- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Methylation-sensitive Amplified Polymorphism as a Tool to Analyze Wild Potato Hybrids


Published: Vol 10, Iss 13, Jul 5, 2020 DOI: 10.21769/BioProtoc.3671 Views: 5223
Reviewed by: Samik BhattacharyaYing FengAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Methylation-Sensitive Amplification Polymorphism (MSAP) is a versatile marker for analyzing DNA methylation patterns in non-model species. The implementation of this technique does not require a reference genome and makes it possible to determine the methylation status of hundreds of anonymous loci distributed throughout the genome. In addition, the inheritance of specific methylation patterns can be studied. Here, we present a protocol for analyzing DNA methylation patterns through MSAP markers in potato interspecific hybrids and their parental genotypes.
Keywords: MSAPBackground
Nucleotide sequences are not the only form of genomic information: DNA methylation, histone proteins, enzymes that modify histones and nucleotide residues on DNA and even RNAs, influence gene activity and provide another layer of instructions to the cell. Epigenetic changes, also called epimutations, can be inherited and have important phenotypic consequences. In plants, methylation reactions modify cytosine residues into 5-methylcytosines. This epigenetic mechanism is essential to maintain the genomic integrity and contributes to regulate gene expression in developmental processes and in response to biotic and abiotic stresses. In addition, changes in DNA methylation are triggered by genomic shocks like hybridization and polyploidization, two vital phenomena in plant evolution (Cara et al., 2019).
There are different alternatives to study changes in DNA methylation. Global cytosine methylation can be assessed by using high-performance liquid chromatography (HPLC), analytical methods that allow to quantify cytosines and 5-methylcytosines and to calculate the percentage of methylated residues in the genome. For studying the DNA methylation at specific positions on the genome two alternatives can be mentioned. One is to use isoschizomers with different sensitivities to methylation at cytosines of the restriction site. For example, Methylation-Sensitive Amplification Polymorphism (MSAP) markers characterize the methylation pattern at anonymous 5′-CCGG sequences from random genomic DNA. This is an adaptation of the original AFLP protocol (Vos et al., 1995) substituting the frequent cutter enzyme MseI by HpaII and MspI. These enzymes recognize the same tetranucleotide restriction site (5′-CCGG), but HpaII is sensitive to full methylation (both strands methylated) and cleaves the hemi-methylated external cytosine, whereas MspI is sensitive only to methylation of the external cytosines of the restriction site. Another possibility to study site-specific methylation status is by using a bisulphite sequencing methodology. Bisulphite treatments on DNA convert cytosines into uracils while 5-methylcytosines remain unchanged. Then, by sequencing amplicons (i.e., a target gene or promoter) of bisulphited and control DNA, it is possible to distinguish between methylated and unmethylated cytosines. With the development of next generation sequencing technologies, whole-genome bisulphite sequencing, or WGBS, can be implemented to infer the position for all 5-methylcytosines in a genome. However, this approach requires a high-quality reference genome to perform epigenetic analyses. Although the increasing number of draft genomes and the reduction in sequencing costs offer possibilities to implement massive methylation analyses in non-model species, the use of MSAP markers continues to be a valuable tool in many laboratories.
Wild potatoes (Solanum, section Petota) are a group of species related to the cultivated potato Solanum tuberosum L. Internal breeding barriers can be incomplete, thus, interspecific hybridization occurs in areas of sympatry (Camadro et al., 2012). Epigenetic changes in response to interspecific hybridization have been documented in synthetic and natural hybrids of wild potato species (Cara et al., 2019). Solanum x rechei H. & H. is a hybrid species that grows in sympatry with its wild progenitors, Solanum kurtzianum B. & W. and Solanum microdontum B. Here, we present a protocol for analyzing DNA methylation patterns through MSAP markers in hybrids and their parental genotypes. Using an R script, fragments present in the synthetic hybrids are categorized as S. microdontum or S. kurtzianum species-specific if they are present on the parental genotypes, S. x rechei species-specific, if they are present in at least one of the S. x rechei evaluated genotypes or as novel if they are only observed in the synthetic hybrids.
Materials and Reagents
- Consumables
- Chemicals
- Tris base (BIOPACK, catalog number: 200016 6800)
- Ethylenediaminetetraacetic acid (EDTA) (BIOPACK, catalog number: 2000964500 )
- Sodium Chloride (BIOPACK, catalog number: 200016 4606)
- Cetyltrimethyl ammonium bromide (CTAB) (Bio Basic INC, catalog number: DB0108 )
- β-mercaptoethanol (BIOPACK, catalog number: 2000954500 )
- Chloroform (BIOPACK, catalog number: 200016 5100)
- Isoamyl alcohol (BIOPACK, catalog number: 2000972500 )
- Ethyl alcohol (BIOPACK, catalog number: 200016 5400)
- Sodium acetate (BIOPACK, catalog number: 200016 8000)
- RNase A (Thermo Fisher Scientific, catalog number: EN0531 )
- Lambda DNA/EcoRI+HindIII Marker (Promega, catalog number: G1731 )
- Glycerol (BIOPACK, catalog number: 200016 2000)
- Bromophenol blue (BIOPACK, catalog number: 2000962200 )
- Xylene cyanol FF (SIGMA, catalog number: X4126 )
- Boric acid (BIOPACK, catalog number: 2000935900 )
- Agarose (TransGen, catalog number: GS201 )
- UltraPureTM Ethidium Bromide (Thermo Fisher Scientific, catalog number: 15585011 )
- EcoRI (New England Biolabs, catalog number: R0101S )
- Bovine Serum Albumin (BSA) (Promega, catalog number: R3961 )
- HpaII (New England Biolabs, catalog number: R0171S )
- MspI (New England Biolabs, catalog number: R0106 )
- T4 DNA Ligase (Promega, catalog number: M1801 )
- Oligonucleotides (Table 1) (IDT, Integrated DNA Technologies, Inc., Iowa, USA)
Table 1. Sequences of adaptors and primers used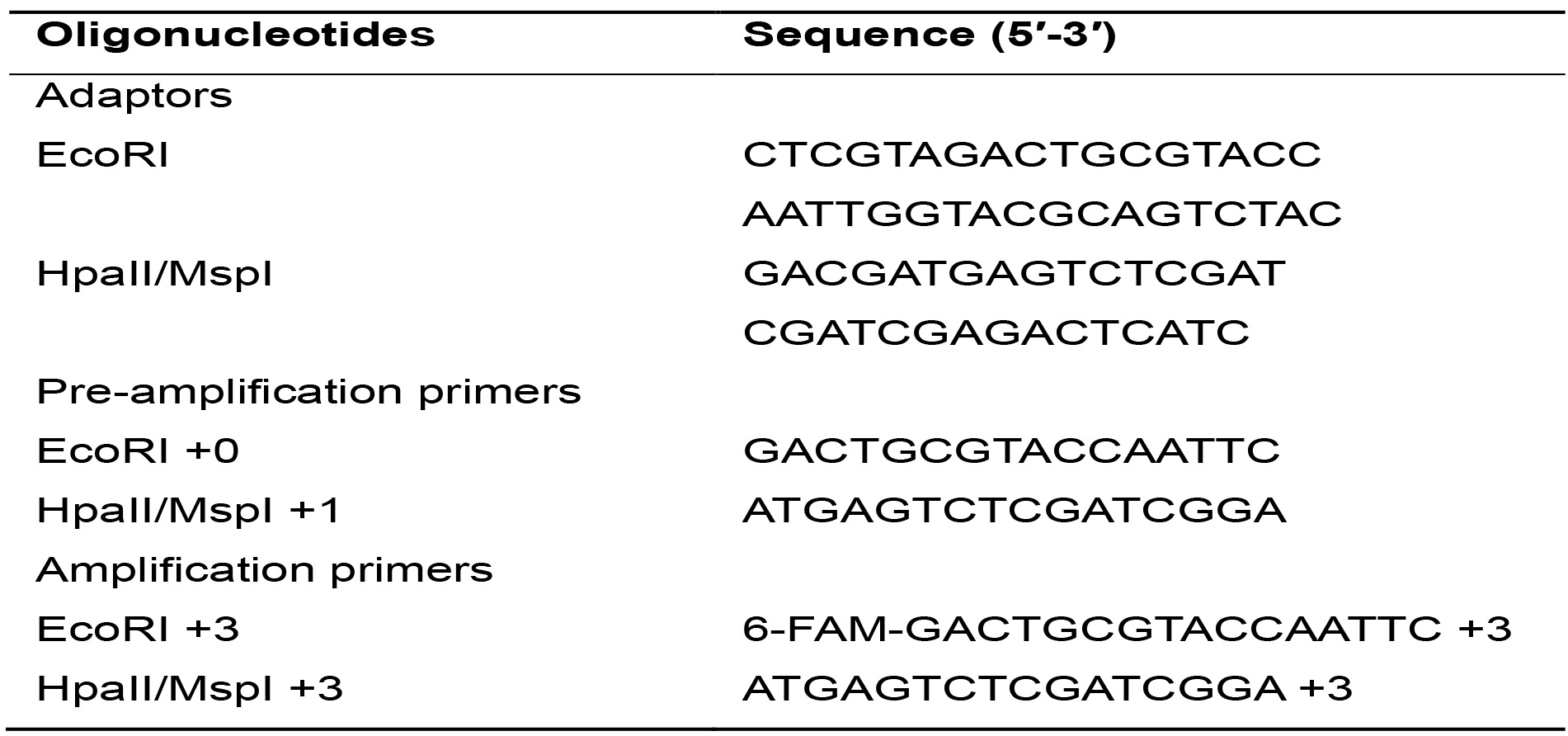
- Taq DNA Polymerase (Thermo Fisher Scientific, catalog number: 11615044 )
- MgCl2 (Thermo Fisher Scientific, catalog number: 11615044 )
- dNTP Set (100 mM) (Thermo Fisher Scientific, catalog number: 10297018 )
- Hi-Di formamide (Thermo Fisher Scientific, catalog number: 4311320 )
- GeneScan 500HD Rox size standard (Thermo Fisher Scientific, catalog number: 401734 )
- ddH2O (sterile)
- Extraction buffer (see Recipes)
- TBE (Tris-Borate-EDTA) buffer (see Recipes)
- 6x DNA loading buffer (see Recipes)
Equipment
- Pipettes (BOECO, Wheaton SOCOREX and Finnpipette)
- Vortex (IKA, model: MS1 )
- UltraCentrifuge (Eppendorf, model: 5804R )
- Thermostatic bath (Vicking, model: Masson )
- Agarose gel electrophoresis system (Bio-Rad Laboratories, model: 1645056 )
- Microwave oven (HITPLUS, model: CM203M )
- Spectrophotometer (AmpliQuant, model: AQ-07 )
- Gel Imager (Bio-Rad Laboratories, model: Gel Doc 1000 )
- Incubator (SANYO, model: MIR 262 )
- Thermocycler Veriti 96 well (Applied Biosystems, model: 9902 )
- Genetic Analyzer (Invitrogen, model: ABI PRISM 3130 )
- Ultra-low Freezer -80 °C (Forma Scientific, model: 8270 )
Software
- GeneMapper v3.7. (Applied Biosystems, Foster City, CA, USA)
- R v2.15.1. (R Foundation for Statistical Computing, Vienna, Austria)
- RStudio v1.1.442 (RStudio Inc., Boston, MA, USA)
Procedure
- DNA extraction procedure
- Collect 50-100 mg of leaf tissue in a 1.5 ml centrifuge microtube. Flash-freeze the tissue in liquid nitrogen and grind it with a plastic pestle.
- Add 300 µl of extraction buffer and mix it vigorously by vortexing (5-10 s) to homogenize the tissue. Incubate the homogenate at 65 °C for 30 min in a thermostatic bath.
- Add 100 µl of chloroform:isoamyl alcohol (24:1) and mix the sample gently by inversion.
- Centrifuge the homogenate at 20,800 x g at 4 °C for 5 min.
- By using a 200 µl micropipette, carefully pipette the aqueous phase and transfer it to a new microtube.
- Add 500 µl of ice-cold ethanol:acetate (24:1) and mix the sample by inverting the microtube 2-3 times to precipitate the DNA, and let it settle at -20 °C for 30 min.
- Centrifuge the sample at 20,800 x g at 4 °C for 10 min to form a DNA pellet at the bottom of the microtube.
- Discard the supernatant and clean the pellet with 100 µl of 70% ethanol. Vortex the tube for 1 min to wash the pellet.
- Centrifuge the sample at 15,300 x g at room temperature for 5 min and let the pellet dry by inverting the microtube onto a paper towel for 30 min until the ethanol evaporates completely or by placing it in a drying chamber for 15 min.
- Resuspend the DNA in 50 µl of ultrapure water with RNase A (50 μg·ml-1). Incubate at room temperature for 30 min.
- Store the DNA sample at 4 °C until use. If the DNA will not be used for a long time, store it at -20 °C.
- Check the integrity and purity of the extraction by loading 2 μl of the DNA solution on a 0.8% agarose gel and running it at 90 V for 30 min. It should show a thick band of high molecular weight, above 21 Kbp, with no smear (Figure 1).
- Measure the concentration of DNA samples in a spectrophotometer and prepare working solutions of ~100 ng·μl-1.
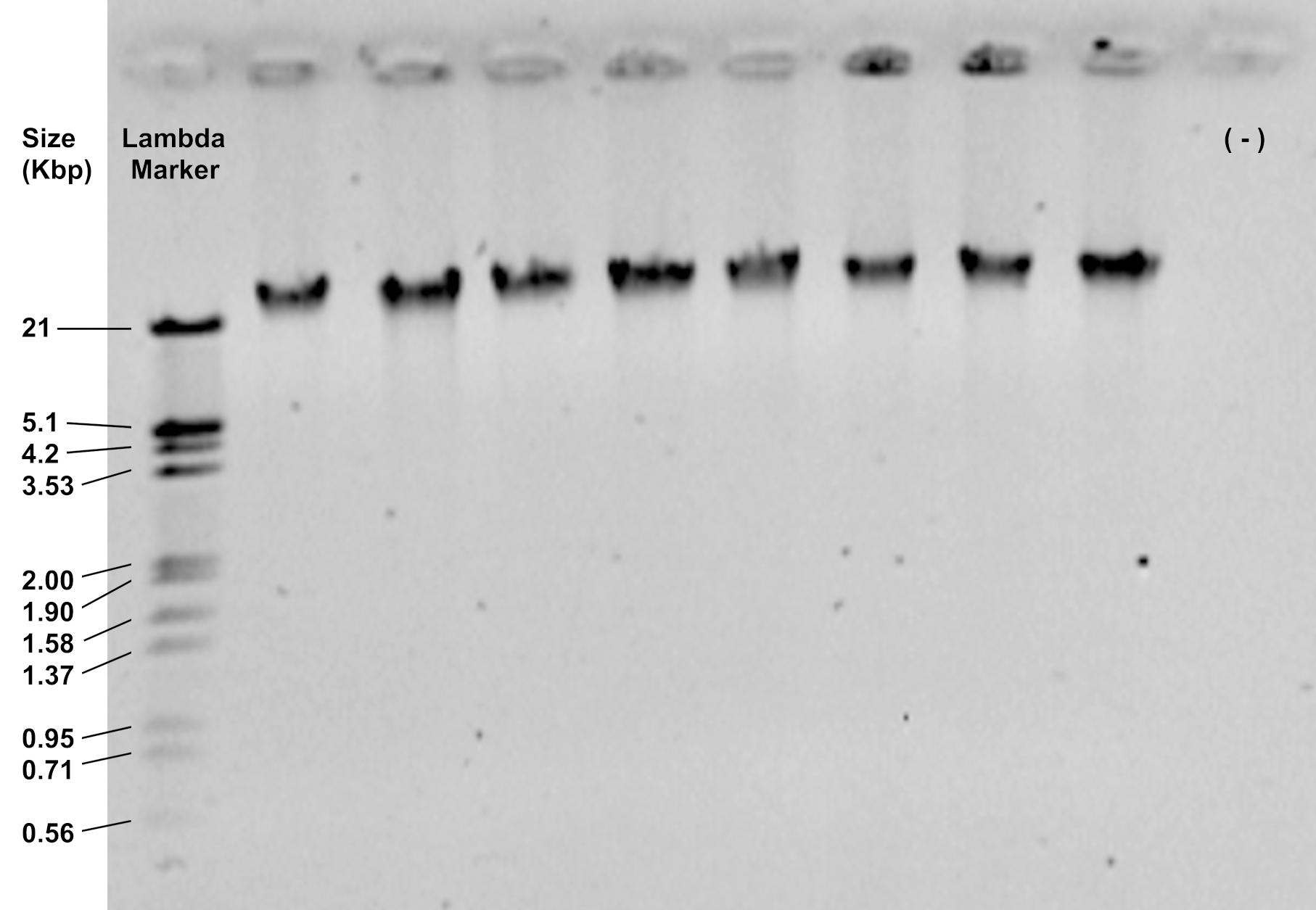
Figure 1. Representative image of DNA samples visualized in a 0.8% agarose gel
- Collect 50-100 mg of leaf tissue in a 1.5 ml centrifuge microtube. Flash-freeze the tissue in liquid nitrogen and grind it with a plastic pestle.
- Digestion with EcoRI
- Calculate the volume of the sample that corresponds to 700 ng of DNA, according to its concentration. However, consider that DNA concentration should be higher than 40.5 ng·μl-1, otherwise it will exceed the reaction’s final volume.
- Calculate the amount of each reagent needed for the total number of samples based on Table 2. To compensate pipetting errors, it is convenient to consider an additional sample to the total number of analyzed samples.
Table 2. Master mix for the enzymatic digestion with EcoRI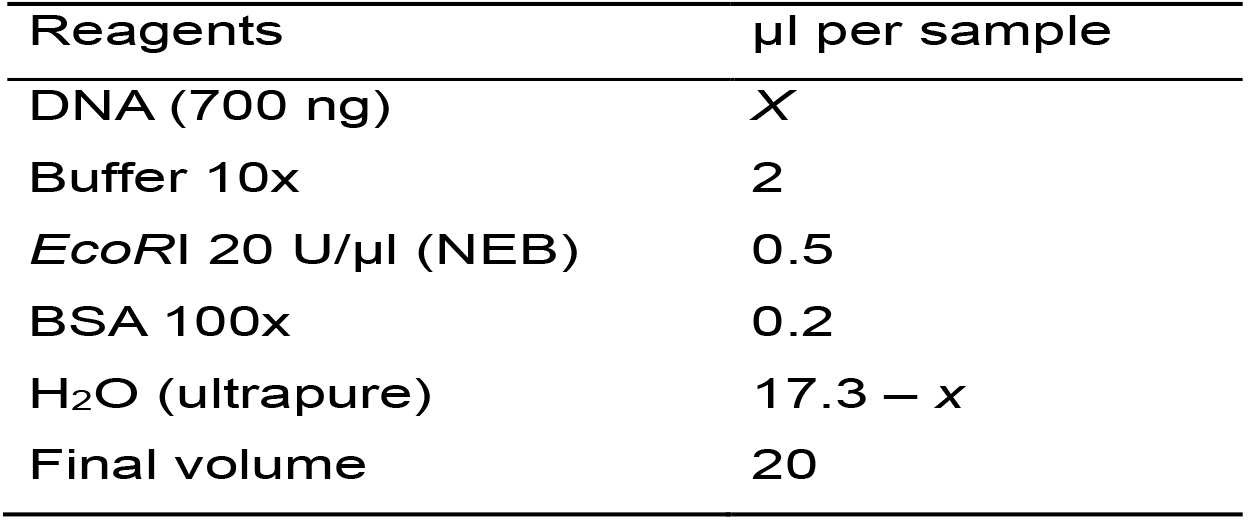
- Prepare the Master mix, homogenize by vortexing (2-3 s) and centrifuge for 10 s.
- Distribute the Master mix in individual tubes and add the DNA. Homogenize gently by inversion or by flicking the tubes (do not vortex) and centrifuge for 10 s.
- Incubate at 37 °C overnight in an incubator.
- Place the tubes on ice if you continue with the next step. Otherwise, store them in freezer at -20 °C until use.
- Digestion with HpaII and MspI
- For each tube from the previous reaction (B), distribute the content into two tubes (10 μl in each tube) and label the new tubes with the sample code, adding the letter "H" to one and the letter "M" to the other. From now on you will work with two tubes for each analyzed sample.
- Two Master mixes will be prepared, only differing on the restriction enzyme, one with HpaII and the other with MspI. For the two reactions, calculate the required amount of each reagent for the total number of samples plus one, based on Table 3.
Table 3. Master mixes for enzymatic digestions with HpaII y MspI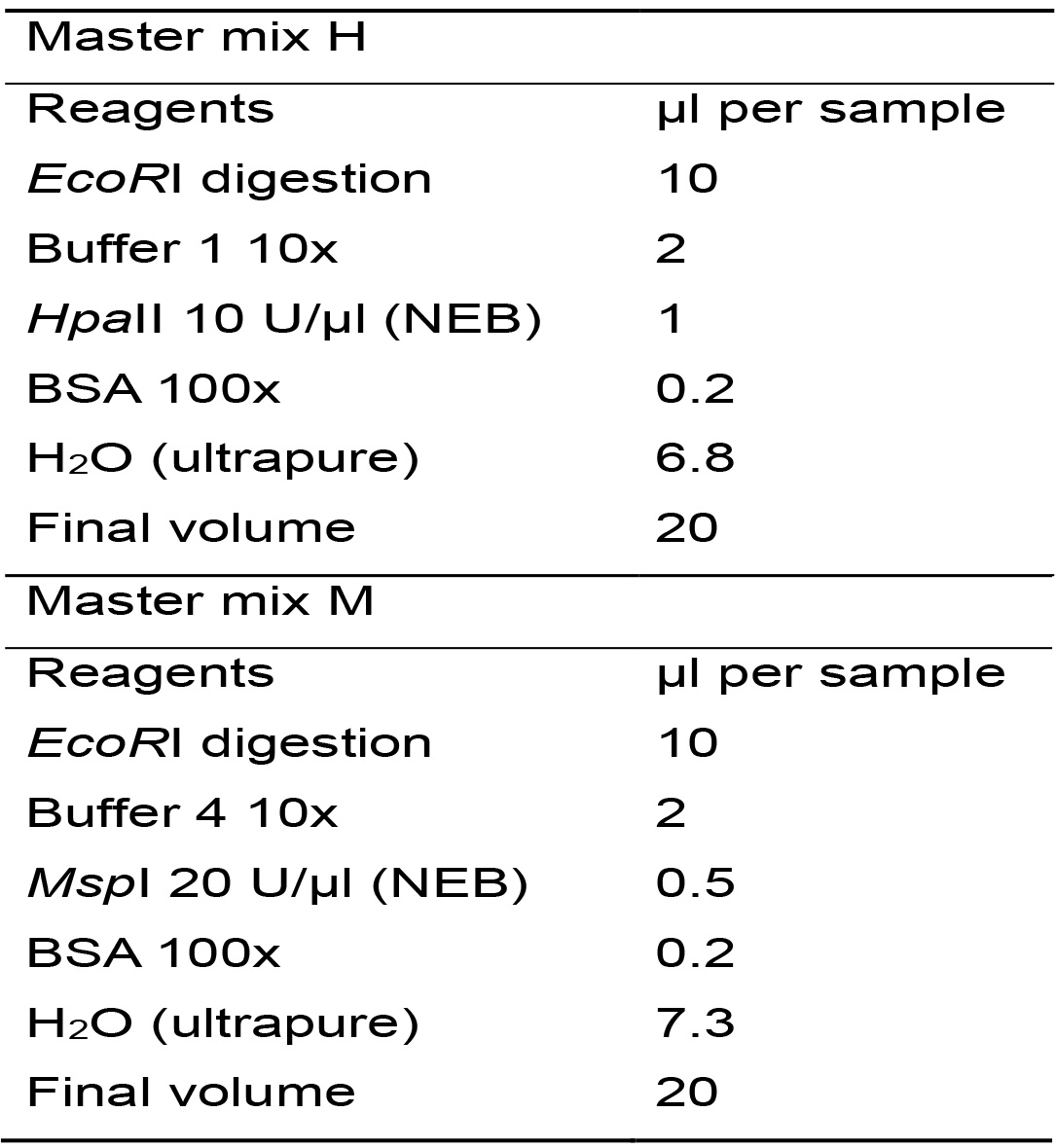
- Prepare the Master mixes, homogenize by vortexing (2-3 s) and centrifuge for 10 s.
- Distribute the Master mix H in the tubes labeled with the letter "H" and the Master mix M in the tubes labeled with the letter "M". From now on you will have two tubes per sample. Homogenize gently by inversion or by flicking the tubes (do not vortex) and centrifuge for 10 s.
- Incubate at 37 °C overnight in an incubator.
- (Optional) Examine the efficiency of both digestions by running 6 μl of the restriction products on a 1.2% agarose gel, at 90 V for 30 min. It should show a subtle smear from 5,100 to 100 bp (Figure 2).
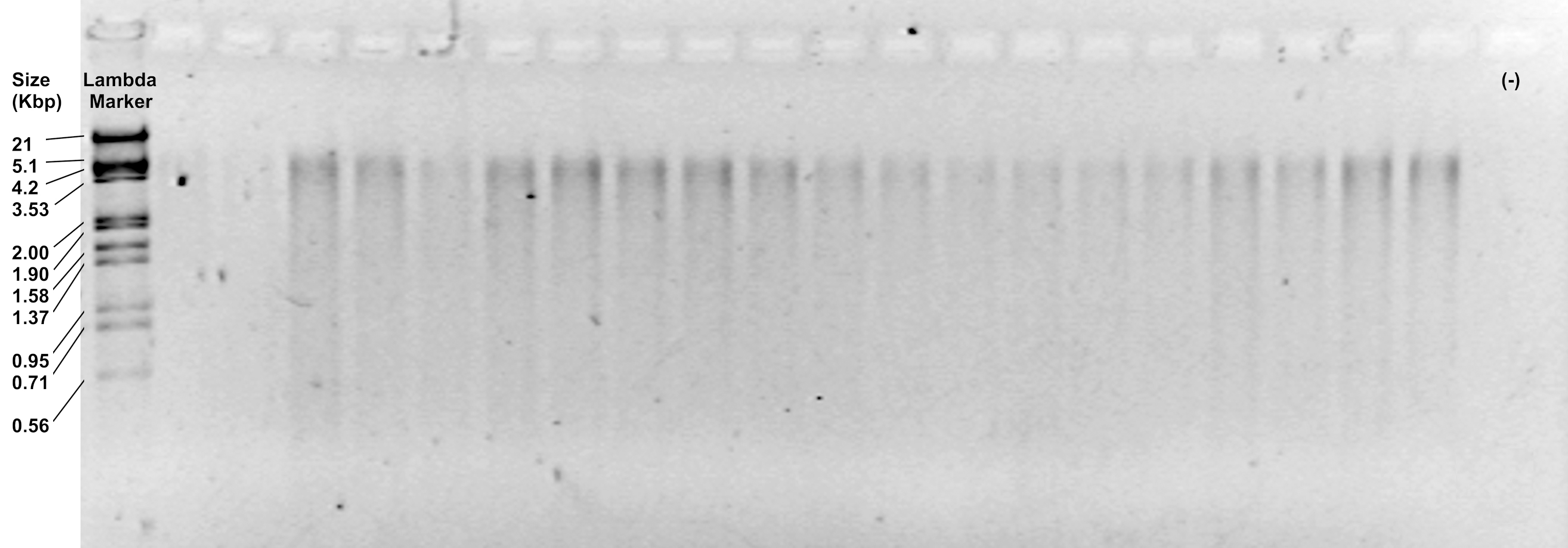
Figure 2. Representative image of restriction products visualized in a 1.2% agarose gel - Keep the tubes on ice if you will continue with the next step. Otherwise, store them in the freezer at -20 °C until use.
- Adaptors ligation
- Prepare the double stranded EcoRI and MspI/HpaII adaptors from the single strand oligonucleotides by adding equal amounts of each oligo in a tube (Table 1). Place the tubes in the thermocycler and run the ADAPTORS program (Table 4) with a hot lid.
Table 4. PCR programs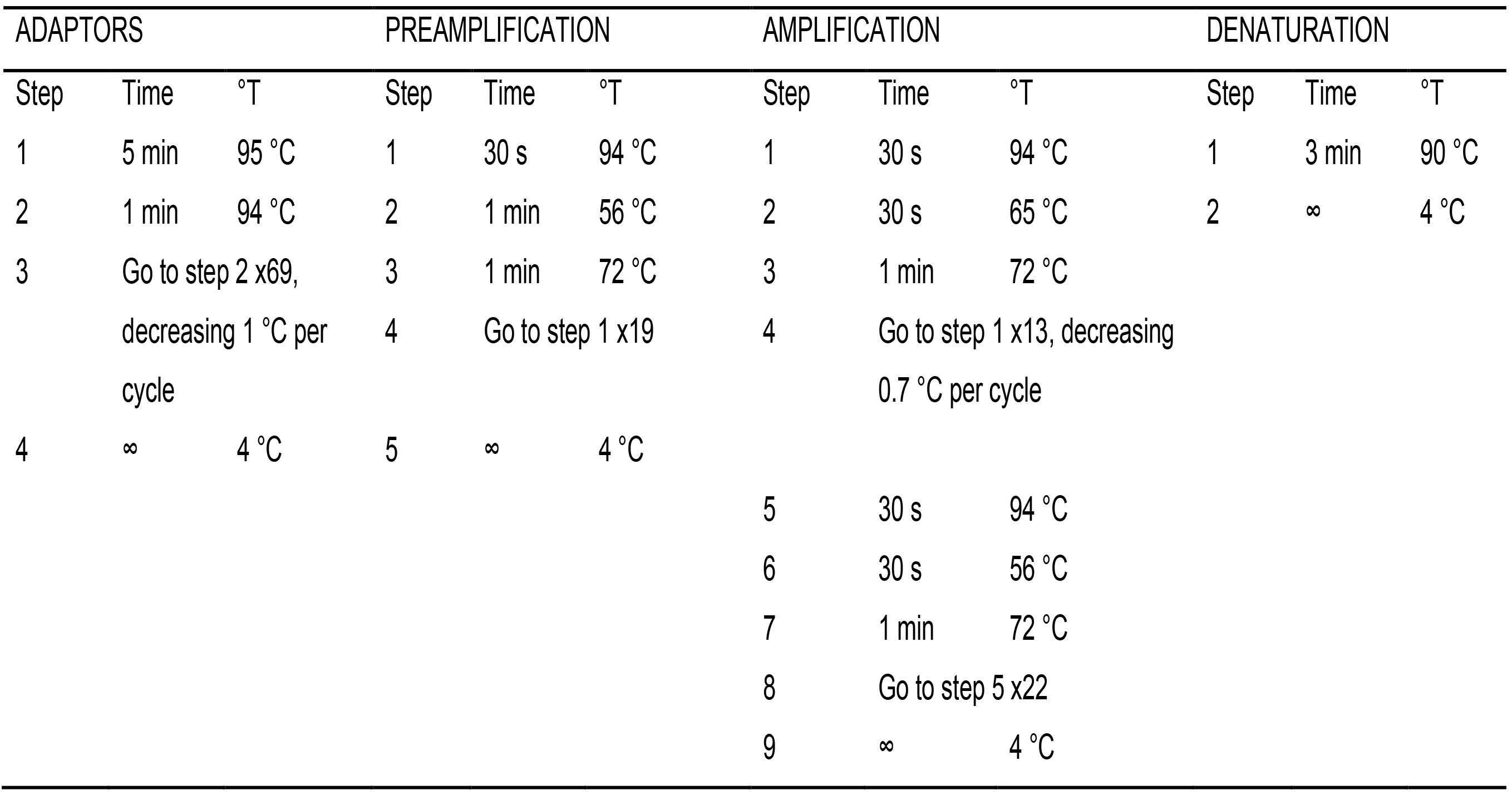
- Based on Table 5, calculate the amount of each reagent for the total number of samples plus one.
Table 5. Master mix for the ligation reaction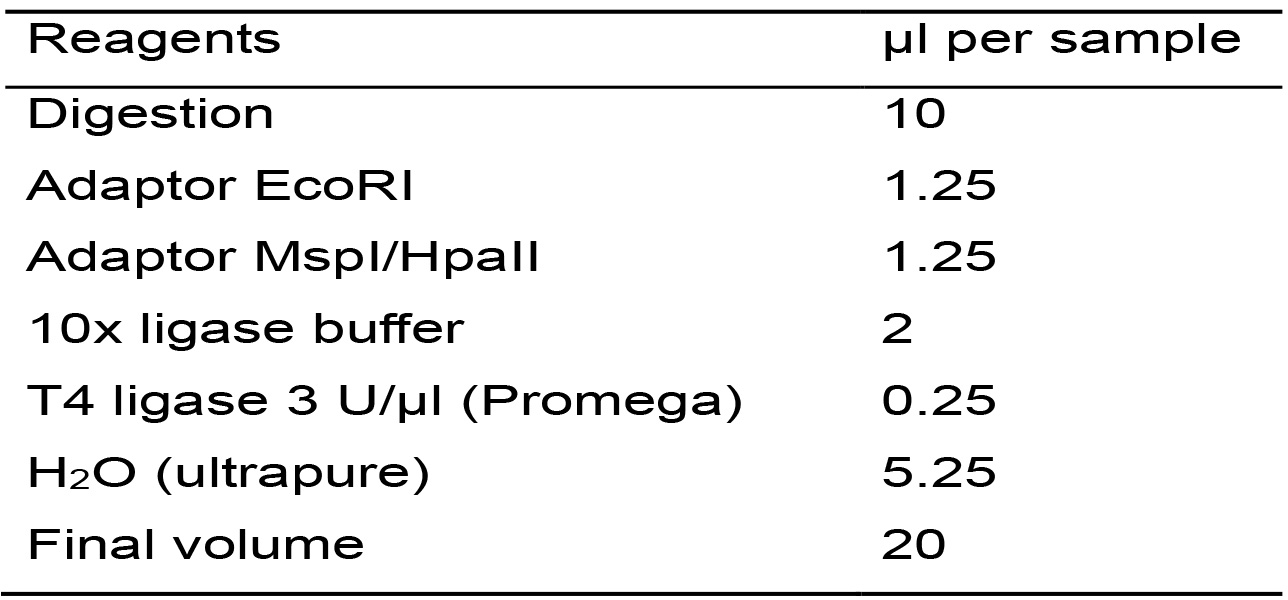
- Prepare the Master mix, homogenize by vortexing (2-3 s) and centrifuge for 10 s.
- Distribute the Master mix in individual tubes and add 10 μl of digestions. Homogenize gently by inversion or by flicking the tubes (do not vortex) and centrifuge for 10 s.
- Incubate at room temperature for 3 h.
- Keep the tubes on ice if you will continue with the next step. Otherwise, store them in a freezer at -20 °C until use.
- Prepare the double stranded EcoRI and MspI/HpaII adaptors from the single strand oligonucleotides by adding equal amounts of each oligo in a tube (Table 1). Place the tubes in the thermocycler and run the ADAPTORS program (Table 4) with a hot lid.
- First amplification (Preamplification)
For this PCR reaction, oligonucleotides with the same sequences as the adaptors plus 1 selective nucleotide are used. These oligonucleotides are identified as Primer EcoRI +1 and Primer H/M +1 (Table 1).- Calculate the necessary amount of each reagent needed for the total samples based on Table 6, considering an additional sample to compensate for pipetting errors.
Table 6. Master mix for the pre-amplification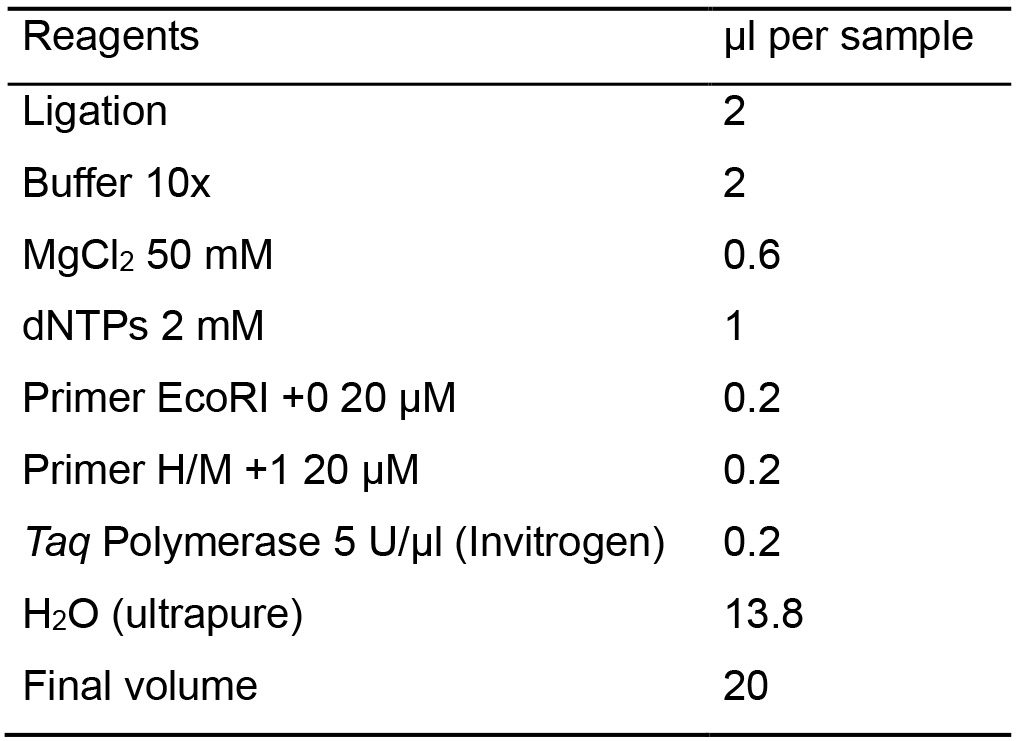
- Prepare the Master mix, homogenize by vortexing (2-3 s) and centrifuge for 10 s.
- Distribute 18 μl of the Master mix in each tube and add 2 μl of the ligations. Add 2 μl of water to the remaining mix to use it as a negative reaction control. Homogenize gently by inversion or flicking (do not vortex) and centrifuge for 10 s.
- Place the samples plus the negative control in the thermocycler and run the PREAMPLIFICATION program (Table 4) with a hot lid.
- Check the efficiency of the reaction by loading 5 μl of the pre-amplifications on a 1.2% agarose gel and running at 90 V for 30 min. It should show an intense smear with few or some diffuse bands from 1,500 to 100 bp (Figure 3).
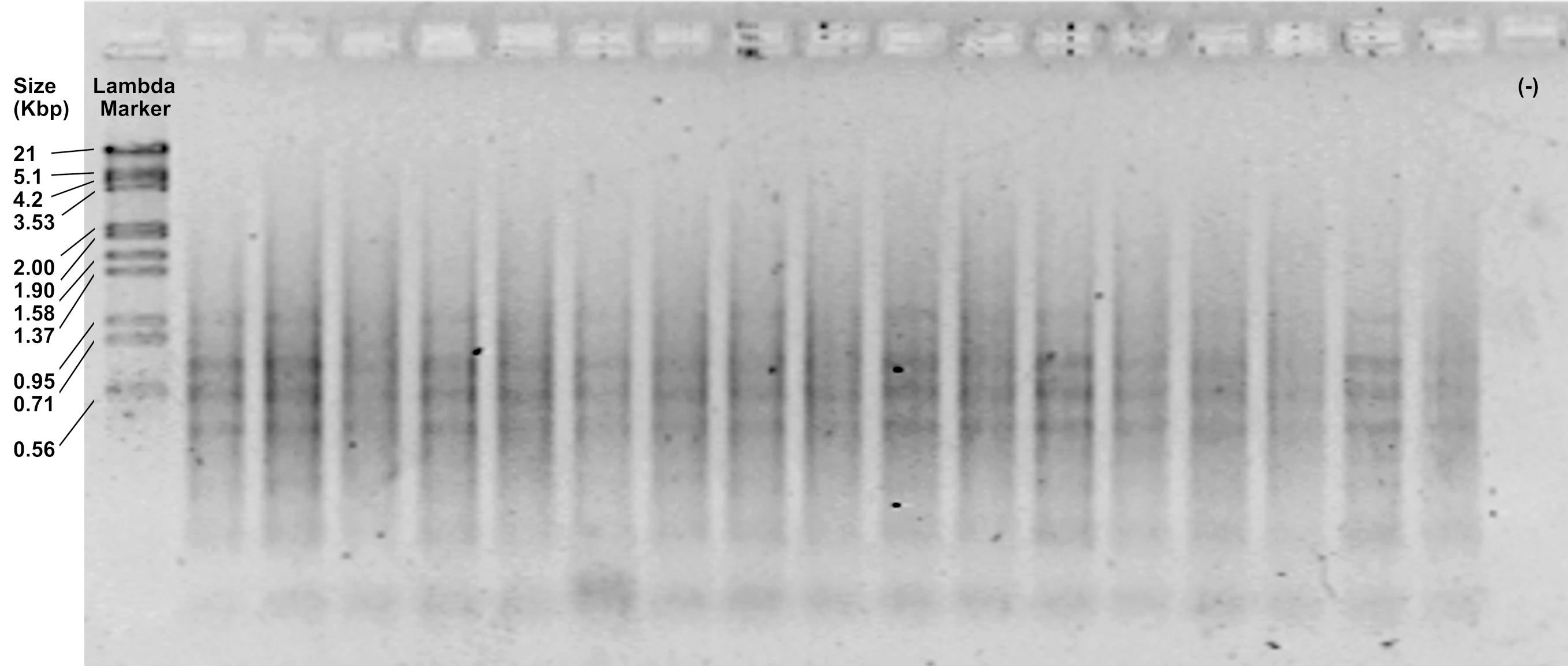
Figure 3. Representative image of pre-amplification products visualized in a 1.2% agarose gel - Dilute the pre-amplifications 1:3. Keep on ice if you continue with the next step or store in a freezer at -20 °C. Preamplifications can be safely stored in a freezer for several months.
- Calculate the necessary amount of each reagent needed for the total samples based on Table 6, considering an additional sample to compensate for pipetting errors.
- Second amplification (Selective amplification)
For this PCR reaction, oligonucleotides with the same sequences as the adaptors plus 3 selective nucleotides are used. These oligonucleotides are identified as Primer EcoRI +3 and Primer H/M +3 (Table 1).- Calculate the necessary amount of each reagent needed for the total number of samples plus one, based on Table 7.
Table 7. Master mix for the amplification reaction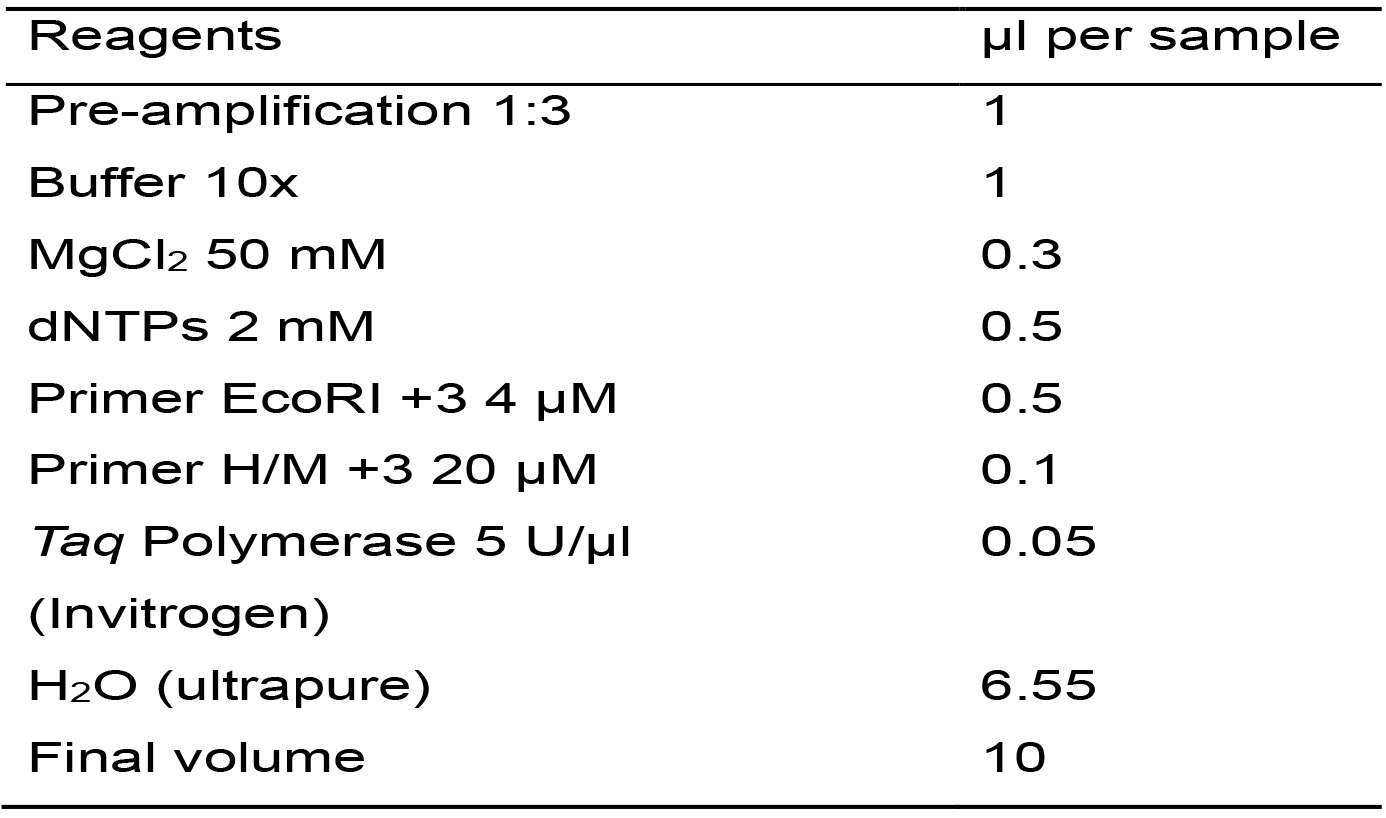
- Prepare the Master mix, homogenize by vortexing (2-3 s) and centrifuge for 10 s.
- Distribute 19 μl of the Master mix in each tube and add 1 μl of 1:3 diluted preamplifications. To the remaining mix add 1 μl of water to use it as a negative reaction control. Homogenize gently by inversion or flicking (do not vortex) and centrifuge for 10 s.
- Place in the thermocycler and run the AMPLIFICATION program (Table 4) with a hot lid.
- Check the efficiency of the reaction by loading 5 μl of the amplification products on a 1.2% agarose gel and running at 90 V for 30 min. Depending of the primer combination, it should show a smear with diffuse bands from 700 to 100 bp (Figure 4).
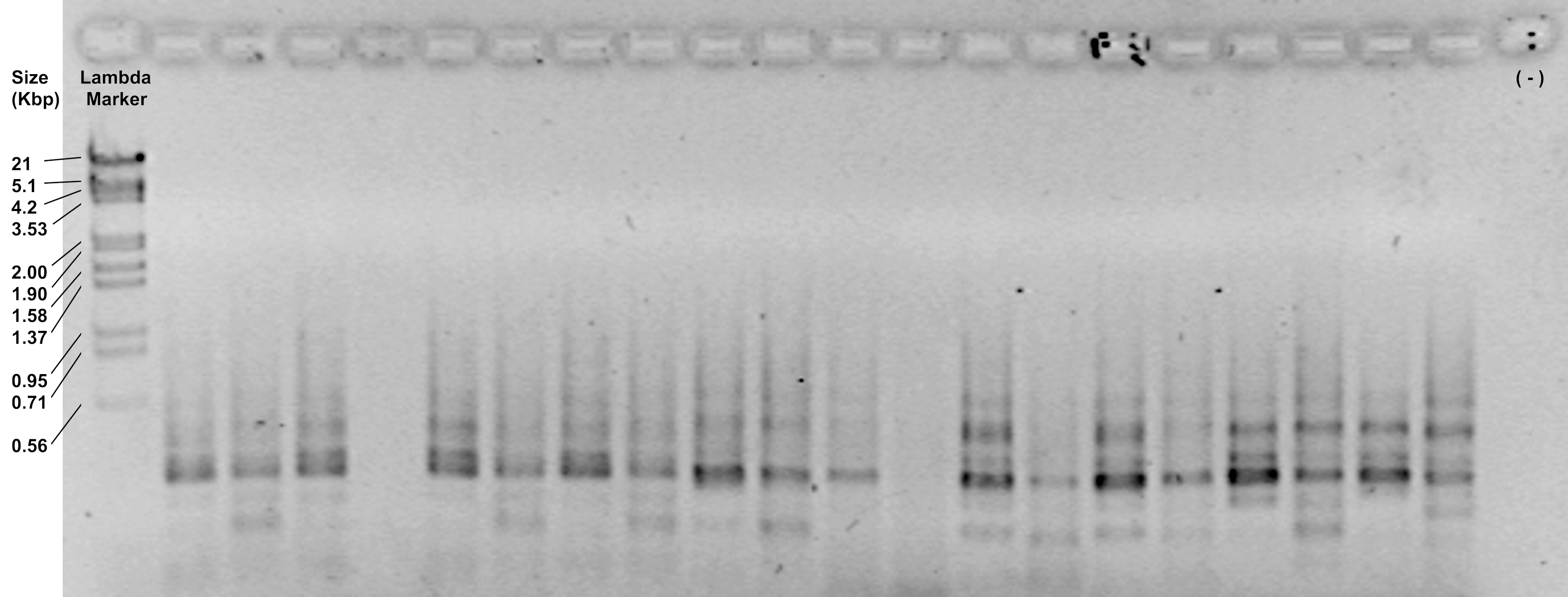
Figure 4. Representative image of amplification products visualized in a 1.2% agarose gel - Keep the tubes on ice if you continue with the next step or store them in a freezer at -20 °C. Fluorescence-labeled amplifications can be safely stored at least for 6 months at -20 °C maintaining their light emission.
- Calculate the necessary amount of each reagent needed for the total number of samples plus one, based on Table 7.
- Electrophoresis
- Calculate the necessary amount of each reagent needed for the total samples based on Table 8, considering an extra sample to compensate for pipetting errors.
Table 8. Master mix for the amplification reaction
- Prepare the Master mix, homogenize by vortexing (2-3 s) and centrifuge for 10 s.
- Distribute 9 μl of the Master mix in each well of the 96-well plate and add 1 μl of amplification products.
- Place the plate in the thermocycler and run the program DENATURATION (Table 4) with a hot lid.
- Take the plate out of the thermocycler and put it on ice.
- Place the plate in the ABI Prism 3,130 DNA sequencer (Applied Biosystems) and run.
- Calculate the necessary amount of each reagent needed for the total samples based on Table 8, considering an extra sample to compensate for pipetting errors.
Data analysis
- Allele calling and matrix generation
- Semi-automated scoring was performed on the resulting electronic profiles using GeneMapper v3.7 (Applied Biosystems). The length range of analyzed fragments was from 100 to 500 bp. Full specifications of the parameters used in GeneMapper are summarized in Cara et al. (2014). GeneMapper’s Report Manager tool was used to retrieve only sample names and allele columns into a Comma Separated Values (.csv) file.
- Patterns of presence/absence between the EcoRI/HpaII and the EcoRI/MspI digests were codified from 0 to 3; then, this codification was converted into a binary matrix for either presence (1) or absence (0) of patterns 3, 2 or 1 (Figure 5). Pattern 0 was not codified in the binary matrix because the absence of fragments in EcoRI/HpaII and EcoRI/MspI digests might be due to either methylation of external cytosines or variations in the nucleotide sequence. The following R script was developed to perform this analysis.
R Script for generating an MSAP binary matrix:
##### R Commands used to generate an MSAP binary matrix from a Genemapper csv file
## The following command makes sure that the working memory is clear
rm(list = ls(all = TRUE))
## Set working directory (PATH will be specific to where data are)
setwd("C:/...")
## Read in dataset. Input file should contain Samples by rows and Loci by columns, including header (loci names) and sample names in the first column
## Each sample digested by HpaII and MspI is identified as 'SampleName_H' and 'SampleName_M', one below the other.
Data = read.csv("MSAP_Genemapper.csv", header=T, row.names = 1, check.names=F)
## Create objects
out <- c()
MSAP_patterns <- matrix(nrow = dim(Data)[2], ncol = 0)
## This is a double loop for j hybrids and i loci. It generates a matrix of MSAP patterns (0, 1, 2 or 3).
for (j in 1:dim(Data)[1]) if (j %% 2 ==1)
{
for (i in 1:dim(Data)[2])
{
out <- append(out, ifelse(Data[j,i]+Data[j+1,i]==0, '0',
ifelse(Data[j,i]+Data[j+1,i]==2, '1',
ifelse(Data[j,i]==1, '2', '3'))))
}
MSAP_patterns <- cbind(MSAP_patterns, matrix(out, dimnames = list(colnames(Data), substr(row.names(Data)[j], 1, nchar(as.character(row.names(Data)[j]))-2))))
out <- c()
}
##This is a loop that creates a binary matrix of presence/absence of each pattern (1, 2 or 3) for i loci.
dummies <- matrix(nrow = dim(MSAP_patterns)[2], ncol = 0)
for (i in 1:dim(MSAP_patterns)[1])
{
dummies <- cbind(dummies, matrix(as.numeric(MSAP_patterns[i,] == 1), dimnames = list(colnames(MSAP_patterns), paste(row.names(MSAP_patterns)[i], "1", sep = "_"))), matrix(as.numeric(MSAP_patterns[i,] == 2), dimnames = list(colnames(MSAP_patterns), paste(row.names(MSAP_patterns)[i], "2", sep = "_"))), matrix(as.numeric(MSAP_patterns[i,] == 3), dimnames = list(colnames(MSAP_patterns), paste(row.names(MSAP_patterns)[i], "3", sep = "_"))))
}
binary_matrix <- dummies[,colSums(dummies != 0) != 0]
## Save output to working directory
write.csv(binary_matrix, file = "MSAP_binary_matrix.csv")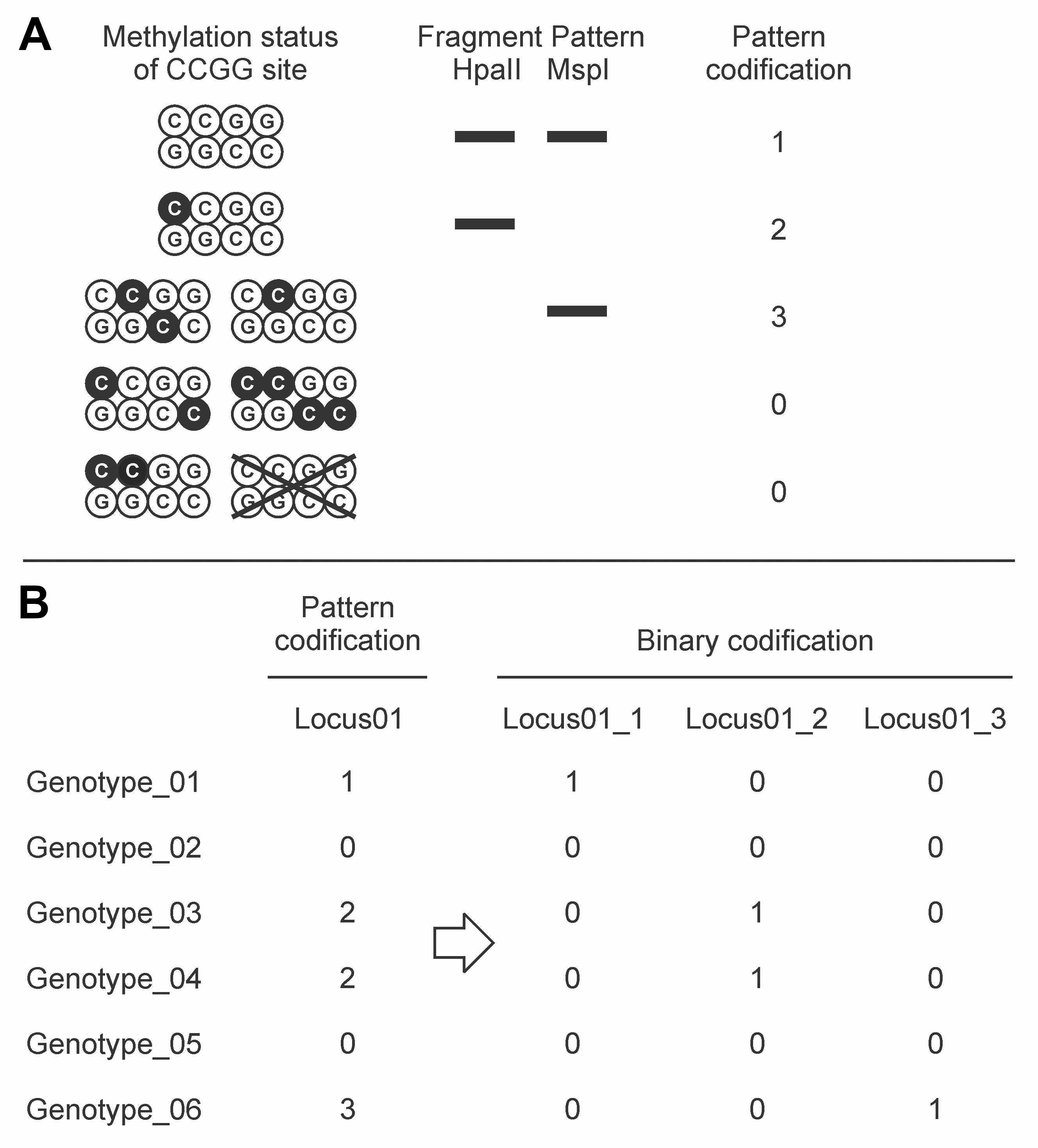
Figure 5. Methylation-sensitive Amplified Polymorphism (MSAP) pattern analysis. A. Inference of methylation status of CCGG restriction sites from the fragment amplification profiles and DNA methylation pattern codification. Black C represents methylated cytosines. B. Binary matrix obtained by codifying the presence (1) or absence (0) of the particular methylation pattern.
- Determination of species-specific and novel fragments
- The amplification patterns observed in the evaluated natural hybrids were compared with the patterns obtained in the synthetic hybrids and their parental genotypes. Fragments present in the synthetic hybrids were categorized as S. microdontum or S. kurtzianum species-specifics. In addition, the presence of novel amplification fragments in the synthetic hybrids (that is present in the hybrids but absent in the genotypes of the parental species S. microdontum and S. kurtzianum) was assessed in the natural hybrids. Novel fragments were classified as S. x rechei species-specific if they were also present in at least one of the S. x rechei evaluated genotypes or as novel if they were only observed in the synthetic hybrids. The following R script was developed to perform this analysis.
R Script for analyzing MSAP epiloci inheritance in hybrids:
#### R Commands used to analyze MSAP epiloci inheritance in Hybrids
## The following command makes sure that the working memory is clear
rm(list = ls(all = TRUE))
## Set working directory (PATH will be specific to where data are)
setwd("C:/...")
## Read in dataset. Data is organized as Samples by rows and Loci by columns
Data = read.csv("MSAP_binary_matrix.csv", header=T, row.names = 1, check.names=F)
## Create objects
out <- c()
results <- matrix(nrow = 4, ncol = 0)
## This is a double loop for j hybrids and i loci. Rows are as follows: 1-5 Hybrids, 6 S. kurtzianum (Parental1), 7 S. microdontum (Parental2), 8-15 S. x rechei (Natural hybrid). Any modifications in these numbers should be applied to the following commands.
for (j in 1:5)
{
for (i in 1:dim(Data)[2])
{
out <- append(out, ifelse(Data[j,i]>0 & Data[6,i]>0 & Data[7,i]==0 & sum(Data[8:15,i])==0, 'A', ## Fragments shared only by hybrids and S. kurtzianum
ifelse(Data[j,i]>0 & Data[6,i]==0 & Data[7,i]>0 & sum(Data[8:15,i])==0, 'B', ## Fragments shared only by hybrids and S. microdontum
ifelse(Data[j,i]>0 & Data[6,i]==0 & Data[7,i]==0 & sum(Data[8:15,i])>0, 'C', ## Fragments shared only by hybrids and S. x rechei
ifelse(Data[j,i]>0 & Data[6,i]==0 & Data[7,i]==0 & sum(Data[8:15,i])==0, 'D', 'E'))))) ## Fragments specific to hybrids
}
results <- cbind(results, matrix(table(out, exclude = "E"),dimnames = list(c("Kurtzianum", "Microdontum", "Rechei", "Hybrid"), row.names(Data)[j])))
out <- c()
}
## Transform counts by hybrid into percentages
percentages <- t(results)/colSums(results)*100
## Save output to working directory
write.csv(percentages, file = "MSAP_Hybrids.csv")
## A basic graphical representation
barplot(t(percentages), horiz = TRUE, las=1, col = c("red", "blue", "green", "yellow"))
- The amplification patterns observed in the evaluated natural hybrids were compared with the patterns obtained in the synthetic hybrids and their parental genotypes. Fragments present in the synthetic hybrids were categorized as S. microdontum or S. kurtzianum species-specifics. In addition, the presence of novel amplification fragments in the synthetic hybrids (that is present in the hybrids but absent in the genotypes of the parental species S. microdontum and S. kurtzianum) was assessed in the natural hybrids. Novel fragments were classified as S. x rechei species-specific if they were also present in at least one of the S. x rechei evaluated genotypes or as novel if they were only observed in the synthetic hybrids. The following R script was developed to perform this analysis.
Recipes
- Extraction buffer
100 mM Tris-HCl (pH 8.0)
20 mM EDTA (pH 8.0)
1.4 M NaCl
0.4% (v/v) β-mercaptoethanol
2% (w/v) CTAB - 1x TBE (Tris-Borate-EDTA) gel running buffer
89 mM Tris base
89 mM Boric acid
2 mM EDTA (pH 8.0) - 6x DNA loading buffer
30% (v/v) Glycerol
0.25% (w/v) Bromophenol blue
0.25% (w/v) Xylene cyanol FF
Notes
Prepare all Master mixes by first adding the larger volumes and then the smaller ones, leaving the enzymes to the end.
Acknowledgments
We thank Dr María Virginia Sanchez Puerta for critical reading of the draft manuscript. This work was supported by Agencia Nacional de Promoción Científica y Tecnológica and Universidad Nacional de Cuyo, Argentina PICT 1243. This protocol was adapted from our previous work (Cara et al., 2019).
Competing interests
The authors have not competing interest.
References
- Camadro, E. L., Erazzu, L. E., Maune, J. F. and Bedogni, M. C. (2012). A genetic approach to the species problem in wild potato. Plant Biol (Stuttg) 14(4): 543-554.
- Cara, N., Ferrer, M. S., Masuelli, R. W., Camadro, E. L. and Marfil, C. F. (2019). Epigenetic consequences of interploidal hybridisation in synthetic and natural interspecific potato hybrids. New Phytol 222(4): 1981-1993.
- Cara, N., Marfil, C. F., García Lampasona, S. C., Masuelli, R. W. (2014). Comparison of two detection systems to reveal AFLP markers in plants. Botany 92: 607-610.
- Vos, P., Hogers, R., Bleeker, M., Reijans, M., van de Lee, T., Hornes, M., Frijters, A., Pot, J., Peleman, J., Kuiper, M. and et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23(21): 4407-4414.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Cara, N., Marfil, C. F., Bertoldi, M. V. and Masuelli, R. W. (2020). Methylation-sensitive Amplified Polymorphism as a Tool to Analyze Wild Potato Hybrids. Bio-protocol 10(13): e3671. DOI: 10.21769/BioProtoc.3671.
Category
Plant Science > Plant molecular biology > Genetic analysis
Molecular Biology > DNA > Genotyping
Molecular Biology > DNA > Electrophoresis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.