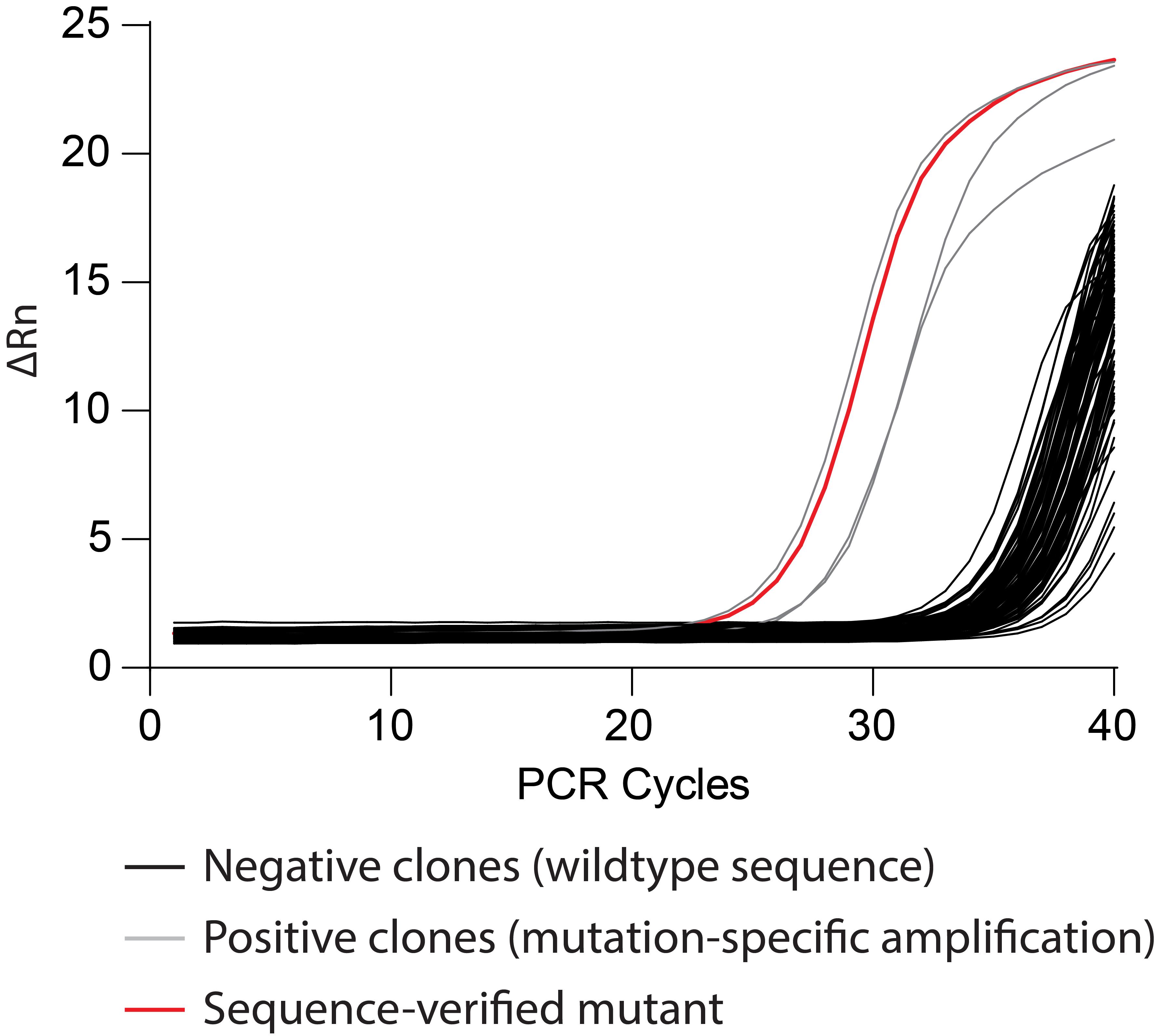
Figure 6. ESC screening by qPCR. Clones are screened using a mutation-specific primer which only amplifies the correctly mutated target sequence. Negative clones (black curves) are excluded due to their high threshold cycle (Ct) value. Potential positive clones (grey/red curves) have lower Ct values. Prospective positive clones are verified by sequencing (potentially in combination with TOPO cloning) to find a clone with all mutations correctly inserted (red curve).For sequencing, the genomic region of interest is amplified with primers flanking the targeted site. The purified PCR product can then directly be used for sequencing. Sequencing data are often difficult to interpret since both alleles of the targeted gene can be altered differently, yielding two overlapping chromatograms that cannot be unequivocally assigned to one or the other allele. This can even be the case if online tools are used, such as CRISPID (Dehairs et al., 2016). In this case, a reliable technique to elucidate the sequence status of the distinct alleles is TOPO cloning of the above-mentioned PCR product. TOPO cloning results in E. coli clones that carry only one or the other allelic fragment, which can then be sequenced separately.
ESC clones with confirmed genomic modification(s) can be recovered by thawing from plate #2 (see Procedure H, step 6, and step 7) and should be characterized with respect to healthy growth and good ESC morphology. Selected ESC clones are expanded and frozen in several aliquots and are now ready for embryo injection for creation of chimeric mice and subsequent germline transmission.
Recipes
10× Annealing buffer
10 mM Tris
1 mM EDTA
50 mM NaCl
pH 7.5
MEF medium
500 mL of DMEM
50 mL of FBS (regular)
500 µL of 50 mM β-Mercaptoethanol
5 mL of NEAA
5 mL of Pen/Strep
ESC medium
500 mL of KnockoutTM DMEM
90 mL of FBS (ESC culture tested)
1.2 mL of 50 mM β-Mercaptoethanol
6 mL of NEAA
6 mL of Glutamax
6 mL of Pen/Strep
90 µL of LIF/ESGRO (1,500 U/mL)
2× MEF Freezing medium
20% DMSO
30% FBS (regular)
50% DMEM (with 1% Pen/Strep)
2× ESC Freezing medium
20% DMSO
30% FBS (ESC culture tested)
50% DMEM (with 1% Pen/Strep)
10× MMC stock
Dissolve MMC powder in MEF medium for a stock concentration of 100 µg/mL, sterile filter, and store at 4°C
10× DNA loading buffer
34% Glycerol
0.5% SDS
10 mM EDTA
2.5 mg/mL Bromophenol blue
2.5 mg/mL Xylene cyanol
Acknowledgments
This protocol was first used in a publication in Science Immunology (O'Neill et al., 2021). This work was supported by Deutsche Forschungsgemeinschaft (ID 210592381 – SFB 1054 A04, ID 360372040 – SFB 1335 P07) and Deutsche Krebshilfe (No. 70112622). D.K. is a scientific advisor of Monopteros Therapeutics Inc., Boston.
Ethics
Pregnant mice used for the generation of Mouse Embryonic Fibroblasts from 12.5 dpc embryos were housed and handled in accordance with the guidelines of the Federation of European Laboratory Animal Science Association.
Competing interests
The authors declare no competing interests.
References
- Cohen, J. (2016). Any idiot can do it. Genome editor CRISPR could put mutant mice in everyone’s reach. Science. https://www.science.org/content/article/any-idiot-can-do-it-genome-editor-crispr-could-put-mutant-mice-everyones-reach.
- Cui, Y., Xu, J., Cheng, M., Liao, X. and Peng, S. (2018). Review of CRISPR/Cas9 sgRNA Design Tools. Interdiscip Sci 10(2): 455-465.
- Dehairs, J., Talebi, A., Cherifi, Y. and Swinnen, J. V. (2016). CRISP-ID: decoding CRISPR mediated indels by Sanger sequencing. Sci Rep 6: 28973.
- Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., Smith, I., Tothova, Z., Wilen, C., Orchard, R., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34(2): 184-191.
- González Castro, N., Bjelic, J., Malhotra, G., Huang, C. and Alsaffar, S. H. (2021). Comparison of the Feasibility, Efficiency, and Safety of Genome Editing Technologies. Int J Mol Sci 22(19): 10355.
- Hall, B., Cho, A., Limaye, A., Cho, K., Khillan, J. and Kulkarni, A. B. (2018). Genome Editing in Mice Using CRISPR/Cas9 Technology. Curr Protoc Cell Biol 81(1): e57.
- Muñoz-Santos, D., Montoliu, L. and Fernandez, A. (2020). Generation of Genetically Modified Mice Using CRISPR/Cas9. Methods Mol Biol 2110: 129-138.
- O'Neill, T. J., Seeholzer, T., Gewies, A., Gehring, T., Giesert, F., Hamp, I., Grass, C., Schmidt, H., Kriegsmann, K., Tofaute, M. J., et al. (2021). TRAF6 prevents fatal inflammation by homeostatic suppression of MALT1 protease. Sci Immunol 6(65): eabh2095.
- Oji, A., Noda, T., Fujihara, Y., Miyata, H., Kim, Y. J., Muto, M., Nozawa, K., Matsumura, T., Isotani, A. and Ikawa, M. (2016). CRISPR/Cas9 mediated genome editing in ES cells and its application for chimeric analysis in mice. Sci Rep 6: 31666.
- Paquet, D., Kwart, D., Chen, A., Sproul, A., Jacob, S., Teo, S., Olsen, K. M., Gregg, A., Noggle, S. and Tessier-Lavigne, M. (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533(7601): 125-129.
- Qin, W., Kutny, P. M., Maser, R. S., Dion, S. L., Lamont, J. D., Zhang, Y., Perry, G. A. and Wang, H. (2016). Generating Mouse Models Using CRISPR-Cas9-Mediated Genome Editing. Curr Protoc Mouse Biol 6(1): 39-66.
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system.Nature Protocols 8(11): 2281-2308.
- Sanson, K. R., Hanna, R. E., Hegde, M., Donovan, K. F., Strand, C., Sullender, M. E., Vaimberg, E. W., Goodale, A., Root, D. E., Piccioni, F., et al. (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 9(1): 5416.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- O'Neill, T. J., Krappmann, D. and Gewies, A. (2022). Optimized CRISPR-Cas9-based Strategy for Complex Gene Targeting in Murine Embryonic Stem Cells for Germline Transmission. Bio-protocol 12(10): e4423. DOI: 10.21769/BioProtoc.4423.
- O'Neill, T. J., Seeholzer, T., Gewies, A., Gehring, T., Giesert, F., Hamp, I., Grass, C., Schmidt, H., Kriegsmann, K., Tofaute, M. J., et al. (2021). TRAF6 prevents fatal inflammation by homeostatic suppression of MALT1 protease. Sci Immunol 6(65): eabh2095.
Category
Stem Cell > Embryonic stem cell
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.