- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative Nucleocytoplasmic Transport Assays in Cellular Models of Neurodegeneration
(*contributed equally to this work) Published: Vol 10, Iss 12, Jun 20, 2020 DOI: 10.21769/BioProtoc.3659 Views: 5318
Reviewed by: Steven BoeynaemsAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Nucleocytoplasmic transport deficits are suggested to play a role in neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS). Given the importance and complexity of this process, understanding when these aberrations occur and which pathways are involved is of great importance. Here, we make use of CRISPR-Cas9 technology to design cell lines stably expressing fluorophore proteins shuttling between the nucleus and cytoplasm by karyopherins of choice. To validate this protocol, we measured an ALS-associated nucleocytoplasmic transport pathway in the presence of the disease-associated peptide poly-PR. This technique allows measuring a particular active nucleocytoplasmic transport pathway in intact cells in a neurodegenerative disease-associated context. Moreover, these experiments can be performed without the need for expensive equipment and have the potential to be upscaled for high-throughput screening purposes.
Keywords: Neurodegenerative diseaseBackground
Nucleocytoplasmic transport is crucial for cellular homeostasis and occurs across the nuclear membrane via large multi-protein complexes that form aqueous channels, called nucleopore complexes (NPCs) (Stoffler et al., 1999; Ryan and Wente, 2000). Although these channels allow small proteins to passively equilibrate across the nuclear membrane (passive transport), most proteins appear to be actively transported by karyopherins, namely importins or exportins (active transport). Cargo proteins that contain a nuclear localization signal (NLS) are targeted by importins. Various receptor-mediated import pathways have been identified, but the best characterized pathway involves importin-β1/importin-α (KPNB1/KPNAx) of which the cargos contain a classical NLS (cNLS), exemplified by the SV40 large T antigen NLS (Lange et al., 2007). Protein export from the nucleus into the cytoplasm is mediated by exportins. Several pathways of nuclear export have been identified. The most common type of nuclear export signal (NES) is a leucine-rich sequence motif recognized by exportin1 (XPO1), of which the NES from protein kinase inhibitor (PKI) is the most often used prototype (Ossareh-Nazari et al., 2001).
Altered subcellular distribution of proteins is a common characteristic among neurodegenerative disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS) (Fahrenkrog and Harel, 2018). Increasing evidence indicates impaired nucleocytoplasmic transport as a possible mechanism explaining this aberrant protein localization in the degenerating neurons. For example, in the majority of ALS patients the RNA binding protein TDP-43 (Arai et al., 2006; Neumann et al., 2006) or, to a lesser extent, FUS (Kwiatkowski et al., 2009; Vance et al., 2009; Tyzack et al., 2019) becomes partially depleted from the nucleus and aggregates in the cytoplasm. Interestingly, the most common genetic cause associated with ALS, a G4C2-repeat expansion in the C9orf72 gene (Majounie et al., 2012), has been suggested to affect nucleocytoplasmic transport in various ways (Boeynaems et al., 2016; Yuva-Aydemir et al., 2018). One possible toxic pathway is the bidirectional translation of the repeat expansion by repeat-associated non-ATG (RAN) translation into dipeptide repeat proteins (DPRs), namely poly-GP, poly-GA, poly-GR, poly-PA and poly-PR (Ash et al., 2013; Zu et al., 2010; Mori et al., 2013a and 2013b). Here, we expressed the DPR poly-PR using a lentiviral vector transduced into our reporter cells, to subsequently measure nucleocytoplasmic transport.
Although there is growing evidence indicating that impaired nucleocytoplasmic transport is a component of neurodegenerative diseases, it is still under large debate whether nucleocytoplasmic transport deficits are a consequence of neurodegeneration or an instigator (Hutten and Dormann, 2019). As nucleocytoplasmic transport is a complex and stress-sensitive process, reliable assays with a minimal interference are essential to answer this question, as we did before (Vanneste et al., 2019). We developed several reporter cell lines stably expressing a shuttling-fluorophore allowing us to consistently measure nucleocytoplasmic import as a function of time and in intact cells without the need for transfection. Moreover, this method is cheap and can easily be up scaled for high throughput screening purposes. By fine-tuning the NLS and NES fused to the reporter, the subcellular localization of the reporter can be modified and different transport pathways of interest can be analyzed. While the experiments here are performed with Hela Kyoto cells, the protocol can easily be adapted for other cell types.
Materials and Reagents
- 6-well plate (Corning Life science, 3516)
- 96-well plate (TTP, catalog number: 92696 )
- T-25 culture flask (TTP, catalog number: 90026 )
- Cover Glass (VWR, catalog number: 631-0150 )
- Nunc CryoTube (Sigma, catalog number: V7884 )
- Hela Kyoto cells (Cellosaurus ID: CVCL_1922)
- HEK-293T cells (ATCC, catalog number: CRL-3216TM)
- pcDNATM 3.1 (+) plasmid (Thermo Fisher Scientific, catalog number: V790-20 )
- pGL4 plasmid (Addgene)
- Plasmid containing Cas9 was a kind gift from Jonathan L. Schmid-Burgk (Schmid-Burgk et al., 2016).
- Plasmid containing the gRNA was a kind gift from Jonathan L. Schmid-Burgk (Schmid-Burgk et al., 2016)
- GibcoTM Dulbecco’s Modified Eagle’s Medium (DMEM), high glucose (Thermo Fisher Scientific, catalog number: 11965092 )
- Fetal Bovine Serum (GE Healthcare, Hyclone, catalog number: SV30160.03 )
- GibcoTM Gentamicin (Thermo Fisher Scientific, catalog number: 15750045 )
- GibcoTM Puromycin Dihydrochloride (Thermo Fisher Scientific, catalog number: A1113802 )
- GibcoTM Trypsin-EDTA (0.05%), phenol red (Thermo Fisher Scientific, catalog number: 253000054 )
- LipofectamineTM 3000 Transfection Reagent (Thermo Fisher Scientific, catalog number: L3000015 )
- Dimethyl sulfoxide (DMSO) (Sigma, catalog number: D2650-100ml )
- Importazole (Selleckchem, catalog number: S8446 )
- Leptomocyin B (Invivogen, catalog number: tlrl-lep )
- NucBlue Live ReadyProbesTM Reagent Hoechst 33342 (Thermo Fisher Scientific, catalog number: R37605 )
- GibcoTM Dulbecco’s Phosphate-Buffered Saline (DPBS), no calcium, no magnesium (Thermo Fisher Scientific, catalog number: 14190250 )
- Formaldehyde 16 % methanol free, Ultra Pure (Polysciences Inc., catalog number: 18814-20 )
- Hexadimethrine bromide (polybrene) (MedChem Express, catalog number: 2878-55-4 )
- X-tremeGENE9 DNA transfection reagent (Sigma-Aldrich/Roche, catalog number: 06 365 809 001 )
- ON-TARGETplus KPNB1 siRNA human (Horizon Discovery, catalog number: L-01523-00-0005 )
- ON-TARGETplus KPNA2 siRNA human (Horizon Discovery, catalog number: L-004702-00-0005 )
- Gene Pulser Electroporation Butter (Bio-Rad, catalog number:165-2677)
- siRNA buffer (Horizon Discovery, catalog number: B-00200-UB-100 )
Equipment
- Incubator
- Forceps
- Mr. Frosty freezing container (Thermo Fisher Scientific, catalog number: 5100-0001 )
- Scanning confocal microscope, e.g., SP8 confocal microscope (Leica, model: SP8 MDi8 )
- CellInsight CX5 high content screening platform (Thermo Fisher Scientific, catalog number: CX51110 )
Software
- ImageJ (Wayne Rasband (retired from), https://imagej.nih.gov/ij/download.html)
- CellProfiler (Broad Institute, https://cellprofiler.org/)
- Snapgene (GSL Biotech LLC, https://www.snapgene.com/)
Procedure
A summary of the protocol is shown in Figure 1. The first part of this protocol describes the generation of three different reporter cell lines (Figure 2), which allows us to investigate the import pathways involved in ALS pathology. The protocol can be easily adapted to investigate other transport pathways by simply changing the nuclear localization or nuclear export signal in the constructs. The second part describes how to measure and analyze nucleocytoplasmic transport.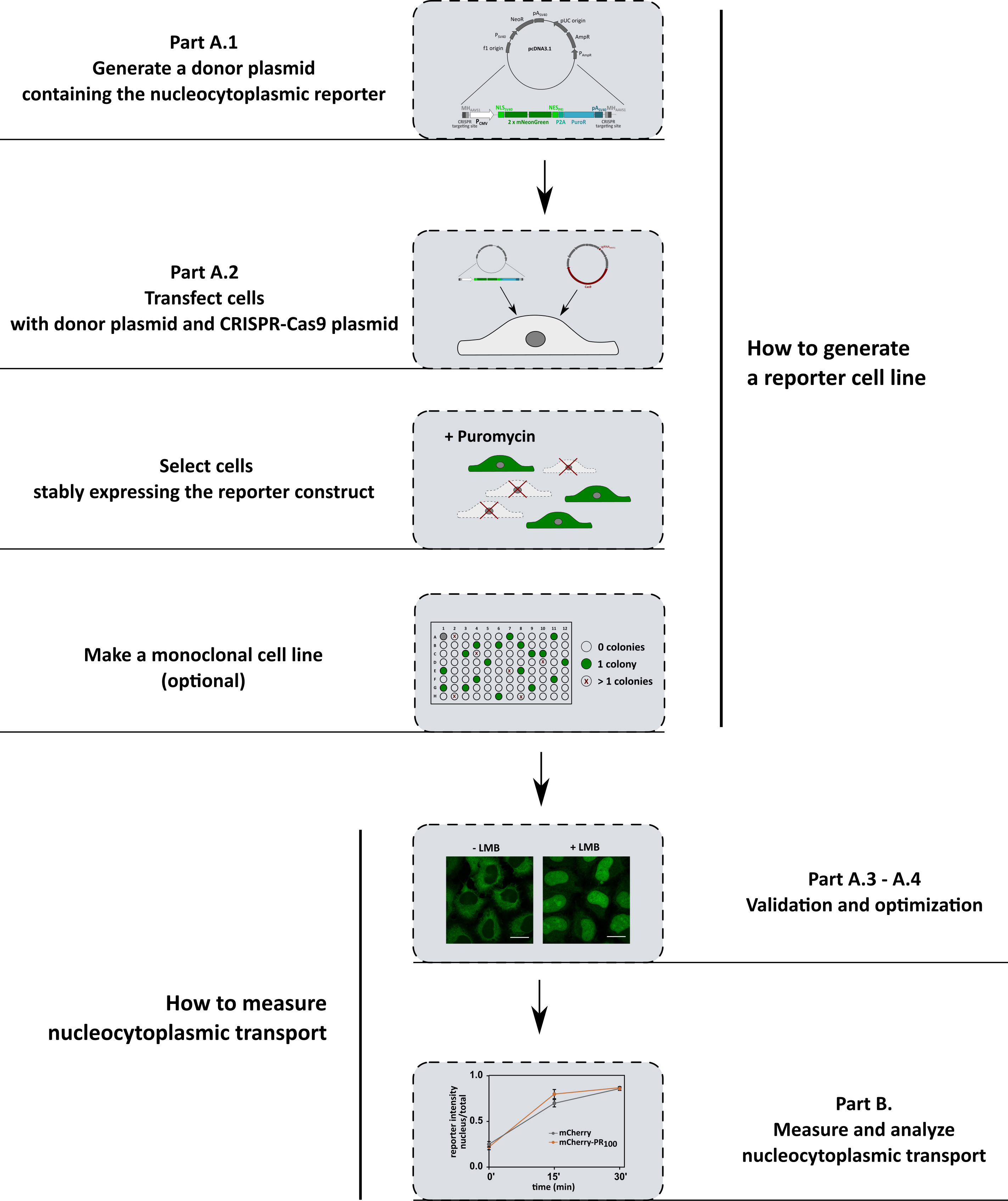
Figure 1. Workflow summary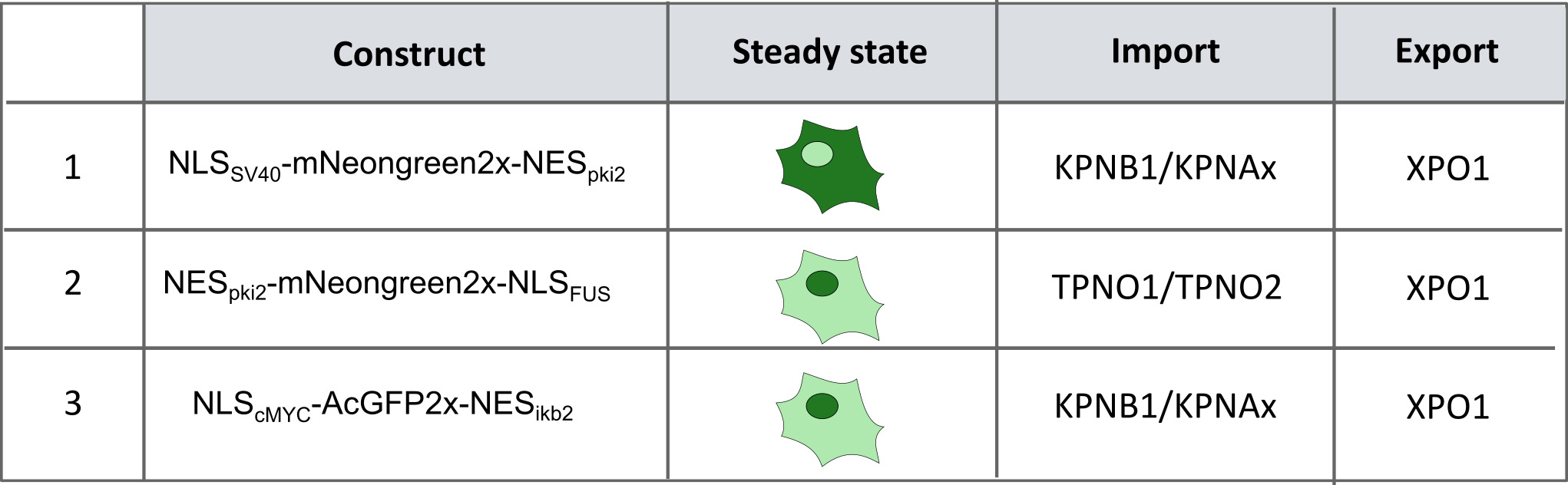
Figure 2. Generated reporter cell lines. Using CRISPR-Cas9 technology three different reporter cell lines were generated that stably express the indicated constructs. This allows us to investigate the import pathways that are involved in ALS: (1 + 3) KPNB1/KPNAx-mediated import pathway (pathway used by TDP43) and (2) the TPNO1-mediated pathway (pathway used by FUS). In cell line 1, the reporter mainly localizes into the cytoplasm. This allows, in combination with an XPO1-inhibitor, to induce a time-dependent transport from the cytoplasm into the nucleus and as such to measure KPNB1/KPNAx-mediated import over time. Cell lines 2 and 3 allow measuring a shift in the balance between import and export in a simple way.
- Generation of cell lines to study nucleocytoplasmic transport
The reporter constructs, driven by a CMV promotor, were integrated in the AAVS1 safe harbor locus of Hela Kyoto cells using a CRISPR-Cas9 knock-in protocol. Three different CRISPR-Cas9 knock-in protocols were used.- CRISPR-Cas9 knock-in protocols for creating nucleocytoplasmic transport cell lines
Protocol 1 (Sakuma et al., 2016):
Use classical PCR to insert the nucleocytoplasmic transport reporter cassette of interest into a pcDNA3.1 plasmid. When designing the insert, make sure the 5' and 3' ends contain two identical targeting sites and micro-homology sequences. The CRISPR-Cas9 targeting sites are needed to cut the insert out of the plasmid in the following step. The microhomology sequences are needed to specifically insert the cassette into the AAVS1 safe harbor site. Here, we cloned a cassette expressing the NLSSV40-mNeongreen2x-NESpki and fused it with P2A-PuromycinR (Figure 3A).
Protocol 2 (Geisinger et al., 2016):
A PuromycinR-P2A-NESpki2-mNeongreen2x-NLSFUS reporter was cloned into a pcDNA3.1 plasmid (Figure 3B) that serves as a template for a PCR reaction with 5′ phosphorothioate modified primers (fw primer: T*C*C*CCTCCACCCCACAGTGGGGCC ACTAACGGGCCAGATATACGCG; rv primer: T*C*C*CCTCCACCCCACAGTGGGGCCACTA CTGGTTCTTTCCGCCTCAGAA, with * denoting a phosphorothioate modified nucleotide). This PCR product is used as the donor for the CRISPR knock-in protocol. The PCR primers contain the 5′ end microhomology sequences to enable specific integration of the insert into the AAVS1 safe harbor site (similar as for protocol 1).
Protocol 3 (Schmid-Burgk et al., 2016):
NLScMYC-AcGFP2x-NESikb2 was cloned into a pGL4-plasmid that contains at the 5′ end the CRISPR-Cas9 targeting side, to linearize it for insertion into the AAVS1 safe harbor site using the CRISPaint knock-in protocol (Figure 3C).
Notes:- Pay attention to the final size of your reporter protein. If your goal is to study active transport, use a fusion protein of two or three times your fluorescent protein reporter (approximately 27 kDa). We used two copies of mNeonGreen to exceed the limit for passive transport (± 50 kDa).
- NESpki2 contains a K to P mutation (MSLNELALPLAGLDI) which weakens the original NESpki export signal. As such, the equilibrium between import and export is shifted towards import and the reporter is localized mainly inside the nucleus.
- Because the Schmid-Burgk protocol is not based on microhomology (Schmid-Burgk et al., 2016), in contrast to 1 and 2, this is the least efficient knock-in protocol. Moreover, the full donor plasmid is integrated in the Hela Kyoto AAVS1 locus in this protocol. The Sakuma protocol (Sakuma et al., 2016) has the highest knock-in efficiency, but is the most difficult one to clone.
- Stable cell line generation (for used plasmids see Figure 3)
Cell culture medium used in all steps consists of Gibco DMEM medium supplemented with 10% fetal bovine serum (FBS) and 50 µg/ml gentamicin.- Day 1: Seed 100,000 Hela Kyoto cells in a 6-well plate.
- Day 2:
Protocol 1: Co-transfect these cells using Lipofectamine 3000 (Thermo Fisher Scientific). Add 500 ng of the respective pcDNA3.1-donor plasmid (Figure 3A), 1,000 ng of a plasmid expressing Cas9 plus a gRNA targeting the AAVS1 locus (GTCACCAATCCTGTCCCTAG) (Figure 3D) and 500 ng of a plasmid expressing a gRNA (GCCAGTACCCAAAAAGCGGG) that cleaves the reporter plasmid at two sites to excise the cassette (Figure 3E).
Protocol 2: Co-transfect these cells using Lipofectamine 3000 (Thermo Fisher Scientific). Add 500 ng of the cassette PCR donor product (Figure 3B) and 1,000 ng of a plasmid expressing Cas9 plus a gRNA targeting the AAVS1 locus (GTCACCAATCCTGTCCCTAG) (Figure 3D).
Protocol 3: Co-transfect these cells using Lipofectamine 3000 (Thermo Fisher Scientific). Add 500 ng of the respective pGL4-donor plasmid (Figure 3C), 1,000 ng of a plasmid expressing Cas9 plus a gRNA targeting the AAVS1 locus (GTCACCAATCCTGTCCCTAG) (Figure 3D) and 500 ng of a plasmid expressing a gRNA (GCCAGTACCCAAAAAGCGGG) that cleaves the reporter plasmid at one site to linearize the donor plasmid (Figure 3E).
Tip: Include a negative control condition by omitting the donor plasmid expressing Cas9/gRNA in one well. - Day 3: Change medium to limit toxicity caused by the transfection reagents.
- Day 4: Trypsinize transfected cells and select with puromycin (1 µg/ml).
- Analyze plates in the next days for discrete colonies that start to appear.
Tip: To exclude falls positives: make sure to only continue when all cells are dead in the negative control (without donor plasmid). In addition, keep cells under selection with antibiotics in the following days. - When the 6-well is completely confluent with this polyclonal cell mixture, monoclonal cells can be grown. Therefore, seed cells in a 96-well plate at a density of 0.5 cell/well. This can be achieved as follows:
- Take off medium and wash cells with 1 ml of 1x PBS.
- Remove PBS and add 500 µl of 0.05% trypsin per well. Return to the incubator for 5 min.
- Confirm that cells are detached. If not, place the cells again in the incubator for several minutes until they do.
- Add 1.5 ml medium per well. It is important to pipette up and down to mix and obtain single cells. Measure the obtained cell number.
- Make a 1/100 dilution (10 µl to 990 µl of medium) to facilitate the next step. Mix well.
- Add a volume that contains 50 cells to 10 ml of medium (10 ml is sufficient for one 96-well plate).
- Add 100 µl per well to obtain a concentration of 0.5 cells/well. As such, the majority of wells will have zero or one cells, with a small number containing two or more cells.
- Scan the plate five days later for the formation of monoclonal colonies. Do not include wells that contain two (or more) separate colonies, as they most probably arose from more than one cell and will therefore not result in a monoclonal culture.
Tip: Pay attention to the sides of the wells, as cells tend to attach there. - When the 96-well is completely full, expand the monoclonal culture.
Tip: Expand gradually by starting with a T-25 culture flask. - Freeze cells at -80 °C before long-term storage in liquid nitrogen. This can be achieved as follows:
- Prepare freezing medium (for a total of 10 ml: 8.2 ml culturing medium + 800 µl DMSO + 1 ml FBS). Place the medium on ice, as DMSO is toxic to your cells at room temperature.
- Label Cryotubes and pre-chill them on ice.
- Take off medium and wash cells with 1x PBS.
- Remove PBS and add 0.05% trypsin. Return to the incubator for 5 min.
- Confirm that cells are detached. If not, return cells to the incubator for several minutes until they do.
- Add medium to inhibit the trypsin and bring everything into a 50 ml tube.
- Count cells.
- Spin off for 5 min at 500 rcf.
- Remove supernatant and resuspend pellet in pre-made freezing medium. Add 1 ml per 1.5 x 106-2 x 106 cells.
- Pipette 1 ml of the freezing medium in each pre-chilled cryotube.
- Place tubes at -80 °C with the help of a Mr. FrostyTM freezing container.
- For long-term storage, move your samples the next day to a liquid nitrogen container.
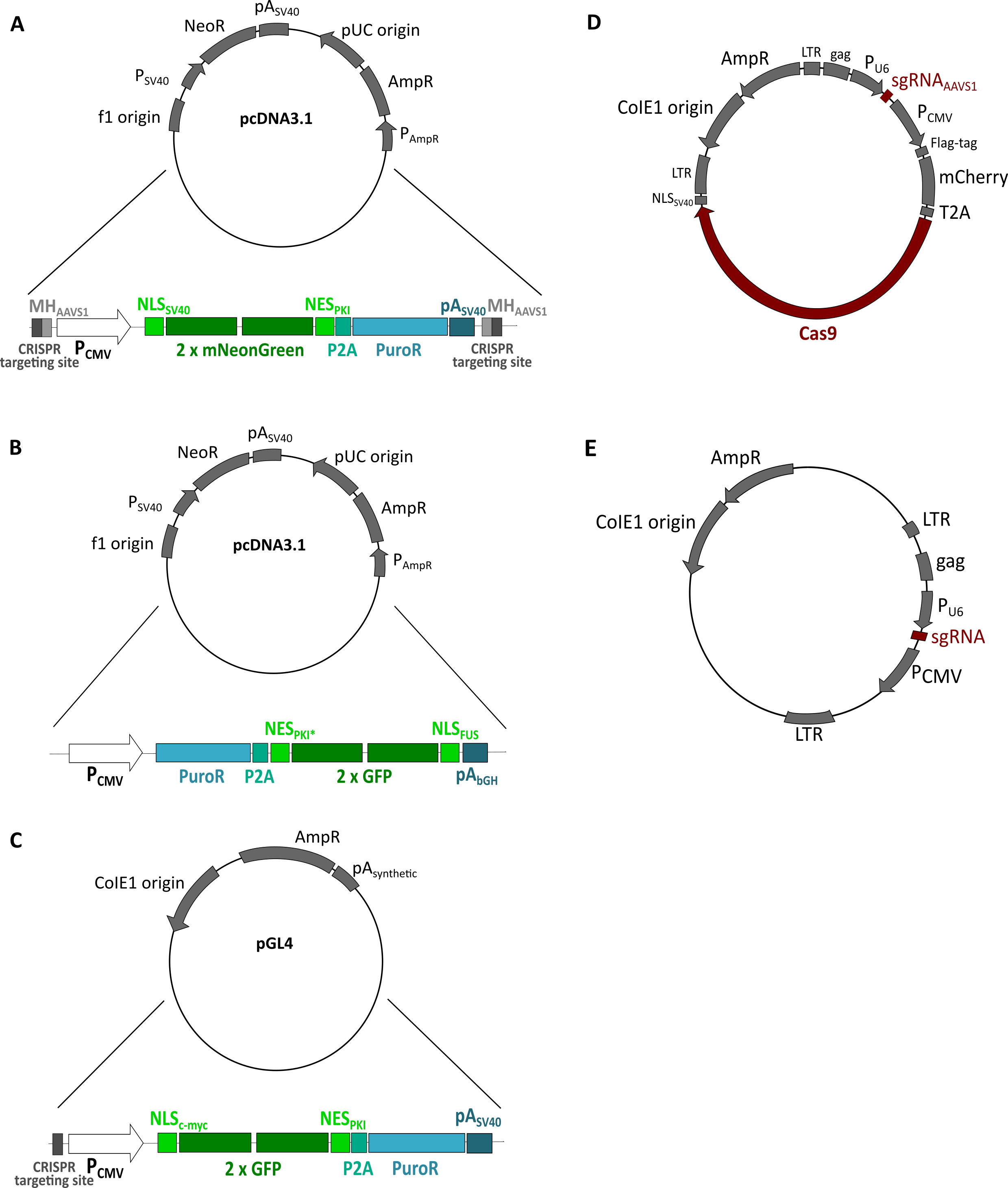
Figure 3. Plasmids used to generate reporter cell lines. A. Donor plasmid containing the reporter NLSSV40-mNeongreen2x-NESpki used in protocol 1. B. Plasmid containing the reporter NESpki2-mNeongreen2x-NLSFUS, which served as a template for the PCR reaction performed in protocol 2. NESpki2* contains a K to P mutation. C. Donor plasmid containing the reporter NLScMYC-AcGFP2x-NESikb2 used in protocol 3. D. Plasmid expressing Cas9 plus a gRNA targeting the AAVS1 locus (Schmid-Burgk et al., 2016) used in protocols 1, 2 and 3. E. Plasmid expressing a gRNA to cleave the donor plasmid containing the reporter (Schmid-Burgk et al., 2016) used in protocols 1 and 3.
- Validation
Note: It is important to validate your reporter. This can be done by:1)Treating your cells with available compounds e.g., leptomycin B to inhibit XPO1-mediated export (Kudo et al., 1999) or importazole to inhibit KPNB1-mediated import (Soderholm et al., 2011).2)Transfecting your cells with appropriate constructs e.g., M9M expression to inhibit TPNO1/TPNO2 (Cansizoglu et al., 2007).3)Knock down of targeted transport receptors e.g., siRNA against KPNB1 or KPNA2.
Note: Cell culture medium used in all steps consists of Gibco DMEM medium supplemented with 10% FBS and 50 µg/ml gentamicin.- Validate XPO1-mediated export with leptomycin B
- Day 1: Seed NLSSV40-mNeongreen2x-NESpki reporter cells at a density to obtain 60-70% confluency the next day.
- Day 2: Change half of the medium in the wells to obtain a final concentration of 50 nM of the XPO1-inhibitor leptomycin B (Kudo et al., 1999).
- Return cells to the incubator for 30 min.
- Fix cells with 4% PFA for 15 min.
- Wash cells three times with 1x PBS.
- Stain nuclei using NucBlue Live Cell Stain reagent (Invitrogen).
- Image cells to confirm increased nuclear localization of the reporter in the leptomycin B treated cells (Figure 4).
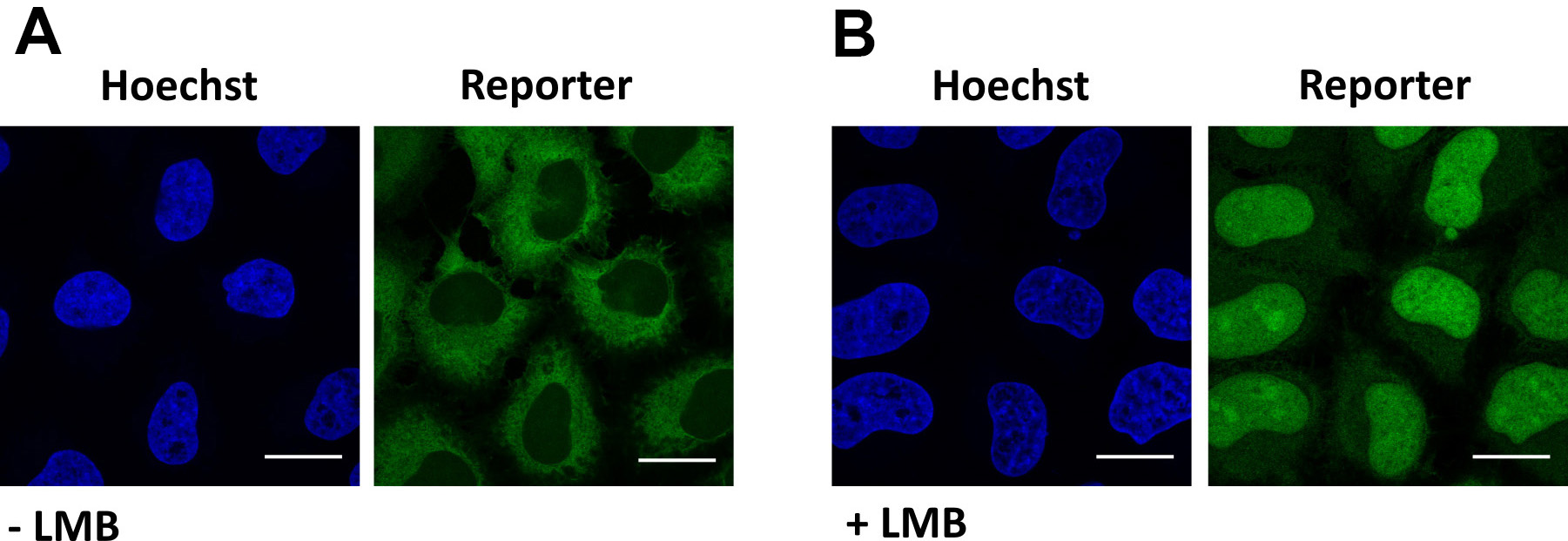
Figure 4. Validation of XPO1-mediated export. Representative images of cells expressing the reporter NLSSV40-mNeongreen2x-NESpki. A. In steady state, the reporter localizes in the cytoplasm of cells. B. Addition of the XPO1-inhibitor leptomcyin B (LMB) results in nuclear localization of the reporter. Scale bars = 20 µm.
- Validate KPNB1/KPNAx-mediated import with importazole
- Day 1: Seed NLSSV40-mNeongreen2x-NESpki reporter cells at a density to obtain 60-70% confluency the next day.
- Day 2: Incubate the cells for 2 h with 50 µM of the KPNB1-inhibitor importazole (Soderholm et al., 2011).
- Change half of the medium in the wells to obtain a final concentration of 50 nM leptomycin B.
Note: The reporter localizes steady state in the cytoplasm of the cells (due to XPO1-mediated export) and serves therefore, in combination with an XPO1-inhibitor (leptomycin B), as an import-reporter for KPNB1/KPNAx-mediated import. - Return cells to the incubator and fix cells with 4% PFA after 30 min.
- Wash cells three times with 1x PBS.
- Stain nuclei using NucBlue Live Cell Stain reagent (Invitrogen).
- Image cells to confirm a decreased nuclear localization of the reporter in the importazole-treated cells (Figure 5).
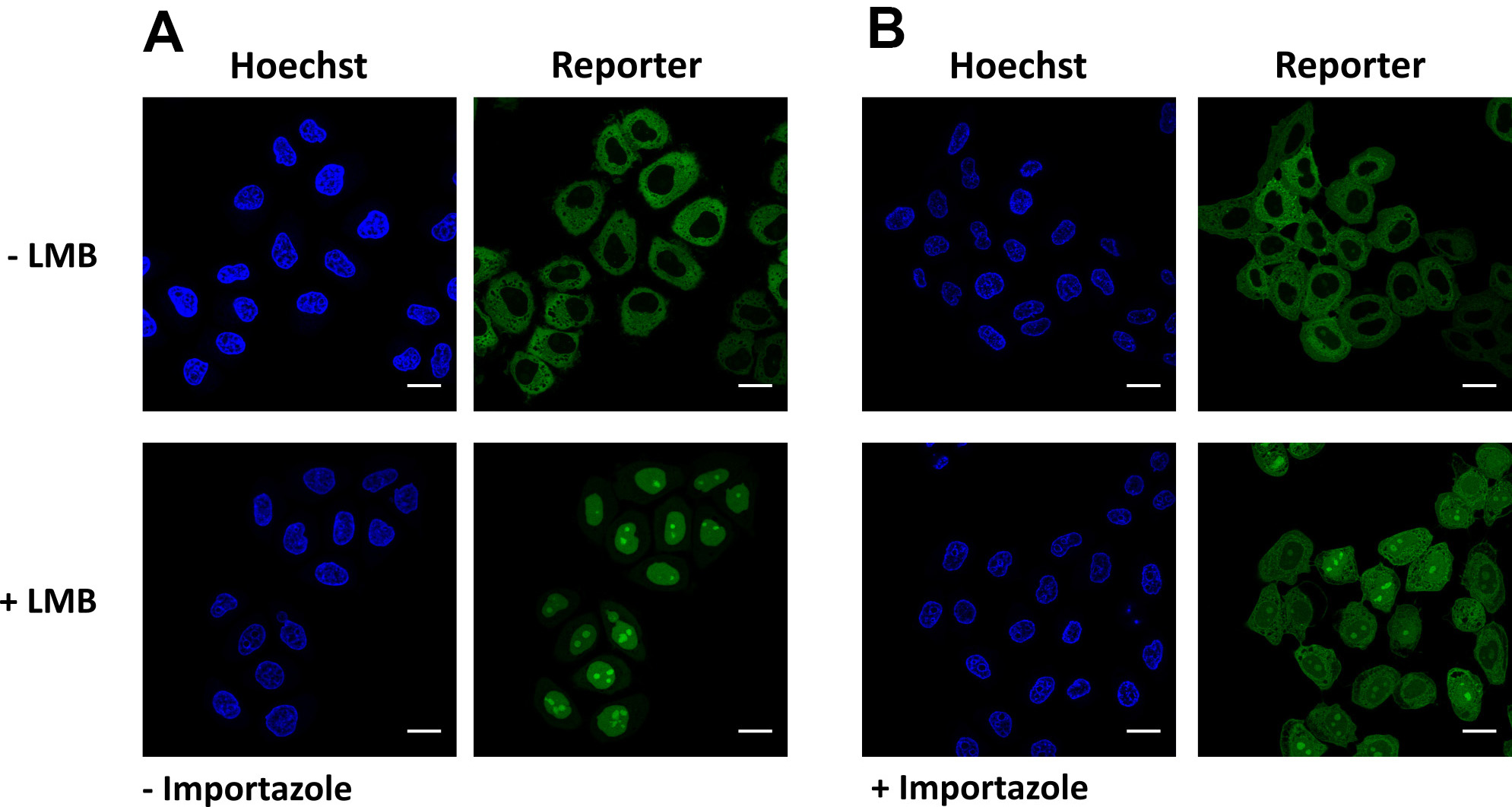
Figure 5. Validation of KPNB1/KPNAx-mediated import. Representative images of cells expressing the reporter NLSSV40-mNeongreen2x-NESpki. A. Addition of leptomycin B results in nuclear localization of the reporter, due to KPNB1/KPNAx-mediated import. B. Pre-incubation with importazole partially abolishes KPNB1/KPNAx-mediated import. Scale bars = 20 µm.
- Validate TPNO1/TPNO2-mediated import through M9M expression
- Day 1: Seed NESpki2-mNeongreen2x-NLSFUS reporter cells at a density to obtain 60-70% confluency the next day.
- Day 2: Transfect cells with a plasmid expressing the peptide M9M (Cansizoglu et al., 2007) using lipofectamine 3000 (Thermo Fisher Scientific).
Note: Express M9M in fusion with a fluorescent protein (e.g., Red fluorescent protein) to identify transfected cells. - Change medium 4 h after transfection to reduce toxicity.
- Incubate cells overnight.
- Day 3: Fix cells with 4% PFA for 15 min.
- Wash three times with 1x PBS.
- Stain nuclei using NucBlue Live Cell Stain reagent (Invitrogen).
- Image cells to confirm decreased nuclear localization of the reporter in the M9M-expressing cells (Figure 6).
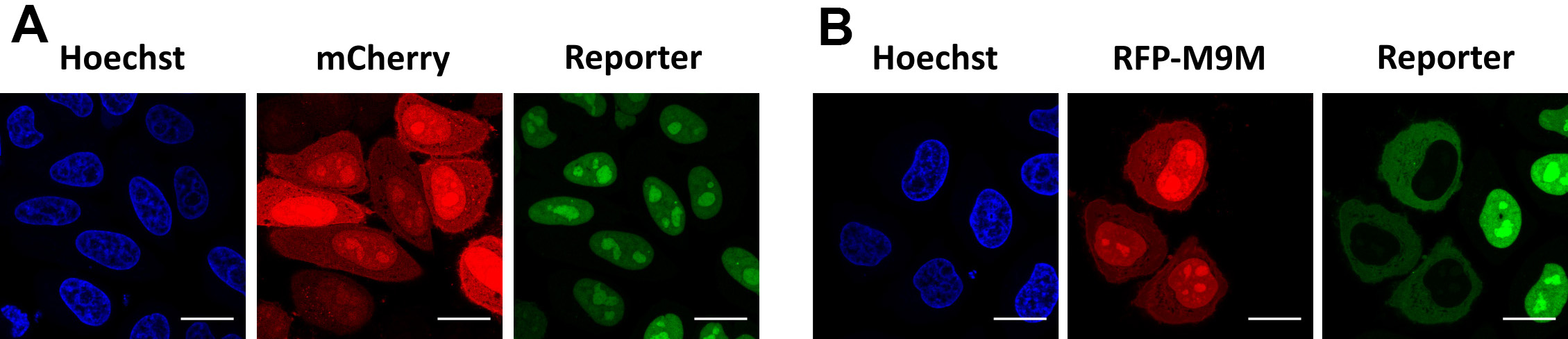
Figure 6. Validation of TPNO1/TPNO2-mediated import. Representative images of cells expressing the reporter NESpki2-mNeongreen2x-NLSFUS. A. In steady state, the reporter localizes inside the nucleus, caused by TPNO1/TPNO2-mediated import. Reporter cells were transfected with an mCherry-plasmid as negative control. B. Reporter cells transfected with an RFP-M9M-plasmid, which inhibits TPNO1/TPNO2-mediated import and results in cytoplasmic redistribution of the reporter. Scale bars = 20 µm.
- Validate KPNB1/KPNAx-mediated import through KPNB1/KPNA2-knock down
- Day 1: Seed NLScMYC-AcGFP2x-NESikb2 reporter cells at a density to obtain 60-70% confluency the next day.
- Day 2: Electroporate cells with siRNA against KPNB1 or KPNA2 (Horizon Discovery siRNA resuspension buffer, Bio-Rad: 230V, 950 µF, ∞Ω, 10,000 ms, #1 pulse).
- Incubate cells for 48 h.
- Fix cells with 4% PFA for 15 min.
- Wash three times with 1x PBS.
- Stain nuclei using NucBlue Live Cell Stain reagent (Invitrogen).
- Image cells to confirm decreased nuclear localization of the reporter (Figure 7).
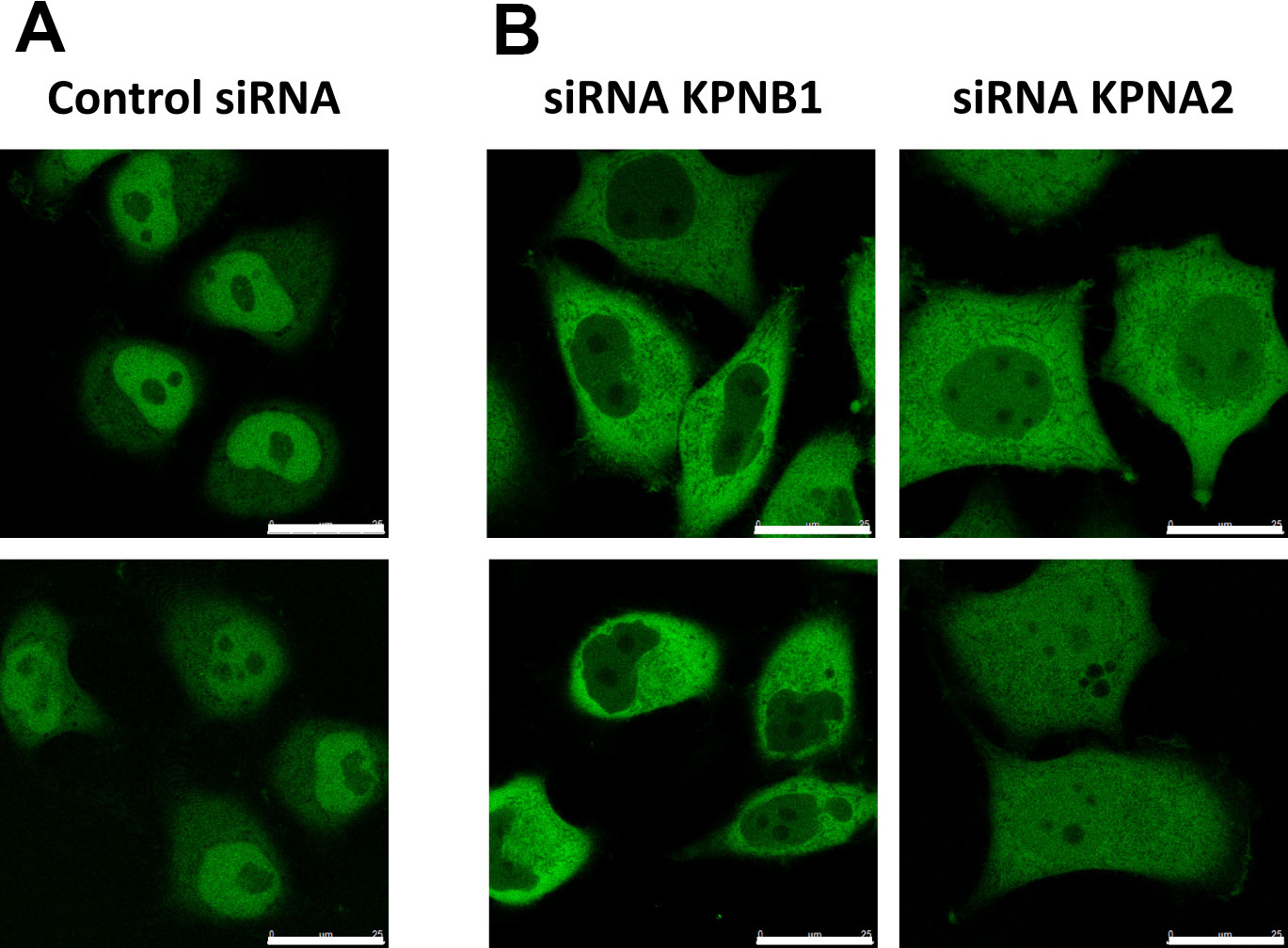
Figure 7. Validation of KPNB1/KPNAx-mediated import. Representative images of cells expressing the reporter NLScMYC-AcGFP2x-NESikb2. A. The reporter localizes steady state in the nucleus of the cells, due to KPNB1/KPNAx-mediated import. B. Expression of siRNA targeting KPNB1 or KPNA2 results in cytoplasmic localization of the reporter. Scale bars = 25 µm.
- Validate XPO1-mediated export with leptomycin B
- Optimization of the NLSSV40-mNeongreen2x-NESpki reporter
In steady state, the NLSSV40-mNeongreen2x-NESpki reporter localizes in the cytoplasm of the cells (caused by XPO1-mediated nuclear export) and serves therefore, in combination with an XPO1-inhibitor (leptomycin B; LMB), as an import-reporter for KPNB1/KPNAx-mediated import.
Note: Because the nuclear intensity results from the balance between export and import, it is important to optimize LMB concentrations to maximize the sensitivity of the assay to measure import deficits. In addition, to minimalize variability it is recommended to work with a LMB concentration that induces a gradual import of the reporter and allows import measurements to occur over longer periods (30 min). Moreover, it is recommended to measure at least two different time points to assure that your measurement falls within the dynamic range of the nuclear import process and saturation is not yet reached.
Note: Cell culture medium used in all steps consists of Gibco DMEM medium supplemented with 10% FBS and 50 µg/ml gentamicin.- Day 1: Seed NLSSV40-mNeongreen2x-NESpki reporter cells in a 96-well plate at a density to obtain 70% confluency the next day.
Note: Keep in mind that you will need three different time points and this in duplicate or triplicate per condition. - Day 2: Replace old medium with 100 µl of medium containing 25 µM of the KPNB1-inhibitor importazole.
- Return cells to the incubator for 1.5 h.
Note: The importazole concentration was chosen with the aim to induce average import inhibition, to not miss potential small import deficits induced by poly-PR. - Meanwhile, prepare different dilutions of LMB (dissolved in ethanol) in pre-warmed medium at a final concentration of 0 nM, 15 nM, 30 nM, 45 nM, 60 nM, 75 nM and 90 nM. Vortex.
Note: Keep in mind the dilution in the next steps. Therefore, prepare twice the final concentration, e.g., 120 nM for a final concentration of 60 nM. - Treat the cells intended for the 30 min time point with the different concentrations of LMB, by adding 100 µl of medium containing LMB to the 100 µl of medium already in the wells.
- Return cells to the incubator for 15 min.
- Treat the cells intended for the 15 min time point with the different concentrations of LMB by adding 100 µl of the medium containing LMB.
- Return cells to the incubator for 15 min.
- Fix with 4% PFA for 15 min.
- Wash cells three times with 1x PBS.
- Stain nuclei using NucBlue Live Cell Stain reagent (Invitrogen).
- Cells can be analyzed automatically by the CellInsight CX5 high content screening platform (Thermo Fisher Scientific) to determine the LMB concentration that induces the largest difference between control cells and importazole-treated cells (Figure 8).
Note: Use a multichannel pipette to reduce variability.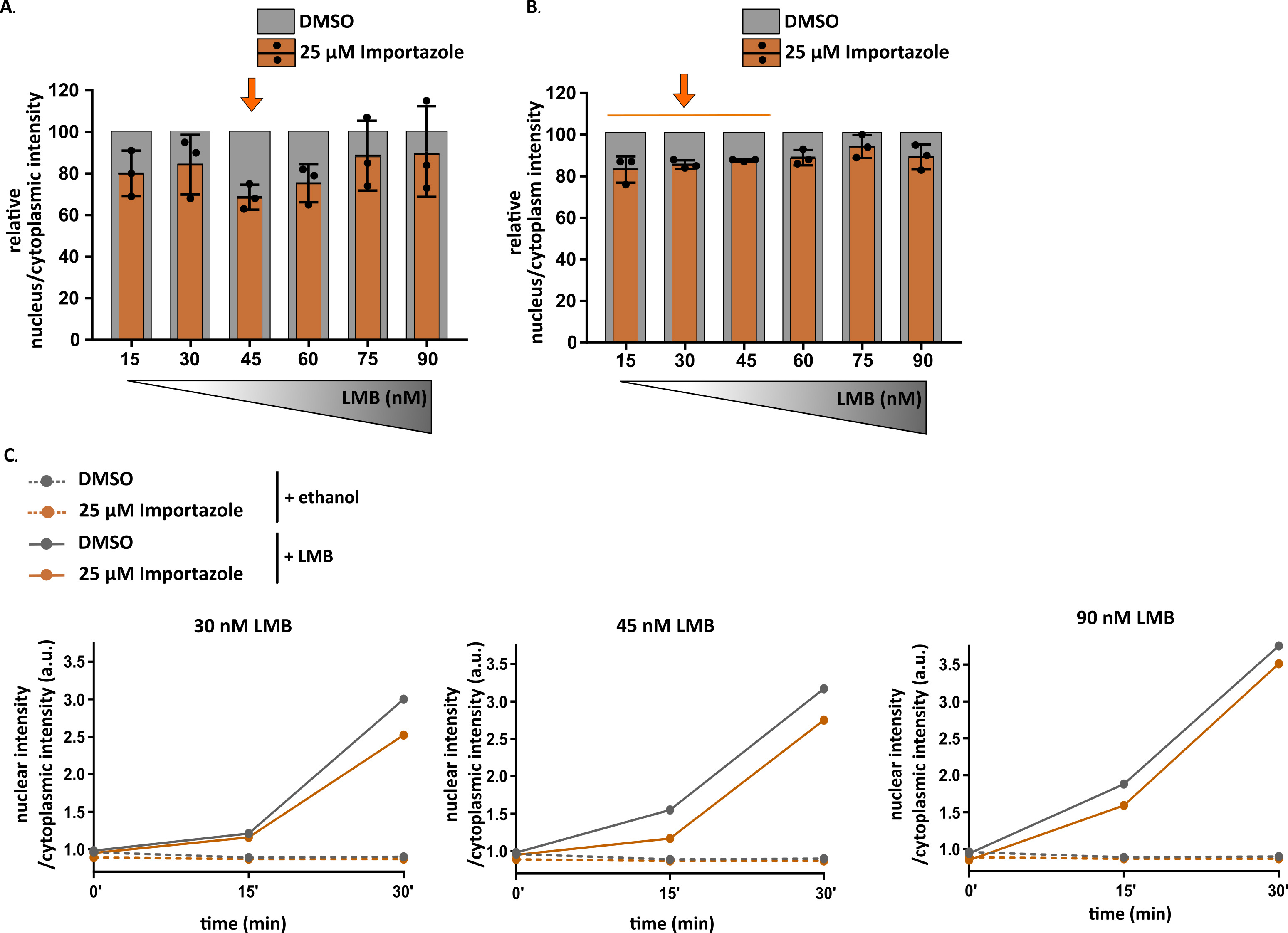
Figure 8. Optimization of KPNB1/KPNAx-mediated import of the NLSSV40-mNeongreen2x-NESpki reporter measured over time. A-B. Import of importazole-treated cells relative to import of DMSO-treated cells for different concentrations of leptomycin B (LMB). A-15 min; B-30 min. A. 45 nM of LMB results in the strongest difference between control cells and cells-treated with importazole. B. Lower concentrations allow measuring import-inhibition, as import becomes saturated at this later time point for higher concentrations of LMB. C. Left graph: lower concentrations (e.g., 30 nM) of LMB result in less overall import and make it therefore harder to observe disturbed import after 15 min. Import defects can be observed at later time points (30 min). Middle graph: 45 nM of LMB revealed import deficits at both time points. Right graph: higher concentrations of LMB (e.g., 90 nM) can push import too hard to observe a minimal impediment of import. A-B. Dot represents the average of one experiment with 2,500-7,000 cells per experiment. Mean + S.D. C. Dot represent the average of one experiment with 2,500-7,000 cells per experiment.
- Day 1: Seed NLSSV40-mNeongreen2x-NESpki reporter cells in a 96-well plate at a density to obtain 70% confluency the next day.
- CRISPR-Cas9 knock-in protocols for creating nucleocytoplasmic transport cell lines
- Measuring nucleocytoplasmic transport in the presence of mutant C9orf72-related peptides
- Production of lentiviral vectors expressing mutant C9orf72-related peptides
DPR100-plasmids were a kind gift of Dr. Daisuke Ito (Department of Neurology, Keio University, Tokyo, Japan). Classical PCR was used to clone mCherry-DPR100 constructs into lentiviral vector plasmids (LentiCrisprV2_Puro from Addgene) (Figure 9). Production of lentiviral vectors was done using HEK-293T cells as previously described (Hart et al., 2017).
Note: We strongly advise to use transduction instead of transfection to express desired disease-related constructs. Transfection is more stressful which can influence nucleocytoplasmic transport measurements.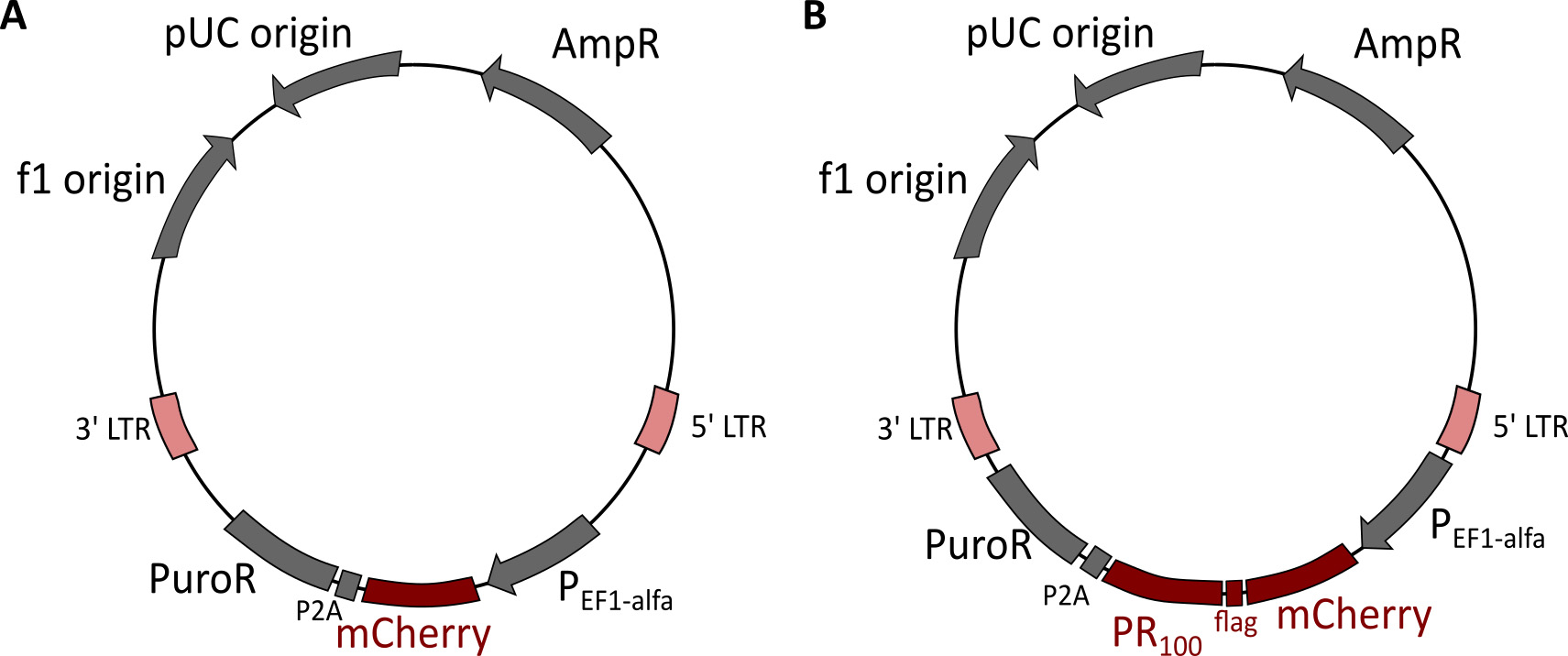
Figure 9. Lentiviral vector plasmids used to express mCherry or mCherry-PR100. A. Lentiviral vector containing mCherry. B. Lentiviral vector containing mCherry-PR100. - Assay
Note: Cell culture medium used in all steps consists of Gibco DMEM medium supplemented with 10% FBS and 50 µg/ml gentamicin.- Passage cells and seed onto cover glasses
- Day 0: Place a single cover glass into each well of a sterile 24-well polystyrene tissue culture plate with the use of sterile forceps.
Notes:1)Pre-coating coverslips with poly-L-lysine and poly-L-ornithine or other cell-surface treatments may be necessary to adhere other cell types to glass. Hela Kyoto cells adhere to glass without additional coating.2)For high throughput screens, cells can be seeded into 96-well plates or other formats.3)Keep in mind that you will need three different time points and this in duplicate or triplicate per condition. - Seed at a density that will result in 70% confluency at the day you plan to perform the nucleocytoplasmic transport assay.
- Incubate cells overnight in a tissue culture incubator (37 °C with 5% CO2).
- Day 0: Place a single cover glass into each well of a sterile 24-well polystyrene tissue culture plate with the use of sterile forceps.
- Transduction of cells to express C9orf72-related peptides
- Day 1: Dilute polybrene in medium at a final concentration of 10 µg/ml. Add lentiviral particles to each well. Leave the viral particles overnight on the cells (about 18 h).
- Day 2: Remove the lentivirus-containing medium and replace with fresh DMEM medium.
- Incubate cell for an additional two days in a tissue culture incubator (37 °C with 5% CO2).
Note: Addition of polybrene can significantly improve transduction efficiency. However, first confirm that polybrene is not toxic to your cells as it can induce cell death in certain cell types (e.g., iPSC-derived neurons).
- Measure nucleocytoplasmic transport
- Day 5: Prepare a desired volume of pre-warmed medium containing 45 nM LMB.
- Treat the cells intended for the 30 min time point with the medium containing LMB.
- Return cells to the incubator for 15 min.
- Treat the cells intended for the 15 min time point with the medium containing LMB.
- Return cells to the incubator for 15 min.
- Fix cells by adding 4% PFA (final concentration) to the medium for 15 min.
- Stain nuclei using NucBlue Live Cell Stain reagent (Invitrogen).
- Use a 20 µl pipette to place a drop of mounting medium on a slide, before placing the cover glass upside down on the slide using forceps. Remove excess mounting medium with a paper towel and seal the slides using nail polish.
- Image cells using a confocal microscope (Figure 10).
Notes:1)Keep in mind to maintain the same setting between different conditions and to focus on the middle of the nuclei (nucleoli are clearly visible).2)For high-throughput screens, intensity can be automatically analyzed by using a high content analyzer (e.g., CellInsight CX5 high content screening platform from Thermo Fisher Scientific).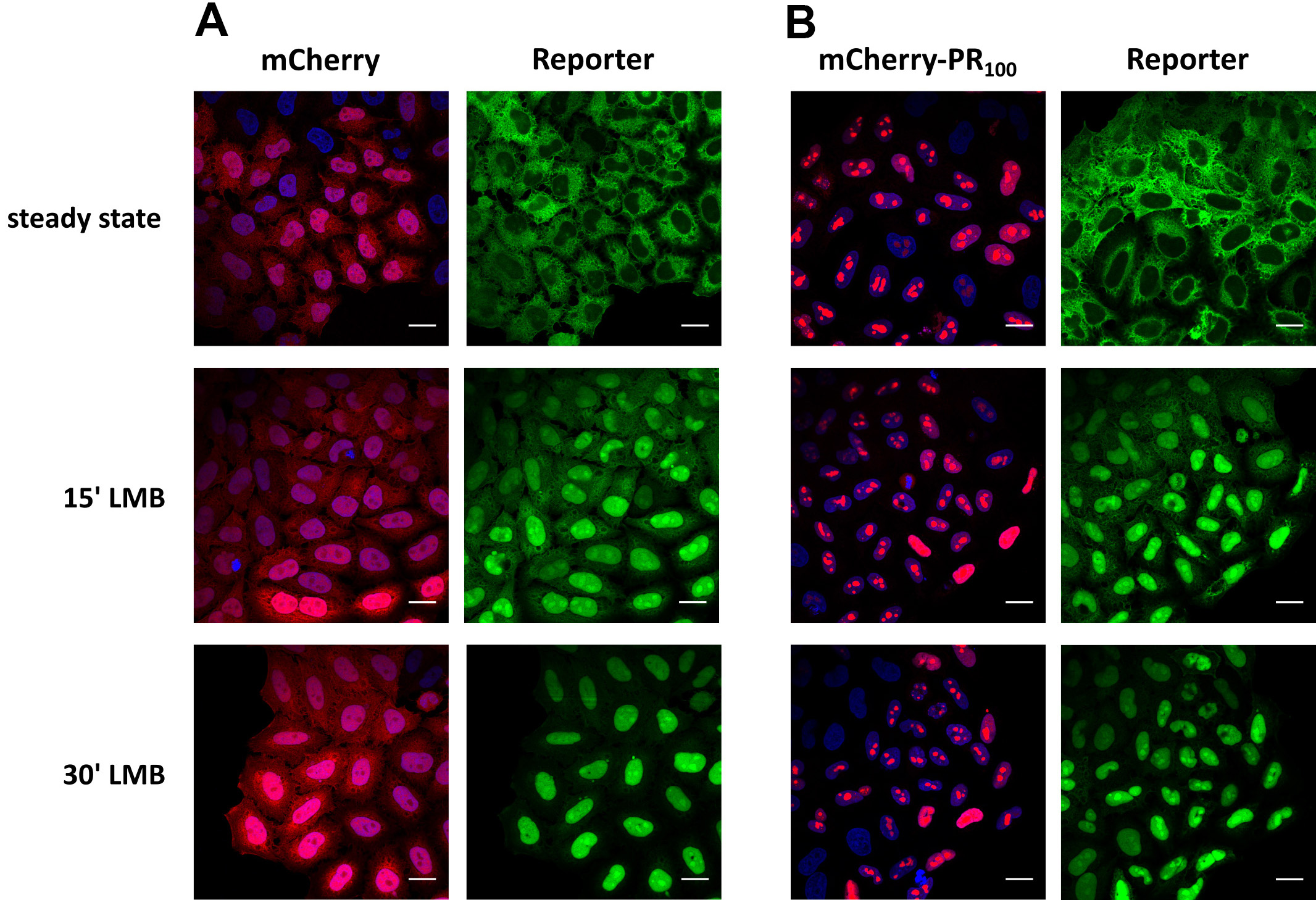
Figure 10. Measuring KPNB1/KPNAx-mediated import over time in a neurodegenerative-associated context. Representative images of cells expressing the reporter NLSSV40-mNeongreen2x-NESpki. The reporter localizes steady state in the cytoplasm of the cells, as a consequence of XPO1-mediated nuclear export. Addition of an XPO1-inhibitor (leptomycin B; LMB) allows to measure KPNB1/KPNAx-mediated import over time. A. Reporter cells were transduced with a mCherry-lentiviral vector as control. B. Reporter cells transduced with a poly-PR100-mCherry-lentiviral vector. Poly-PR dipeptides have been linked to the neurodegenerative disease amyotrophic lateral sclerosis. Scale bar = 20 µm.
- Passage cells and seed onto cover glasses
- Production of lentiviral vectors expressing mutant C9orf72-related peptides
Data analysis
- Manual
- ImageJ
Images taken on the confocal microscope can be manually analyzed with the free-online available software ImageJ.- Import images in ImageJ.
Note: It is recommended to analyze the images in 64-bit format. - Select region of interest + Ctrl M (Figure 11).
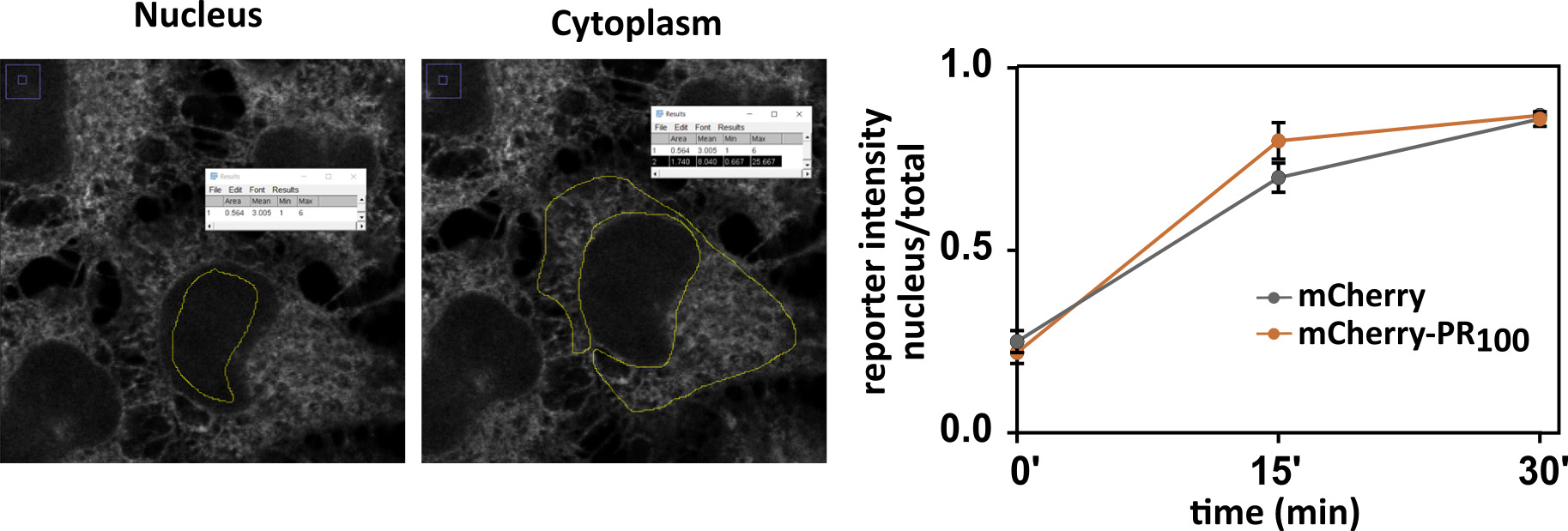
Figure 11. Manual analysis in ImageJ. Images are representative for NLSSV40-mNeongreen2x-NESpki reporter cells before addition of LMB and transfected with mCherry. Graph contains analyzed data of images from Figure 10. Mean ± S.D. are shown.
- Import images in ImageJ.
- ImageJ
- Automated
- Cell profiler
Cells imaged on the confocal microscope can be automatically analyzed with the free-online available software Cell profiler (Figure 12).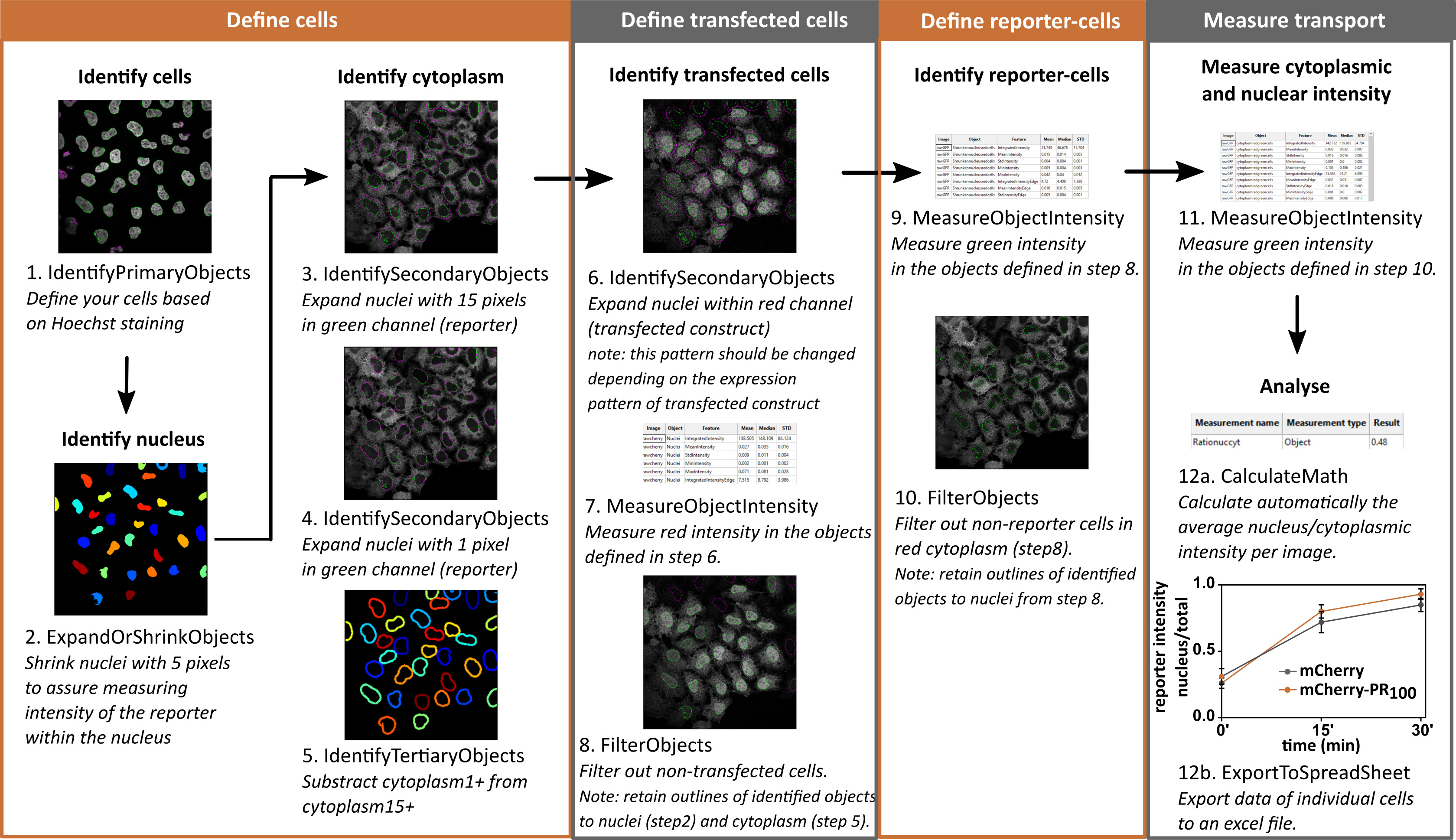
Figure 12. Workflow for automatic analysis in Cell profiler. Images are representative for NLSsv40-mNeongreen2x-NESpki reporter cells before addition of LMB and transfected with mCherry. Graph contains analyzed data of images from Figure 10. Mean ± S.D. are shown.
Statistical analysis- We recommend to analyze nuclear-to-total (= cytoplasm + nucleus) ratios as nucleus-to-cytoplasm ratios may be susceptible to large variations. The difference between nuclear and cytoplasmic values can become very large, especially before addition of LMB (very low nuclear intensity) or at the end of the assay (very low cytoplasmic intensity). In addition, when the data have to be normalized to the initial intensity, a small difference in the nuclear and cytoplasmic intensity might create a large difference in the ratio.
- Ratios are intrinsically asymmetric, meaning that all decreases are expressed as ratios between zero and one, and all increases are expressed as ratios greater than 1.0. Therefore, we recommend analyzing the logarithm of the ratios. As such, no change will be zero (the logarithm of 1.0), increases will be positive and decreases will be negative.
- Cell profiler
Acknowledgments
We thank Liesbeth Mercelis and Bob Massant for technical assistance. This work was supported by the KU Leuven (C1 and Opening the Future Fund); Fund for Scientific Research Flanders (FWO-Vlaanderen GOB3318N – G098314N), the ALS Liga (Belgium). J.V. is doctoral fellow of FWO. P.V.D. holds a senior clinical investigator ship of FWO-Vlaanderen and is supported by Fund ‘Een hart voor ALS’ and Laevers Fund for ALS Research.
Competing interests
The authors declare they have no financial and non-financial competing interests.
References
- Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., Mann, D., Tsuchiya, K., Yoshida, M., Hashizume, Y. and Oda, T. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351(3): 602-611.
- Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., van Blitterswijk, M. M., Jansen-West, K., Paul, J. W., 3rd, Rademakers, R., Boylan, K. B., Dickson, D. W. and Petrucelli, L. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77(4): 639-646.
- Boeynaems, S., Bogaert, E., Van Damme, P. and Van Den Bosch, L. (2016). Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol 132(2): 159-173.
- Cansizoglu, A. E., Lee, B. J., Zhang, Z. C., Fontoura, B. M. and Chook, Y. M. (2007). Structure-based design of a pathway-specific nuclear import inhibitor. Nat Struct Mol Biol 14(5): 452-454.
- Fahrenkrog, B. and Harel, A. (2018). Perturbations in traffic: aberrant nucleocytoplasmic transport at the heart of neurodegeneration. Cells 7(12). DOI: 10.3390/cells7120232.
- Geisinger, J. M., Turan, S., Hernandez, S., Spector, L. P. and Calos, M. P. (2016). In vivo blunt-end cloning through CRISPR/Cas9-facilitated non-homologous end-joining. Nucleic Acids Res 44(8): e76.
- Hart, T., Tong, A. H. Y., Chan, K., Van Leeuwen, J., Seetharaman, A., Aregger, M., Chandrashekhar, M., Hustedt, N., Seth, S., Noonan, A., Habsid, A., Sizova, O., Nedyalkova, L., Climie, R., Tworzyanski, L., Lawson, K., Sartori, M. A., Alibeh, S., Tieu, D., Masud, S., Mero, P., Weiss, A., Brown, K. R., Usaj, M., Billmann, M., Rahman, M., Constanzo, M., Myers, C. L., Andrews, B. J., Boone, C., Durocher, D. and Moffat, J. (2017). Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens. G3 (Bethesda) 7(8): 2719-2727.
- Hutten, S. and Dormann, D. (2019). Nucleocytoplasmic transport defects in neurodegeneration - Cause or consequence? Semin Cell Dev Biol S1084-9521 (18). 301090-301093.
- Kudo, N., Matsumori, N., Taoka, H., Fujiwara, D., Schreiner, E. P., Wolff, B., Yoshida, M. and Horinouchi, S. (1999). Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A 96(16): 9112-9117.
- Kwiatkowski, T. J., Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., Davis, A., Gilchrist, J., Kasarskis, E. J., Munsat, T., Valdmanis, P., Rouleau, G. A., Hosler, B. A., Cortelli, P., de Jong, P. J., Yoshinaga, Y., Haines, J. L., Pericak-Vance, M. A., Yan, J., Ticozzi, N., Siddique, T., McKenna-Yasek, D., Sapp, P. C., Horvitz, H. R., Landers, J. E. and Brown, R. H., Jr. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918): 1205-1208.
- Lange, A., Mills, R. E., Lange, C. J., Stewart, M., Devine, S. E. and Corbett, A. H. (2007). Classical nuclear localization signals: definition, function, and interaction with importin α. J Biol Chem 282(8): 5101-5105.
- Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., Chio, A., Restagno, G., Nicolaou, N., Simon-Sanchez, J., van Swieten, J. C., Abramzon, Y., Johnson, J. O., Sendtner, M., Pamphlett, R., Orrell, R. W., Mead, S., Sidle, K. C., Houlden, H., Rohrer, J. D., Morrison, K. E., Pall, H., Talbot, K., Ansorge, O., Chromosome, A. L. S. F. T. D. C., French research network on, F. F. A., Consortium, I., Hernandez, D. G., Arepalli, S., Sabatelli, M., Mora, G., Corbo, M., Giannini, F., Calvo, A., Englund, E., Borghero, G., Floris, G. L., Remes, A. M., Laaksovirta, H., McCluskey, L., Trojanowski, J. Q., Van Deerlin, V. M., Schellenberg, G. D., Nalls, M. A., Drory, V. E., Lu, C. S., Yeh, T. H., Ishiura, H., Takahashi, Y., Tsuji, S., Le Ber, I., Brice, A., Drepper, C., Williams, N., Kirby, J., Shaw, P., Hardy, J., Tienari, P. J., Heutink, P., Morris, H. R., Pickering-Brown, S. and Traynor, B. J. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11(4): 323-330.
- Mori, K., Arzberger, T., Grasser, F. A., Gijselinck, I., May, S., Rentzsch, K., Weng, S. M., Schludi, M. H., van der Zee, J., Cruts, M., Van Broeckhoven, C., Kremmer, E., Kretzschmar, H. A., Haass, C. and Edbauer, D. (2013a). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126(6): 881-893.
- Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., Schmid, B., Kretzschmar, H. A., Cruts, M., Van Broeckhoven, C., Haass, C. and Edbauer, D. (2013b). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339(6125): 1335-1338.
- Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., Bruce, J., Schuck, T., Grossman, M., Clark, C. M., McCluskey, L. F., Miller, B. L., Masliah, E., Mackenzie, I. R., Feldman, H., Feiden, W., Kretzschmar, H. A., Trojanowski, J. Q. and Lee, V. M. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796): 130-133.
- Ossareh-Nazari, B., Gwizdek, C. and Dargemont, C. (2001). Protein export from the nucleus. Traffic 2(10): 684-689.
- Ryan, K. J. and Wente, S. R. (2000). The nuclear pore complex: a protein machine bridging the nucleus and cytoplasm. Curr Opin Cell Biol 12(3): 361-371.
- Sakuma, T., Nakade, S., Sakane, Y., Suzuki, K. T. and Yamamoto, T. (2016). MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat Protoc 11(1): 118-133.
- Schmid-Burgk, J. L., Höning, K., Ebert, T. S. and Hornung, V. (2016). CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat Commun 7: 12338.
- Soderholm, J. F., Bird, S. L., Kalab, P., Sampathkumar, Y., Hasegawa, K., Uehara-Bingen, M., Weis, K. and Heald, R. (2011). Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chem Biol 6(7): 700-708.
- Stoffler, D., Fahrenkrog, B. and Aebi, U. (1999). The nuclear pore complex: from molecular architecture to functional dynamics. Current Opinion Cell Biology 11(3): 391-401.
- Tyzack, G. E., Luisier, R., Taha, D. M., Neeves, J., Modic, M., Mitchell, J. S., Meyer, I., Greensmith, L., Newcombe, J., Ule, J., Luscombe, N. M. and Patani, R. (2019). Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain 142(9): 2572-2580.
- Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., Hu, X., Smith, B., Ruddy, D., Wright, P., Ganesalingam, J., Williams, K. L., Tripathi, V., Al-Saraj, S., Al-Chalabi, A., Leigh, P. N., Blair, I. P., Nicholson, G., de Belleroche, J., Gallo, J. M., Miller, C. C. and Shaw, C. E. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918): 1208-1211.
- Vanneste, J., Vercruysse, T., Boeynaems, S., Sicart, A., Van Damme, P., Daelemans, D. and Van Den Bosch, L. (2019). C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci Rep 9(1): 15728.
- Yuva-Aydemir, Y., Almeida, S. and Gao, F. B. (2018). Insights into C9ORF72-Related ALS/FTD from Drosophila and iPSC Models. Trends Neurosci 41(7): 457-469.
- Zu, T., Gibbens, B., Doty, N. S., Gomes-Pereira, M., Huguet, A., Stone, M. D., Margolis, J., Peterson, M., Markowski, T. W., Ingram, M. A., Nan, Z., Forster, C., Low, W. C., Schoser, B., Somia, N. V., Clark, H. B., Schmechel, S., Bitterman, P. B., Gourdon, G., Swanson, M. S., Moseley, M. and Ranum, L. P. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108(1): 260-265.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Vanneste, J., Vercruysse, T., Van Damme, P., Van Den Bosch, L. and Daelemans, D. (2020). Quantitative Nucleocytoplasmic Transport Assays in Cellular Models of Neurodegeneration. Bio-protocol 10(12): e3659. DOI: 10.21769/BioProtoc.3659.
Category
Neuroscience > Nervous system disorders > Neurodegeneration
Molecular Biology > Protein > Protein shuttling
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.