- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification and Proteomic Analysis of Alphavirus Particles from Sindbis Virus Grown in Mammalian and Insect Cells

(*contributed equally to this work) Published: Vol 9, Iss 10, May 20, 2019 DOI: 10.21769/BioProtoc.3239 Views: 7506
Reviewed by: David PaulJinping ZhaoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Current mass spectrometry (MS) methods and new instrumentation now allow for more accurate identification of proteins in low abundance than previous protein fractionation and identification methods. It was of interest if this method could serve to define the virus proteome of a membrane-containing virus. To evaluate the efficacy of mass spec to determine the proteome of medically important viruses, Sindbis virus (SINV), the prototypical alphavirus was chosen for evaluation. This model system was chosen specifically because the alphaviruses contain members which are human pathogens, this virus is well defined biochemically and structurally, and grows to high titers in both vertebrate and non-vertebrate host cells. The SINV proteome was investigated using this method to determine if host proteins are specifically packaged into infectious virions. It was also of interest if the SINV proteome, when grown in multiple host cells representing vertebrate and mosquito hosts, incorporated specific host proteins from all hosts. Observation of recurrent or distinctive proteins in the virus proteome aided in the determination of proteins incorporated into the virion as opposed to those bound to the particle exterior. Mass spectrometry analysis identified the total protein content of purified virions within limits of detection. The most significant finding was that in addition to the host proteins, SINV non-structural protein 2 (nsP2) was detected within virions grown in all host cells examined. This analysis identified host factors not previously associated with alphavirus entry, replication, or egress, identifying at least one host factor integrally involved in alphavirus replication. Key to the success of this analysis is the method of virus purification which must deliver measurably infectious virus free of high levels of contaminants. For SINV and other members of the alphavirus family, this is accomplished by isopycnic centrifugation through potassium tartrate, followed by a high salt wash.
Keywords: Sindbis virusBackground
The terms proteome and proteomics were coined by Marc Wilkins to describe the systematic evaluation of proteins in a model system using a detailed study of structure, function and regulation of its biology including aberrations which lead to disease (Wilkins et al., 1996 and 2009). However, virus proteomes have been under investigation long before the field of proteomics evolved in an attempt to understand the mechanisms of virus-host interactions in vitro and to evaluate virus pathogenesis of the animal host. To this end it was of interest to evaluate the virus proteome of a model system in a family of viruses which contained medically significant pathogens. Sindbis virus was chosen for this investigation because it is a member of the Alphavirus genus, Family Togaviridae, which contains a significant number of human pathogens of medical importance, it is structurally stable, grows to high titers, is well described in the literature and can be grown in vertebrate and non-vertebrate host cells. Sindbis virus has been the subject of many studies because it is a relatively simple membrane-containing +RNA icosahedral virus (Figure 1). The viral 42S genome is infectious and serves as the template for a 26S subgenomic RNA which encodes the structural genes, organized in the sequence C-PE2(E3/E2)-6K(TF)-E1 3’UTR-polyA (Figure 2). The genomic strand encodes the non-structural proteins in the order 5’UTR-nsP1-nsP2-nsP3-nsP4 (Strauss, J. H. and Strauss, E. G., 1994). The process of virus assembly has marked differences when occurring in vertebrate or invertebrate cells (Brown, 1986). In vertebrate cells the nucleocapsid is pre-assembled in the cytoplasm with the genomic RNA prior to association with the viral protein modified plasma membrane and virus budding from the plasma membrane. In invertebrate cells the virus nucleocapsid and fully matured virus are assembled in endosomes termed “virus factories” prior to fusion of the virus-containing endosomes with the plasma membrane, releasing infectious particles. Depending on the virus’ host cells origin, the glycosylation patterns of the glycoproteins will be that specified by the cellular biochemistry and the specific lipid bilayer will be that of the host cell. The virus particles released from either host can be equally infectious and if the SVHR strain of Sindbis virus is used close to 100% viability is achieved (Vancini et al., 2013). This is an important detail because virus infectivity requires all of its components to be in their metastable conformation and native physical state to be functional that is, infectious (Hernandez et al., 2014). Thus, not all virus particles will be amenable to this method of analysis because the particle structure must be 1) of regular stoichiometry 2) capable of rigorous purification without the loss of infectivity and 3) express a very low particle to PFU ratio. These factors are important to be able to discern any protein contaminants from proteins that are carried within the virus particle. Because of the symmetry of the virus particle and the stoichiometry, the protein concentration can be used to calculate the number of physical particles from the number of infectious particles using the plaque assay to determine infectious particle titer. This specific analysis cannot be applied to non-symmetrical viruses.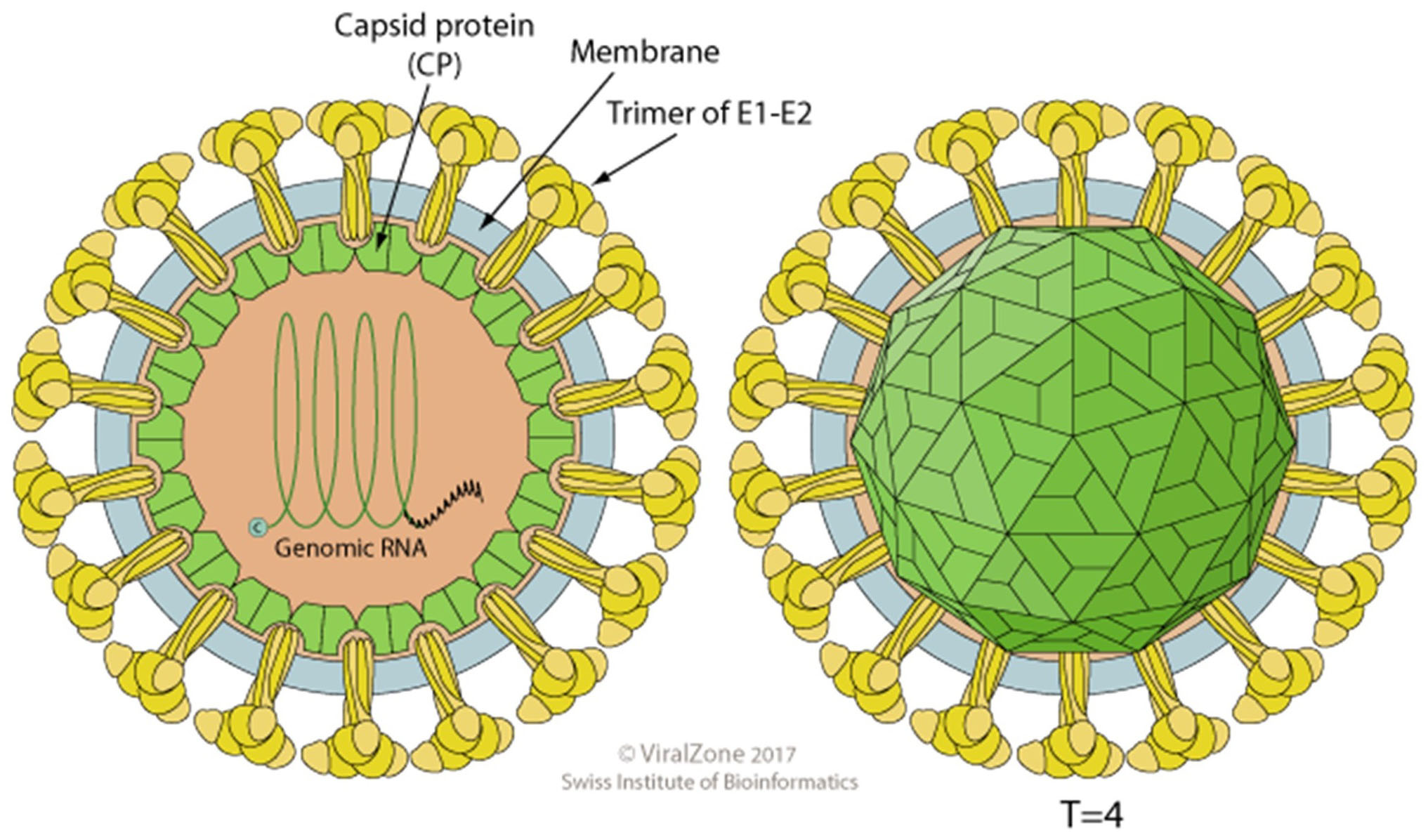
Figure 1. A cartoon of an alphavirus. The cartoon depicts an enveloped, spherical, icosahedral, 65-70 nm in diameter, capsid with a T = 4 icosahedral symmetry made of 240 monomers 1:1:1 stoichiometric ratio of E1:E2:C. The envelope contains 80 spikes, each spike is a trimer of E1/E2 protein dimers. Reprinted with permission from ViralZone, SBI Swiss Institute of Bioinformatics. Figures from ViralZone, with permission.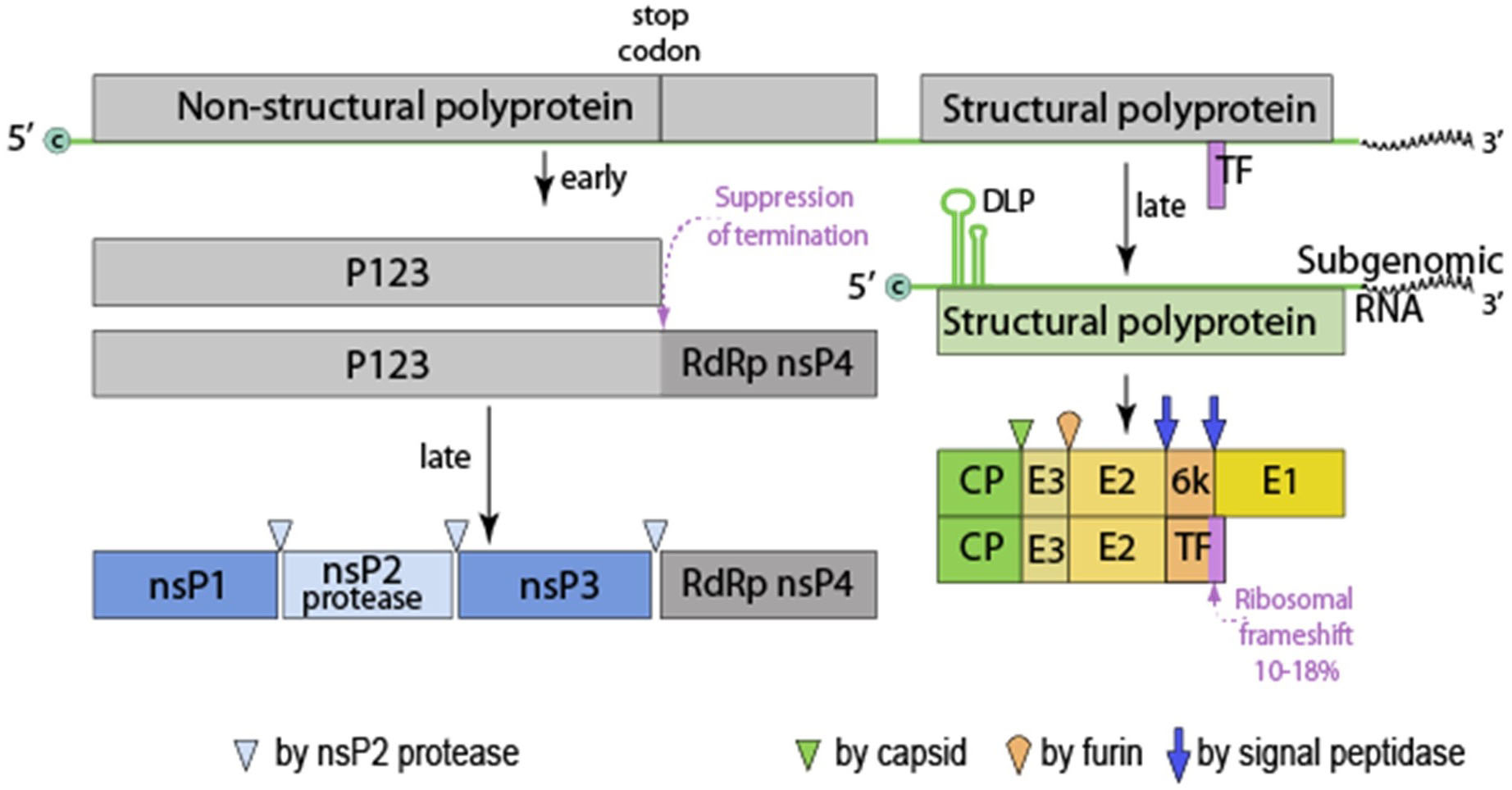
Figure 2. Alphavirus genomic organization. This genome is monopartite, linear, ssRNA(+) genome of 11-12 kb. The genome is capped (c) and polyadenylated. P123, precursor to nsP1, 2 and 3. Virus proteins are matured by both host and viral proteases. RdRp, RNA dependent RNA polymerase, TF, transframe protein (made from ribosomal frame shift in 6k protein) and DLP (downstream loop), stable RNA structure in the coding sequence of the 5’ region of the subgenomic RNA. Figures from ViralZone, with permission.
Stock Virus growth in BHK cells or C7-10 cells
The heat resistant SINV (SVHR) strain was used in this study. This strain was isolated by Burge and Pfefferkorn in 1966 by collecting virus that was resistant to heating to 54 °C. The choice of virus strain is important because this strain produces high titers (1010 PFU/ml) and low particle/PFU (~1 particle/PFU), ratios of highly infectious and physically stable virus. BHK and C7-10 (Aedes albopictus) cells were obtained from internal collections and are the favored cells to grow SVHR. Virus was harvested from 10 T-75 flasks (Corning) which produces enough virus to form a large visible band in a 30 ml potassium tartrate gradient and sufficient material for the mass spec analysis. Cells were infected at an MOI of 10 PFU/ml, for Sindbis virus infections and allowed to replicate for a single cycle and harvested at 18 h post infection to ensure that no cell lysis took place. A single cycle of SINV growth from mammalian cells is ~12 h and from C7-10 cells is about 24 h. The supernatants were clarified by low speed centrifugation (Sorvall RC-5B Super speed centrifuge at 2,000 rpm, 700 x g). Twenty microliter of the resulting virus supernatant was loaded onto a 15-35% linear potassium tartrate gradient and twice purified by isopycnic ultracentrifugation (Beckman SW-28 rotor, 18 h at 10,000 x g). The resulting band of purified virus was collected and washed twice by pelleting the virus in 5 ml 1x PBS in an SW-40 Beckman-40 rotor at 45,000 rpm (12,000 x g) for 30 min and collecting the pellet.
Virus titration by Plaque assay
The assay of virus titer by plaque formation, “plaque assay” is the most accurate method for measuring of the amount of infectious virus. This assay is used to determine the titer, in plaque-forming units (PFU) per ml, of virus by infecting a standardized monolayer of cells with a known volume of a known dilution of a virus-containing solution. The infection is contained under agarose which only allows diffusion of the virus to adjacent cells. Virus from a single initially infected cell infects adjacent cells producing a “plaque” or clearing (formed by lysed cells) localized to the original site of infection by an overlay of 1% agarose in 1x EMEM. Plaques of SVHR are visible to the naked eye after neutral red staining within 2-3 days of incubation at 37 °C. Begin with an estimate of what the titer could be, if the titer is estimated to be around 108 PFU/ml, a flask infected with a dilution of 10-6 would show 20-200 plaques; in this case, infecting flasks with dilutions of 10-5, 10-6, and 10-7 PFU/ml should give adequate data to make a relatively accurate calculation. If the titer of the virus is completely unknown, it may be necessary to infect flasks or plates with a wide range of dilutions (10-1 to 10-8). The number and quality of the plaques seen in a given assay can be influenced by a number of factors, including the pH and/or temperature of media, dilution buffer, agarose overlay, or the condition of the cell monolayer. Due to the sensitivity of this assay, it is important to include both positive and negative controls within each assay. The negative control is a flask that is inoculated with diluent only, and the positive control consists of one dilution of SVHR stock virus of known titer sufficient to give ~20 plaques. This number of plaques is significant statistically.
It is important that when going from a high concentration to a low concentration the pipet tip is changed to avoid “carry over” contamination. However, when pipetting from a low concentration to a higher concentration, as is done when the wells are inoculated, a single pipet tip can be used.
Materials and Reagents
Note: These can be from any supplier that offers Cell Culture grade materials or reagents except where specified. All culture flasks and plates are standard and can be from any supplier except where noted.
All fetal bovine serum (FBS) should be heat inactivated at 56 °C for 30 min.
- Sub-culture of vertebrate and invertebrate cells
Sub-culture of BHK-21 cells- 6-well plates
- Sterile tissue culture supplies (pipets, vented or plug cap flasks and 15 ml conical tubes)
- Heat-inactivated fetal bovine serum (FBS)
- Tryptose phosphate broth
- L-glutamine
- Gentamicin sulfate
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2 or CaCl2·2H2O
- MgCl2·6H2O
- Complete E-MEM, warmed to 37 °C (Earl’s salts minimal essential medium, completed with 10% heat inactivated fetal bovine serum, FBS)
- 1x PBS-D (see Recipes)
- Trypsin stock, 0.25%, warmed to 37 °C (see Recipes)
Sub-culture of C7-10 cells- Sterile tissue culture supplies (pipets, vented or plug cap flasks and 15 ml conical tubes)
- 70% ethanol
- 1x MEM
- Virus growth and titration
Stock virus preparation- Serological pipettes (5 ml, 10 ml and 25 ml)
- FBS (fetal bovine serum)
- Sterile Tissue culture grade water: for a detailed discussion of purified water and specifications for cell culture and other grades of water see Laboratory Water (The national institutes of health, March, 2013)
- Gentamicin sulfate
- L-glutamine
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2 or CaCl2·2H2O
- MgCl2·6H2O
- Neutral red
- Complete E-MEM, 1x and 2x (see Recipes)
- Gentamicin sulfate, 100x (mammalian cells only) (see Recipes)
- L-glutamine 100x (see Recipes)
- HEPES (pH 7.2-7.4), 1 M (see Recipes)
- 2% Neutral red stock solution (see Recipes)
- PBS-D (10x) (see Recipes)
- SVHR diluent (see Recipes)
- Tryptose phosphate broth (TPB) (optional for ATCC cells) (see Recipes)
Virus titration by plaque formation on BHK cells
BHK cells preparation- Sterile tissue culture supplies (pipets, vented or plug cap flasks and 15 ml conical tubes)
- Heat-inactivated fetal bovine serum (FBS)
- Tryptose phosphate broth
- L-glutamine
- Gentamicin sulfate
- Trypsin
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2
- CaCl2·2H2O
- KH2PO4
- MgCl2·6H2O
- Complete E-MEM, warmed to 37 °C (see Recipes)
- 1x PBS-D (see Recipes)
- Trypsin stock, 0.25%, warmed to 37 °C (see Recipes)
Virus titration by Plaque assay- 2% Agarose
- Heat inactivated Fetal Bovine Serum (FBS)
- 100% glycerol
- Heat-inactivated fetal bovine serum (FBS)
- Tryptose phosphate broth
- L-glutamine
- Gentamicin sulfate
- Trypsin
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2 or CaCl2·2H2O
- KH2PO4
- Neutral Red
- Complete E-MEM 2x (see Recipes)
- 2% Neutral Red (see Recipes)
- SVHR Diluent (see Recipes)
- 1x PBS-D (see Recipes)
- 1 M HEPES (pH 7.4) (see Recipes)
- Sindbis virus infection of vertebrate and invertebrate cells
Sindbis virus infection of BHK cells- Centrifuge tubes (Corning, 15 ml Fisherbrand, catalog number: 05-539-5)
- Sindbis virus infection of C7-10 cells
- 100% glycerol (sterile)
- Tissue culture grade water
- Heat-inactivated fetal bovine serum (FBS)
- Tryptose phosphate broth
- L-glutamine
- Gentamicin sulfate
- Trypsin
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2 or CaCl2·2H2O
- KH2PO4
- MgCl2·6H2O
- Tryptose phosphate broth (TPB; Difco)
- Complete E-MEM (see Recipes)
- Gentamicin sulfate 100x (see Recipes)
- L-glutamine 100x (see Recipes)
- 1 M HEPES (see Recipes)
- TPB-tryptose phosphate broth (see Recipes)
- PBS-D (10x) (see Recipes)
Sindbis virus infection of C7-10 cells- Glycerol
- Liquid N2
- Heat-inactivated fetal bovine serum (FBS)
- Tryptose phosphate broth
- L-glutamine
- Gentamicin sulfate
- Trypsin
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2 or CaCl2·2H2O
- KH2PO4
- MgCl2·6H2O
- Completed E-MEM medium (see Recipes)
- 1x PBS-D (see Recipes)
- Purification of Sindbis virus
Purification and Concentration by isopycnic centrifugation- Centrifuge tubes (ultra-clear)
- Small tube clamp
- Waste beaker
- 2 ml cryotube
- 15% potassium tartrate (dibasic, hemihydrate) in PBS-D (filter sterilize to store and store at 4 °C)
- 35% potassium tartrate in PBS-D (filter sterilize to store and store at 4 °C)
- BCA assay (PierceTM Rapid Gold BCA Protein Assay Kit) (Thermo Fisher Scientific, catalog number: A53225)
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2 or CaCl2·2H2O
- KH2PO4
- MgCl2·6H2O
- PBS-D pH 7.4 (see Recipes)
Concentration of virus by Polyethylene glycol (PEG) precipitation- Kimax high strength glass centrifuge tube (Kimax, catalog number: #Z2514888) (plain 30 ml)
- Polyallomer centrifuge tubes
- Polyethylene glycol-8000 (PEG-8000)
- 15% potassium tartrate (dibasic, hemihydrate) in PBS-D
- 35% potassium tartrate in PBS-D
- BCA assay (PierceTM Rapid Gold BCA Protein Assay Kit) (Thermo Fisher Scientific, catalog number: A53225)
- KCl
- KH2PO4
- NaCl
- Na2HPO4 or Na2HPO4·7H2O
- CaCl2 or CaCl2·2H2O
- KH2PO4
- MgCl2·6H2O
- Tris Buffer, pH 7.0
- EDTA
- PBS-D, pH 7.4 (see Recipes)
- PEG Buffer (see Recipes)
- 2 M NaCl (see Recipes)
- Calculation of particle to PFU ratio
None - Mass spectrometry and proteomic analysis
Note: All reagents should be LCV-MS grade.
Protein Extraction and Digestion- Lo-bind Centrifuge tubes (Eppendorf)
- Spin filter (Millipore-Ultracell YM-30)
- Pierce C18 spin columns (Thermo Fisher Scientific)
- Parafilm
- Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, catalog number: A53225)
- Mammalian Protein Extraction Reagent (M-PER) supplemented with 50 mM dithiothreitol (Thermo Fisher Scientific, catalog number: 78501)
- 0.05 M iodoacetamide in UA buffer
- Trypsin/Lys-C prepared in 100 mM TEAB to 10 µg/ml
- DTT
- CaCl2
- CaCl2·2H2O
- KH2PO4
- MgCl2·6H2O
- NaCl
- Urea
- Tris-HCl pH 8.5
- TEAB (Sigma, catalog number: T7408-100ML)
- 100% trifluoroacetic acid
- IAA powder
- Sterile 1x PBS (see Recipes)
- UA buffer (see Recipes)
- 100 mM triethylammonium bicarbonate (TEAB) (see Recipes)
- 0.5 M NaCl (see Recipes)
- 10% trifluoroacetic acid (TFA) (see Recipes)
- M-PER supplemented with 50 mM dithiothreitol (DTT)
- Iodoacetamide (IAA) solution (0.05 M IAA in UA buffer) (see Recipes)
- Trypsin digestion solution (see Recipes)
LC-MS/MS Data Acquisition- Picofrit 15 cm x 75 µm ID HPLC column packed with 5 µm BioBasic C18 particles 300Å (New Objective)
- 3% acetonitrile/0.1% formic acid
- 100% formic acid (Thermo Scientific, catalog number: 28905)
- 100% acetonitrile
- A Buffer (see Recipes)
- B Buffer (see Recipes)
Equipment
- Sub-culture of vertebrate and invertebrate cells
- 25 cm2 flask
- 75 cm2 flask
- CO2 Cell culture incubator (Any supplier is adequate)
Note: Any supplier is adequate; the choices are price/quality and capacity. All that is necessary is a water-jacketed incubator which can be regulated to 5% CO2 in a water-saturated atmosphere. This will require a medical grade CO2 tank (liquid gas under pressure) and a pressure regulator specific for that tank. Two incubators will be required if the cells are to be grown simultaneously because the vertebrate cells are incubated at 38 °C and invertebrate cells at 28 °C. - Biological safety cell culture hood
- Inverted microscope
- Hemacytometer
- Virus growth and titration
Stock virus preparation- CO2 Cell culture incubator
Virus titration by Plaque assay- Rocker platform (Bellco Biotechnology)
- 37 °C water bath
- Dilution tube rack
- Ice bucket
- Sindbis virus infection of vertebrate and invertebrate cells
Sindbis virus infection of BHK cells- Platform rocker (Bellco Biotechnology)
- Hemocytometer
- 75 cm2 flasks
- Centrifuge
- Hemocytometer
- Purification of Sindbis virus
Purification and Concentration by isopycnic centrifugation- Inverted microscope
- Platform rocker (Bellco Biotechnology)
- Hemocytometer or cell counting device
- CorningTM polypropylene tubes (micro centrifuge tubes, self-standing and conical)
- Sorvall RC-5B Super speed centrifuge
- Beckman ultracentrifuge
- Beckman SW-28 rotor
- Polyallomer 38 ml tubes
- Beckman SW-55Ti
- Polyallomer 5 ml tubes
- Ring stand
- Small tube clamp
- Hand held low-intensity lamp
- 30-100 ml gradient former (Bio-Rad, model 385)
- Medium stir plate
- Sorvall RC-5B Super speed centrifuge
- Beckman ultracentrifuge and appropriate rotor and tubes for the volume used
- 30-100 ml gradient former (Bio-Rad, model 385)
- Calculation of particle to PFU ratio
None - Mass spectrometry and proteomic analysis
Protein Extraction and Digestion- Tabletop centrifuge capable of reaching up to 14,000 x g
- Shaker heat block
- Vortex
- SpeedVac Concentrator
LC-MS/MS Data Acquisition- Orbitrap ELITE mass spectrometer (or equivalent; e.g., Orbitrap QE+, Orbitrap Fusion, Orbitrap Fusion Lumos)
- Easy-nLC II liquid chromatography system (or equivalent HPLC/UHPLC nanoflow pump system)
Software
Data Processing:
- High performance computer meeting the minimum specifications to run Proteome Discoverer Software (Currently available version: 2.2; Thermo Fisher Scientific)
- PANTHER classification system (http://www.pantherdb.org/)
Procedure
Protocol outline:
- Sub-culture of vertebrate and invertebrate cells
- Sub-culture of BHK cells
- Sub-culture of C7-10 cells
- Virus growth and titration
- Preparation of stock virus
- Virus titration by plaque assay
- Sindbis virus infection of vertebrate and invertebrate cells
- Sindbis virus infection of BHK cells
- Sindbis virus infection of C7-10 cells
- Purification of Sindbis virus
- Concentration by isopycnic centrifugation
- Concentration by polyethylene glycol (PEG) precipitation
- Calculation of particle/PFU ratio
- Mass Spectrometry and Proteomic Analysis
- Protein Extraction and Digestion
- LC-MS/MS Data Acquisition
- Data Processing
- Sub-culture of vertebrate and invertebrate cells
Sub-culture of BHK-21 cells
Individual stocks of BHK cells may require different passage schedules and may have different levels of viable passages. It is good practice to keep track of the number of passages that an individual culture can be sub-cultured so that a schedule of cell thawing and storage can be established.
Fresh newly thawed cells can be passaged up to 30 times. If you are not going to split them immediately, cells can be incubated at 28 °C for 2 to 3 days but must be split at least once to recover normal growth before use in experiments.- Wash a confluent BHK cell monolayer once with 1x PBS-D, using 5 ml for a 25 cm2 flask or 15 ml for a 75 cm2 flask.
- Decant PBS-D and add trypsin to the monolayer. Add 2 ml to a 25 cm2 flasks, or 5 ml to a 75 cm2 flask. Incubate at room temperature until the cells begin to detach from the flask. Disrupt cell clumps by pipetting up and down using a 5 to 10 ml serological pipet.
- Add BHK cell culture medium to a volume of 1:1 to stop trypsin. Resuspend cells in the trypsin solution and check for cell clumps by under the microscope. If there are significant remaining clumps, pipet up and down using a 5 to 10 ml serological pipet.
- Pipet cell solution into a conical tube of the appropriate size.
- Spin tube(s) at 500 x g (medium speed on a tabletop centrifuge) for 3 to 5 min to form a firm pellet. Do not overfill tube. Do not invert tube to mix.
- Decant supernatant and replace with the appropriate amount of medium.
- For preparation of a single 25-cm2 flask: resuspend in 3 ml E-MEM. Add 1 ml of cells to 10 ml of medium in the flask. Up to 3 flasks can be prepared from one initial 25 cm2 flask, at a 1:3 split ratio (area of the flask).
- For preparation of a large number of 25-cm2 flasks (plaque flasks or plates): To a sterile bottle, add 10 ml E-MEM per each flask to be prepared. If you are using 6-well plates use 2 ml/well. Remove some medium from the bottle and resuspend cells. Add back to the rest of the medium and mix well. To prepare multiple flasks or plates aliquot 10 ml of the cell mixture into each flask 25-cm2 flasks, swirling continuously. One 75 cm2 flask will make 9-25 cm2 flasks or 4- 6-well plates are (9.5 cm2/well x 6 wells = 57 cm2 total) using 2 ml /well.
Note: This protocol ensures that the cell monolayers will be uniform and contain approximately the same number of cells. Care should be taken when placing the flasks into the incubator so as not to disturb the cell suspension. - For preparation of a single 75-cm2 flask: Aliquot 1 ml of cells from a single 25 cm2 flask into 30 ml complete E-MEM.
- Incubate cells in flasks at 37 °C for 24 h or until confluent.
- If medium does not hold pH of < 8, adjust pH with 1 M HEPES to a final concentration of 8 mM HEPES. This is normally not necessary however if the pH needs adjusting then it should be between pH 7 and 8.
Sub-culture of C7-10 cells- Maintain mosquito cell lines in semi-suspension in tissue culture flasks at 28 °C in a 5% CO2 humidified environment. Subculture up to every other day as described below. The cells need to be split when they begin to clump or float. These cells like to be concentrated; do not split more than 1/3x the area of the monolayer.
Notes:- Semi-suspension cells adhere loosely to the substrate initially, and then begin to float as they age. Some cells may stick tightly to the substrate.
- Do not attempt to completely disrupt the clumps or scrape the cells, this damages the cells.
- Clean the area under the hood with 70% ethanol. Clean all materials going under the hood with 70% ethanol and remove unneeded materials.
- Place media at room temperature to warm.
- Remove cell flasks from the incubator. Observe cell media for changes in color (red) and clarity.
- Visualize cells under a microscope at 20x magnification to check monolayer for irregularities, and confluency. Cells need to be 95%-100% confluent prior to passage. Cells will be at a density of ~ 2 x 108 cells/75 cm2 flask in 25 ml of medium.
- Hit the side of the flask sharply on any soft surface several times to loosen any cells that are stuck to the surface of the flask.
Notes:- If the cells are aggregated into clumps, try to break up the clumps by pipetting up and down several times using a 5-10 ml pipet, before transferring the cells to a new flask.
- Small clumps are acceptable.
- Transfer one-third of the 25 ml of the medium of the cell suspension to each of three flasks. To each of these flasks, add fresh media to increase the volume to the original 25 ml. Add 25 ml of fresh 1x MEM to the original flask if there are many adherent cells remaining if the cells are needed. If not, omit this step.
- Cap flasks tightly if the caps are vented, leave loose for non-vented caps.
- Return flasks to the incubator set no higher than 26 °C. Mosquito cells will go into heat-shock at temperatures higher than 34°C.
- Virus growth and titration
Stock virus preparation
It is standard practice to grow a stock of virus from which additional virus stocks will be grown prior to any additional work with the virus. This practice avoids the production of defective interfering particles which will accumulate upon successive serial passage of high concentrations of virus. Generally, an MOI (multiplicity of infection) of 0.01 PFU (plaque forming units) per cell is required for production of stock virus. To calculate the correct MOI, the number of cells to be infected must be known. Then multiply the number of cells by the required MOI, e.g., 106 total cells to be infected x MOI of 0.01 = 104 PFU of virus inoculum is needed.
If your stock virus is 1 x 109 PFU/ml and you need a 104 PFU inoculum, do the following:- Make a serial dilution of the stock virus, begin by making a 1:10 dilution of the stock virus to a total of 1 ml. This will be 100 µl of virus supernatant in 900 µl of PBS-D + 3% FBS. This is a 10-1 virus dilution.
- Make a serial dilution of this sample so that you get -1 through -4 dilutions in 5 separate tubes (see Figure 3). The amount of virus that you need is in the -4 dilution tube, 1 ml of virus of 10-4 PFU/ml.
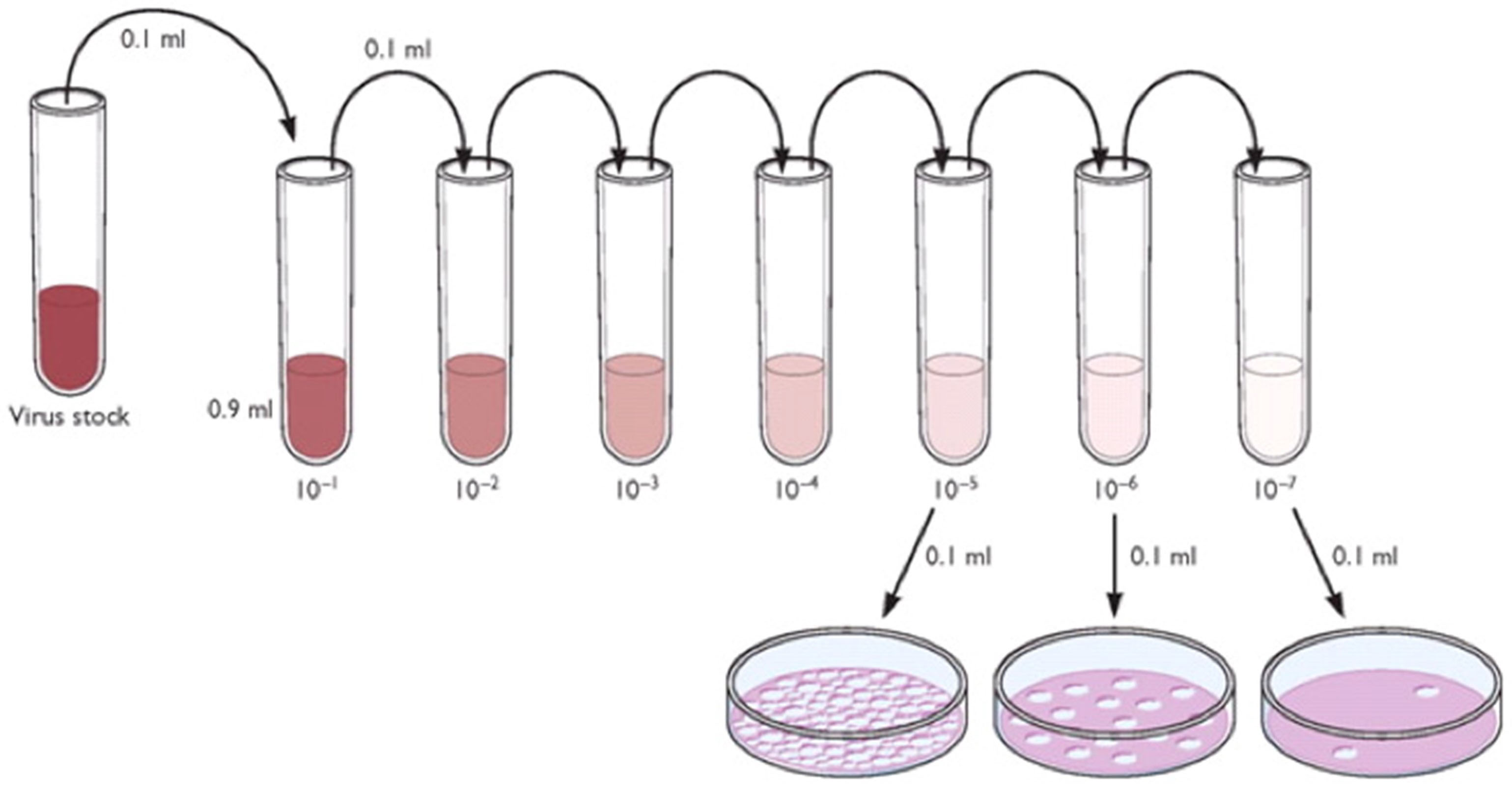
Figure 3. Serial passage scheme. The figure is modified from http://www.virology.ws/2009/07/06/detecting-viruses-the-plaque-assay/. This work is licensed under a <a rel="license" href="http://creativecommons.org/licenses/by/3.0/">Creative Commons Attribution 3.0 Unported License</a>. - Make as much of this dilution as you need to infect all of your flasks with 1 ml/75 cm2 flask. Do not be tempted to use an inoculum of less than a 50 µl measurement of virus because the only correct way to dilute virus is to make a serial dilution. Virus particles do not go into true solution and care must be taken to suspend them correctly, otherwise the counts will be artificially high or low. Alternatively, you can use 100 µl of the -5 dilution up to a volume of 1 ml or use the entire -5 dilution to make 10 ml of the virus and arrive at the correct MOI.
- Refreeze your initial stock virus; this is your primary stock from which you will grow subsequent stocks. Virus is quantitated by plaque assay on the cells from which the virus produces CPE which is usually the cell line from which it is produced (in this case BHK). However Sindbis virus does not produce CPE in many insect cells (e.g., U4.4 of C7-10 cells), which cannot be used for the plaque assay.
Notes:- Stock virus can be grown from vertebrate or invertebrate cells.
- Sindbis virus is very sticky and will bind to glass and plastic surfaces. We have found that CorningTM polypropylene tubes (microcentrifuge tubes, self-standing and conical) bind fewer virus particles than other brands of plastic. Disposable glass tubes are used to make the virus dilutions required for the plaque assay.
- Tissue culture grade reagents from all major suppliers have been found suitable for the cell culture and virus production portions of this protocol.
Preparation of BHK cells
Note: Individual stocks of BHK cells, e.g., from ATCC, may require different passage schedules and may have different numbers of viable passages.- Wash a confluent BHK cell monolayer once with 1x PBS-D, using 5 ml for a 25 cm2 flask or 15 ml for a 75 cm2 flask.
- Decant PBS-D and add trypsin to the monolayer. Add 2 ml to a 25 cm2 flasks, or 5 ml to a 75 cm2 flask. Incubate at room temperature until the cells begin to detach from the flask. Disrupt cell clumps by pipetting up and down using a 5 to 10 ml serological pipet.
- Add BHK medium 1:1 to stop trypsin. Resuspend cells in the trypsin solution and check for cell clumps by under the microscope. If there are significant remaining clumps, pipet up and down using a 5 to 10 ml serological pipet.
- Pipet cell solution into a conical tube of the appropriate size, e.g., 10 or 50 ml tubes.
- Spin tube(s) at 500 x g (medium speed on a tabletop centrifuge) for 3 to 5 min to form a firm pellet. Do not overfill tube. Do not invert tube to mix.
- Decant supernatant and replace with the appropriate amount of medium 3 ml for 25-cm2 flask and 9 ml for a 75 cm2 flask.
- For preparation of a single 25-cm2 flask: Resuspend in 3 ml E-MEM. Add 1 ml of cells to 10 ml of medium in the flask. Up to 3 flasks can be prepared from one initial 25 cm2 flask, at a 1:3 split ratio (area of the flask). Cap flasks tightly (vented caps, leave loose for non-vented caps).
- 9-25 cm2 flasks can be prepared from 1-75 cm2 flask.
Many of the materials used for the plaque assays are used for both mammalian and mosquito cell assays. It is important to make the serial dilutions of virus immediately prior to infection of the plaque assay flasks/plates to limit the amount of virus binding to the tubes.- Determine the number of plates/flasks required, with two wells per dilution of virus (plated in duplicate), plus a few control plates or wells. The day before the plaque assay, split BHK cells into 6-well plates containing ~6 x 106 cell/plate = 1 x 106 cells/well. In general, the use of 6-well plates has replaced the use of individual flasks; however when learning the technique the use of flasks in much simpler.
Note: The number of cells required is determined by the confluency required at the time of the assay. Because confluency is related to the area the cells cover, this number is manipulated by knowing the area of the vessel the cells will grow on. Thus, the area of the flasks (rectangular) or wells (circular) will vary but the number of cells/cm2 is constant. (e.g., 1 x 106 cells. For a 6- well plate, each well is 9.6 cm2 seeded with 1 x 106 cells/well x 6 wells for a total of ~58 cm2 onto which a total of ~6 x 106 cells is seeded. These numbers are relative to every specific culture of any cell line and may need to be adjusted up or down. The important thing is the % confluency on the day of the assay during which the cells should still be in log phase. - Allow cell monolayers to become ~80%-90% confluent by the time of the plaque assay.
Note: If the cells are ready in the morning and the assay is to be done in the afternoon, the cells can be stored at room temperature with the caps closed tightly, or moved to a 28 °C incubator to slow growth. - Remove the virus to be titered from the freezer and thaw on ice.
- Fill the required number of dilution tubes with 900 μl diluent (SVHR diluent; 1x PBS-D/3% FBS (see Recipes). You need one tube/dilution. One tube for the control (not infected) and one for the positive infected control.
- Prepare serial dilutions of the virus by adding 100 μl virus to the first tube containing 900 µl of diluent (10-1), vortex that dilution at full speed, removing 100 μl, and adding it to the next dilution tube (10-2). Continue this process until the desired dilutions have been made. Change pipet tips each time to avoid “carry-over” effect of contaminating virus.
Note: Dilutions should be made immediately prior to use. Virus and dilutions should be kept on ice at all times. - Once the dilutions have been made, pour growth media out of flasks (into a sterile waste beaker) and pipet out of plates with a serological pipet (leave enough liquid to cover the monolayer) and infect each flask or well of a 6-well plate with 200 μl of the appropriate dilution. In this case, the area of the vessel no longer matters because the calculation will convert plaques at each dilution to PFU/ml.
- Do not let monolayers dry out while adding virus–i.e., do not pour/pipette media off too many flasks/plates at one time or try to drain every drop. DRY MONOLAYERS ARE DEAD MONOLAYERS.
- Place flasks on a rocking platform at room temperature for one hour. Do not rock plates; the liquid will only swirl around the edge of the well. Place the plates at the appropriate temperature, 38 °C for BHK cells and hand rock every 15 min.
Note: The caps on the flasks should be tightened while they are rocking since they are not in the appropriate CO2 environment unless they are vented, then place these in the incubator. - After an hour of rocking/incubating, remove the inoculum (pipette) and overlay the monolayer with 7 ml of a 1:1 mixture of 2% agarose in dH2O and complete 2x EMEM with 7 ml for T-25 cm2 flasks and 2 ml for each well of a 6-well plate.
Notes:- The agarose should be hot enough that it will not solidify too quickly, but cool enough to allow one to touch the bottle before mixing with the medium (~60-70 °C). Bottles of media at the correct temperature should not feel excessively hot to the touch. Do not try to move flasks/plates before the overlay has solidified, because the cell monolayer will tear. Solidified agar will appear cloudy.
- Not all agarose is tolerated by cells in culture. In general, agarose formulated for gel electrophoresis or chromatography is not suitable for tissue culture. Use Sigma agarose () (Sigma-Aldrich, St Louis, MO, catalog number: A6013).
- Incubate flasks for 2 days at 37 °C, 5% CO2.
Note: Before staining monolayers, hold the flasks up to the light to see if plaques are visible. The plaques will appear more opaque than the rest of the monolayer. Visible plaques are an indication that the assay has worked up to this step. - To stain the monolayer, add 5 ml of 1:1 mixture of 2% agarose in dH2O and 2x PBS-D with 3% neutral red stain (3% of the total amount of agarose required). Cover the flasks to protect cells from light.
Note: Cells become light sensitive after they take up neutral red and should be protected from light. - If necessary, when the plaques are faint, return flasks (plates) to 37 °C for 4 h or 28 °C overnight prior to counting plaques.
Note: You should expect to see clear plaques surrounded by red, living cells. The number of plaques per flask should roughly follow the dilutions made (e.g., 1 plaque on 10-6 flask, 10 plaques on 10-5 flask, and 100 plaques on 10-4 flask). Plates can be used to produce duplicates (6-well plates) or triplicates (24-well plates). If there are no plaques, check the monolayers under the microscope, the cells may be lysed. - Count plaques and calculate the titer.
- To calculate the virus titer: e.g., with 2 plaques at -7 dilution and an inoculum of 200 µl, convert this to plaques/ml or 2 plaques x 1/200 µl x 103 µl/1 ml = 10 plaques/ml from a 1/10-7 dilution = 10 x 107 or 1 x 108 PFU/ml. Thus the equation is #plaques/dilution factor (ml) x volume of inoculum (ml) = PFU/ml. Unless the number of virus particles = number of plaques/ml then this unit is a plaque forming unit. This will be explained in the section calculating the particle to PFU ratio.
Note: If there are no plaques but the positive control worked then the sample has no virus or the incorrect dilutions were plated. If there are no plaques in the control sample check the cells under the microscope for lysis, lysed cells will not take up stain or produce a plaque. Cell lysis will occur if the osmolarity and tonicity of the cultures are not correct. Crystals of stain mean that the agarose was too hot when it was added. Agarose that is too hot will also kill the cells.
- Sindbis virus infection of vertebrate and invertebrate cells
Sindbis virus infection of BHK cells- Subculture BHK cells the day prior to infection such that the monolayer is ~90% confluent at the time of infection.
- Calculate the amount of virus needed for the desired multiplicity of infection (MOI).
No. cells x MOI = PFU needed.
For a single cycle MOI = 10, stock virus MOI = 0.01
For 75 cm2 flask, the number of cells is ~ 2 x 107cells
For 25 cm2 flask, the number of cells is ~6 x 106 cells
For a 6-well plate, the number of cells is about 1 x 106 cells/plate.
Plates are also used if a small amount of inoculum is used, e.g., stock virus volume titer is too low to infect too many cells and the virus must be amplified before use. - Remove the virus from -80 °C freezer and thaw the vial on ice. Dilute inoculum to the desired concentration in 1x PBS-D/3% FBS. Refreeze unused stock virus.
Notes:- A total volume of at least 1 ml is required for infection of 75 cm2 monolayer, and a minimum of 200 μl is required for a 25 cm2 monolayer. A minimum of 200 μl is required for a 6-well plate. Close the cap tightly and secure to rocker platform. Do not rock plates.
- SVHR is heat stable however, many other strains and mutants of Sindbis are not, and thus must be thawed on ice to retain infectivity.
- Place flask on a rocking platform for 1 h at room temperature, with caps tightened. Alternatively, infect in the incubator at 37 °C with intermittent hand rocking.
- Remove inoculum and add fresh, complete 1x E-MEM media. Add 5-7 ml media to a 75-cm2 monolayer or 3 ml to a 25-cm2 monolayer. Leave the caps loosened during incubation to allow for CO2 exchange. If more concentrated virus is desired, add 3 ml medium to a 75-cm2 monolayer. Do not allow the monolayer to dry out.
- Virus should be harvested once CPE becomes evident, which is generally between 18 (BHK) and 24 h (C7-10) post-infection.
Note: Cell lines will differ in the time period in which CPE is demonstrated. Some indicators of CPE in BHK cells include the cells becoming long and thin, cytoplasm with a “foamy” appearance, aggregations of cell nuclei, and, in advanced stages, cell lysis. Do not allow the cells to lyse for any type of virus analysis work. - Remove medium from flasks to conical centrifuge tubes (Corning, 15 ml Fisherbrand Cat # 05-539-5 or 50 ml centrifuge tubes, Fisherbrand Cat # 05-539-6). Combine like samples if desired. Spin samples for 10 min in a tabletop centrifuge at ~700 x g to remove cell debris. Decant virus to a new tube and bring the solution to 10% glycerol which acts as a cryoprotectant. Aliquot the virus supernatant into tubes appropriate for freezing and flash freeze virus using liquid N2.
- Store virus at -80 °C.
Note: Membrane containing viruses will undergo a freeze-thaw at -65 °C due to phase transition in ice. Thus a freezer failure resulting in warming to -65 °C should be considered a thaw. Sindbis virus will lose some titer (~ 0.5 logs) upon freeze-thaw but is still infectious.
Sindbis virus infection of C7-10 cells
Mosquito cells are used when a comparative analysis of virus titers from the invertebrate host are required. To infect mosquito cells, a confluent monolayer of single cells (no clumps) must be formed. Mosquito cells do not adhere well to the substrate, and tend to lift from the flasks regardless of the medium used. To temporarily circumvent this problem, mosquito cultures can be starved for serum for one hour, which causes the cells to adhere to the substrate more tightly. Mosquito cultures in our lab have been adapted to use in E-MEM, however, all other treatments of the cells are consistent with cells which are in other insect media.- Spin down for 5 min at 200-500 x g approximately 1½ -75 cm2 flasks of cells per 1-75 cm2 flask needed for infection.
Note: Mosquito cells grow as semi-suspension cultures and are easily removed from the flask by tapping the flask against a hard surface, or alternatively by pipetting up and down with a 5-10 ml pipet. Do not spin excessively as the cells will die during pelleting or resuspension. Some cells will be lost during the process. - Wash the cell pellet once with 1x PBS-D to remove any remaining medium.
- Resuspend the final cell pellet in 1 ml/flask needed for infection, of E-MEM medium deficient in FBS.
- Count the cells using a hemocytometer.
- Aliquot approximately 6 x 107 cells per 75-cm2 flask with enough medium (without FBS) to cover the monolayer.
- Incubate the flask of cells at 28 °C for one hour, or until cells are well attached to the surface of the flask.
- Once cells are attached, remove medium and add the desired amount of virus to the monolayer. Close caps tightly.
- Rock the flask slowly at room temperature for one hour.
Note: Rocking slowly is critical to keep a minimum number of cells from lifting from the flask. - Most of the cells should remain attached to the flask after rocking. If cells have lifted from the monolayer they should be removed with the inoculum.
- Fresh, complete E-MEM medium should be added to each flask.
Note: To increase the concentration of virus, minimize the volume of fresh medium added to the flasks. A minimum of 3 ml medium/75 cm2 is required to cover the cells and support metabolism. Take care that the flasks are level to ensure complete coverage of the monolayers. - Virus may be harvested from 24 to 72 hpi depending on the multiplicity (MOI) used. Cytopathic effect (CPE) will not be evident in mosquito cells, in which case virus will be harvested based on time post-infection rather than CPE.
- For normal storage: Harvest virus and store in sterile 10% glycerol. For best preservation of the virus titer quick freeze tubes in liquid N2.
Note: Virus frozen and thawed will lose 1/3 to 1/5 log of titer. This is not a significant loss for use to infect cells. For preservation of the highest infectivity level, virus can be stored at 4 °C for up to 5 days in neutral pH buffer. This method preserves the most infectivity and is used for virus to be analyzed in structural studies and for the calculation of particle to PFU ratios.
- Purification of Sindbis virus
Purification and concentration by isopycnic centrifugation
The virus supernatant is harvested into conical tubes and cell debris is removed from the supernatant by low-speed centrifugation. This is a critical step and if it is omitted, too much debris will interfere with the following gradient purification steps. Clarify virus supernatant by centrifugation in the appropriate size conical tube for 10 min at 1,800 x g.- Virus supernatant is combined into an appropriate size container omitting addition of the glycerol.
Note: From this step forward, you should not consider your samples sterile. This is not usually a problem and antibiotic is added to the 2x E-MEM media. - Determine the number of gradients which will be needed.
Typically, 20 ml of virus is layered onto the initial potassium tartrate step gradient in the 38 ml tubes. This gradient is formed by carefully layering 12 ml of 15% potassium tartrate onto a layer of 6 ml of 35% potassium tartrate, or making a 10% to 35% continuous potassium tartrate gradient. Read the manufacturer’s manual for instructions on use of the gradient former. The tubes are placed into the rotor buckets and weighed upright (we use a small beaker cushioned with tissue). PBS-D is added drop- wise to balance the tubes. Take special care to weigh the buckets and tubes, and load the centrifuge rotor according to the manufacturer’s tolerances and specifications found in the manual. Failure to do so can result in severe damage to the rotor and the instrument.
Note: The rotor, buckets and solutions should be pre-chilled to 4 °C. If an odd number of gradients are required a “blank” gradient is made and overlaid with buffer to balance the rotor. - The gradients are centrifuged overnight (this is purely for convenience) at 24,000 rpm in an SW-28 Beckman rotor (or 100,000 x g) and 4 °C.
- After the run is completed, remove a tube carefully from the bucket and attach to a ring stand using a small tube clamp. A virus band of iridescent blue should be visible at the potassium tartrate 15%-35% step interface when the gradient is illuminated from the side with a hand held light of 20 watts. See Figure 4 Density gradient purified Sindbis virus band. Repeat for each of the gradients.
- Collect the band by puncturing the bottom of the tube and letting the tartrate solution flow into a waste beaker, while collecting the virus band into a 2 ml cryotube. Discard the remaining solution. Alternatively, the band can be collected from the side of the tube using a needle and syringe.
- Pool the virus-containing samples and dilute with 1.5 volumes of ice-cold PBS-D. This solution should be less dense than 15% potassium tartrate. To check that your concentration is correct, your virus solution should not sink into an aliquot of 15% potassium tartrate.
- This diluted virus is then layered over a continuous 15%-35% potassium tartrate linear gradient of a volume not less than ¾ of the added virus sample. The first gradient may be a step or linear gradient but the second should be a linear one. Do not overfill.
- The continuous gradients are run for 2 h at 26,000 rpm in an SW 40ti Beckman rotor (120,000 x g) and the virus band collected as described in Step D6 above. The virus band should appear about 1/3 down the length of the tube. The virus band may be stored at 4 °C in the tartrate solution. The refractive index of the final virus-tartrate solution is 1.3665 which is ~28% tartrate (Rumble, 2005).
- Collect a sample of the virus to titrate by plaque assay and to determine protein concentration by BCA assay (PierceTM Rapid Gold BCA Protein Assay Kit, Thermo Fisher).
Note: Properly grown and purified SVHR should give a particle/PFU value of ~1.
Figure 4. Shown is the opalescent blue band of purified Sindbis virus in a 15-35% potassium tartrate gradient. Note the flocculent pellet at the bottom of the tube. This will be removed after the second K-tartrate gradient.
Note: This is an alternative purification step used to concentrate the virus particles but infectivity will be lost.- Infect BHK cells with Sindbis virus as previously described for virus purification. The number of flasks infected will vary depending on how much virus you need.
- Pour culture medium into a centrifuge tube.
- Spin supernatant for clarification, removing cells and cell debris (from this step proceed as in Steps D2-D9 in virus purification protocol.
- Transfer supernatant to a fresh tube.
- Add 0.25 volumes of 40% polyethylene glycol-8000 in 2 M NaCl and mix well.
- Incubate about 24 h at 4 °C for virus precipitation.
- Centrifuge at 10,000 rpm (12,000 x g) for 10 min.
- Discard the supernatant into a waste beaker and drain well (invert the tube and use a kimwipe to absorb any traces of PEG).
- Re-dissolve the pellet in 5 ml of 1x PEG buffer.
- Transfer to a Kimax glass centrifuge tube.
- Centrifuge at 12,000 x g for 10 min (Sorval)–this is a further clarification step.
- Use the supernatant for gradient purification as described (from this step proceed as in Steps D2-D9 in virus purification protocol.
- We did not use PEG concentration of Sindbis virus in our proteomic protocol but include the results in this protocol to demonstrate the importance of purification of infectious virus particles compared to non-infectious particles to extrapolate conclusions on functions of the virus system in question.
- We find that using the SVHR strain of Sindbis virus, PEG concentration followed by isopycnic purification increases the particle/PFU ratio by 10 fold. This increase in the number of non-infectious particles in the interpretation of the experimental outcomes should be seriously evaluated and noted in the results.
- Virus supernatant is combined into an appropriate size container omitting addition of the glycerol.
- Calculation of particle to PFU ratio
- A precise protein concentration of the purified virus is required for this calculated value to be accurate. The tartrate solution or PEG will interfere with this assay; the sample must be diluted 1:10.
- SVHR virus proteins MW in Daltons = total combined weight of structural proteins (C, E2 and E1) = 130 KDa (by direct BCA measurement, use this measured number and not the calculated value).
- This value is multiplied by 240 copies/virus particle (T = 4 particle:60 T = 240 copies). See Johnson and Speir (1997) for T calculation.
130 KDa (total virus protein, see Step E2) x 240 (copies/particle see Step E3 above) = 3.12 x 107 Da of protein/virus particle
Convert from Da to grams [1 Dalton = 1.66 x 10-24 g (CRC tables)]
3.12 x 107 Da/virus particle x 1.66 x 10-24 g/Da = 5.18 x 10-17 g/particle
e.g., if you have 1011 PFU/ml SVHR you will have 208 µg/ml of protein (measured by BCA assay)
= 2.08 x 10-4 g/ml.
Note: PFU and particles are not the same unit. PFU or plaque forming units is a measure of infectivity of the virus because all virus particles are not infectious. The number of particles refers to the number of physical particles in a solution, which is normally greater than the number of PFU.
Calculate particles/ml:
2.08 x 10-4 g/ml/5.18 x 10-17 g/particle = 0.4 x 1013 particles/ml
0.4 x 1013 particles/ml/1 x 1011 PFU/ml = 0.4 x 102 particles/PFU = 40 particles/PFU
Note: For Sindbis SVHR the particle/PFU ratio from BHK or C7-10 cells should be ~1 part/PFU.
- Mass spectrometry and proteomic analysis
- To visually determine if the sample contains contamination a sample of the purified population was then visualized by transmission electron microscopy to check for contaminating cellular organelles or membrane fragments (Coombs and Brown, 1987). This method is beyond the scope of this protocol but detailed in the reference provided above.
- Additionally, to check for co-purified protein contaminants, check protein content by running ~10 μl of virus on a 4-12% Bis-Tris SDS-PAGE gradient gel (Invitrogen, Novex) as described by the manufacturer.
- Stain the gel with silver in the method of Wray or with coomassie blue (Wray et al., 1981). Excise the visualized bands (silver stain only) and perform an in-gel digestion prior to LC-MS/MS analysis for protein identification (Glaros et al., 2015).
Viral preparations and their respective negative controls, medium from the cell monolayers harvested prior to infection were processed for LC-MS/MS analysis using the filter-aided sample preparation (FASP) method (Wiśniewski et al., 2009).- Following purification, determine the total protein concentration using a Pierce BCA protein assay kit, following the manufacturer’s instructions.
- Normalize all preparations using sterile PBS to 0.5 µg/µl; aliquot out 10 µg of each sample into Lo-bind Eppendorf tubes.
- For each sample, mix 10 µg of total protein (20 µl) 1:1 with M-PER supplemented with 50 mM dithiothreitol (DTT) and heat at 95 °C in a heat block at 400 rpm for 10 min to reduce and denature proteins.
- Cool samples to room temperature, and mix with 200 µl of UA buffer (8 M urea, 100 mM Tris-HCl, pH 8.5).
- Apply each preparation to a 30 kDa filter spin column (Millipore-Ultracell YM-30), and centrifuge at 14,000 x g for 30 min at room temperature to collect all proteins onto the filter membrane.
- Alkylate proteins by adding 100 µl of iodoacetamide (IAA) solution (0.05 M IAA in UA buffer) to each filter membrane and incubate at room temperature in the dark for 20 min. Following alkylation, centrifuge the samples at 14,000 x g for 20 min to remove the alkylation solution.
- Wash each sample by applying 100 µl of UA buffer to the filter and centrifuging at room temperature at 14,000 x g for 30 min. Repeat this washing step two more times for a total of three washes. Then wash three more times each with 100 µl of 100 mM triethylammonium bicarbonate (TEAB).
- To digest the captured protein, apply 100 µl of a trypsin digestion solution (Trypsin/Lys-C prepared in 100 mM TEAB to 10 µg/ml) on each membrane and incubate in sealed tubes overnight (wrap the closed tube caps with parafilm for a better seal) in a heat block at 37 °C, 400 rpm.
- After incubation, collect the peptides for LC-MS/MS analysis. Place filter units in a new, clean tube and centrifuge each tube for 30 min at 14,000 x g at room temperature. Wash the membrane by centrifugation one time with 50 µl of 100 mM TEAB and once with 50 µl of 0.5 M NaCl. Collect both of these washes and pool with the final peptide eluate.
- Acidify each sample with 10% trifluoroacetic acid until the final pH is ~2-3.
- Prior to mass spectrometric analysis, desalt each sample using Pierce C18 spin columns according to the manufacturer’s directions. Eluate from the desalting process should be dried to completeness in a clean Lo-bind Eppendorf microcentrifuge tube using a SpeedVac at 30 °C and stored at -80 °C until LC-MS/MS analysis.
- Reconstitute dried peptides in “A Buffer” (3% acetonitrile/0.1% formic acid) and resolve on VIRGIN Picofrit 15 cm x 75 µm ID HPLC column packed with 5 µm BioBasic C18 particles 300Å using a multistep gradient [e.g., 130 min; 0-5 min: 5-10% B, 6-110 min: 10-35% B, and 111-130 min: 35-95% B]. A virgin or unused column is important to ensure that all peptides identified are from the sample and not from a previous LC-MS/MS run which could result from ‘carry over’ if using a column that already had a sample applied. The liquid chromatography gradient should be chosen to allow for adequate separation without excessive time. These parameters depend upon the column and instrument used for analysis. For the gradient, the A buffer is 3% acetonitrile/0.1% formic acid and the B buffer is 95% acetonitrile/0.1% formic acid. Tryptic peptides should be analyzed in at least technical triplicates.
- For the Orbitrap ELITE the following configurations were used. This should serve as a guide for similar instrument parameters if using other mass spectrometers. These parameters are instrument specific and you should consult the instrument’s manufacture or the literature for recommendations. Generic settings established from the literature used for bottom up (tryptic peptide analysis) proteomics are likely adequate. A variety of instrument methods have been curated and peer-reviewed which are available at www.massspectrometrymethods.org.
Example instrument settings used for an orbitrap ELITE- Orbitrap MS1 scans are performed at a resolution of 120,000 at 400 m/z, with a scan range of 110-2,000 m/z.
- The top 20 precursors are selected for MS2 data-dependent fragmentation. An MS2 spectrum is acquired using the iontrap scanning in normal mode (Top 20 Method).
- The minimum signal required to trigger a data-dependent scan is 5000.
- Collision-induced dissociation (CID) is used to generate MS2 spectra with the following settings: normalized collision energy 35%, default charge state 2, isolation width 2 m/z, and activation time 10 ms.
- AGC target is set to 1 x 106 for MS and 5 x 104 for MS/MS with a maximum accumulation time of 200 ms.
- Dynamic exclusion is set for 60 s for up to 500 targets with a 5 ppm mass window.
- A lock mass of 445.120025 is used for internal calibration to improve mass accuracy.
- Process spectra data using Proteome Discoverer with the embedded SEQUEST search algorithm against a Sindbis virus polyprotein database (Uniprot ID: P03317) merged with either Homo sapiens (RefSeq Tax ID: 9606) or Cricetulus griseus (RefSeq Tax ID: 10029). Download the Aedes albopictus-deducted proteome (Chen et al., 2015) data from VectorBase (Giraldo-Calderón et al., 2015) and additional peptides from the NCBI TSA database.
- Organize the non-redundant proteomes on excel spreadsheets (Ribeiro et al., 2004) and annotate as described previously (Karim et al., 2011). Merge the resulting FASTA file with the SINV polyprotein and use it to search against the SINV mosquito preparations.
- Set dynamic modifications for carbamidomethylation of cysteine [+57.02 Da], oxidation of methionine [+15.99 Da], and N-terminal acetylation [+42.011].
- MS/MS spectra are searched with a precursor mass tolerance of 10 ppm and a fragment mass tolerance of 0.6 Da.
- Trypsin is specified as the protease with a maximum number of missed cleavages set to 2.
- False discovery rate using PERCOLATOR (Kall et al., 2007) is set to < 1% to score high confidence peptide identifications.
- Perform grouping and functional analysis using the PANTHER classification system for the human background only with each protein id’s accession number (Mi et al., 2016).
Recipes
- Complete E-MEM, 1x and 2x
Note: Do not warm 2x MEM until the supplements have been added, it will precipitate.
10% heat-inactivated fetal bovine serum (FBS; any suitable FBS, we test sample lots of FBS prior to purchase)
5% tryptose phosphate broth (TPB; see Recipe 11; This is only added to the BHK cells)
0.02% L-glutamine (see Recipe 3 for 100x)
1x gentamicin sulfate (see Recipe 2 for 100x)
For 1x medium
Mix the following:
400 ml 2x E-MEM (see Recipe 1)
400 ml TC H2O
80 ml FBS
8 ml L-glutamine
*8 ml gentamicin sulfate
Store up to 2 weeks at 4 °C
*Note: Gentamicin is added to medium only for virus growth and not for general maintenance of any of the cell cultures.
For 2x medium (used to titer viruses by plaque assay)
Prepare 536 ml medium by mixing the following:
400 ml 2x E-MEM (incomplete, serum-free; see Recipe 1)
80 ml FBS
40 ml TPB
8 ml 100x L-glutamine
8 ml 100x gentamicin sulfate
Store up to 6 months
IMPORTANT NOTE: Complete medium with supplements are not true percent solutions. The percentage indicated represents the percentage of the original volume of medium. This also holds for any other supplements added to the medium. For example, if the initial volume of the medium is 400 ml, 10% FBS is 40 ml. Addition of other supplements, such as 5% TPB would be 20 ml TPB, bringing the total volume to 460 ml. If problems arise with the cell cultures, growth of the virus, or plaque formation, discard old solutions and make fresh solutions. - Gentamicin sulfate, 100x
Dilute 0.5 g gentamicin sulfate up to 100 ml in tissue-culture-grade water
Store up to 6 months at 4 °C
Dilution to 1x will give a final concentration of 50 μg/ml. If necessary, 100 μg/ml may be used
CAUTION: Antibiotics can become contaminated. If persistent problems are experienced with contaminated cultures, test all solutions in the absence of antibiotics. - L-glutamine, 100x
Weigh out 2.92 g L-glutamine and dissolve in a total volume of 100 ml 1x PBS-D (see Recipe 7 for 10x)
Filter sterilize and divide into 50-ml aliquots in 100-ml bottles
Store frozen up to 6 months; once thawed, discard after 2 weeks
Note: Glutamine is essential and very labile. It is thus added to already complete medium as a precaution. - HEPES (pH 7.2-7.4), 1 M
Weigh out 238.3 g HEPES and dissolve in a total volume of 1 L tissue-culture grade water
Adjust pH to 7.2-7.4 with NaOH
Autoclave for 30 min to sterilize
Store up to 1 year at room temperature - PEG Buffer
0.4 M NaCl
0.01 M Tris Buffer pH 7.0
0.001 M EDTA - Neutral red stock solution, 2%
Dissolve 2 g neutral red and adjust the volume to 100 ml with tissue-culture-grade water
Stir overnight at room temperature and filter sterilize
Store up to 1 year at room temperature
Notes:- Some stain will be lost in the filtration process and each batch may differ. This stain is quite viscous and may require more than one filter to sterilize the entire quantity.
- Alternatively, neutral red stock solution may be purchased ready made from many suppliers.
- PBS-D, 10x
2.0 g KCl
2.0 g KH2PO4
80.0 g NaCl
11.3 g Na2HPO4 or 21.6 g Na2HPO4·7H2O
Tissue-culture-grade H2O to 1 L
Autoclave for 20 min
Dilute to 1x with sterile tissue-culture-grade water
Store up to 1 year at room temperature
Note: PBS-D is PBS (phosphate buffered saline) deficient in calcium and magnesium. This can be made as a 1x solution. Also see annotations to the recipe for PBS with calcium and magnesium. It is critical that the pH of the 1x solution is ~7.4 because low pH will inactivate all Alphaviruses. - PBS with calcium and magnesium, 1x
Prepare solutions 1 and 2 (which are 10x stock solutions) and autoclave separately for 20 min
Note: PBS cannot be made as a concentrated solution, it will precipitate.
Solution 1:
1.0 g CaCl2
1.3 g CaCl2·2H2O
2.0 g KH2PO4 (dissolve separately, then add to mixture)
1.0 g MgCl2·6H2O
80.0 g NaCl
Adjust volume to 1 L with tissue-culture-grade H2O
Solution 2:
21.6 g Na2HPO4·7H2O or 11.3 g anhydrous Na2HPO4
Adjust volume to 1 L with tissue-culture-grade H2O
For 1x solution:
Add 50 ml solution 1 to 400 ml tissue-culture-grade water
Mix, then add 50 ml solution 2
Do not directly mix solutions 1 and 2 together: salts will precipitate
Store solutions at room temperature up to 1 year or until salts begin to precipitate
Check that the final pH of the 1x solution is ~7.2 to 7.4
IMPORTANT NOTE: Do not adjust the pH of the 10x solutions 1 and 2, because this will result in the wrong pH for the 1x solution. It is critical that all solutions that Sindbis virus comes in contact with are neutral pH. Acidic pH quickly inactivates the virus. - Phenol red, 0.5%
Note: Do not confuse with neutral red.
Dissolve 1 g phenol red in 200 ml tissue-culture-grade water
Filter sterilize and store up to 1 year at room temperature - SVHR diluent (used to dilute virus to be titrated by plaque assay)
Supplement 1x PBS-D (see Recipe 7 for 10x) to 3% FBS with heat-inactivated fetal bovine serum (FBS)
Store up to 2 months at 4 °C - Trypsin stock, 0.25%
0.5 g trypsin
0.2 g disodium EDTA
0.6 g phenol red
Adjust volume to 200 ml with 1x PBS-D (see Recipe 7 for 10x)
Adjust pH with 1 N NaOH until a cherry red color is achieved
Divide into 25-ml aliquots
Store frozen up to 6 months at -20 °C
Note: Prior to use, thaw in a 37 °C water bath and dilute to 100 ml with versene solution (see Recipe 13). Do not heat trypsin at 37 °C for > 20 min, as auto-digestion of the enzyme will occur. - Tryptose phosphate broth (TPB)
Weigh out 29.5 g tryptose phosphate broth (TPB; Difco)
Dissolve in a total volume of 1 L tissue culture grade water
Autoclave TPB for 20 min in two separate 500-ml volumes to sterilize
Do not leave in the autoclave for longer periods since this solution will caramelize
Cool and store up to 6 months at 4 °C - Versene solution
500 ml 1x PBS-D (see Recipe 7 for 10x)
5 ml 0.1 M EDTA (see Recipe 14)
1.5 ml 0.5% phenol red (see Recipe 9)
Adjust pH to 7.4 with NaOH
Filter sterilize (if sterile stock solutions were not used)
Store up to 6 months at room temperature - 0.1 M EDTA solution
Use the disodium salt form 3.36 g/100 ml
The pH will drop below 5.3 and solution will remain cloudy until its pH is adjusted to 7.0. Adjust the pH to 8.0 with NaOH
Autoclave to sterilize - M-PER Supplemented with 50 mM dithiothreitol (DTT)
Notes:- You will need a minimum of 20 μl of DTT-supplemented M-PER for every sample you process.
- Leftover 1 M DTT stock can be stored at -20 °C for up to 30 days (it is best make 50-100 μl aliquots to prevent any freeze/thaw cycles).
- UA buffer (8 M urea, 100 mM Tris-HCl pH 8.5) (10 ml)
Note: You will need around 500 μl of UA buffer per sample.
Add 4.8048 g urea (Thermo Scientific) to 6 ml mass spec grade water and dissolve (the solution will become cold, so it may be necessary to heat the solution gently in a water bath to get the urea to dissolve completely). Add 1 ml 1 M Tris-HCl pH 8.5 and add mass spec grade water to 10 ml - IAA solution (0.05 M iodoacetamide in UA buffer) (1 ml)
- 100 mM triethylammonium bicarbonate (TEAB) (3 ml)
Add 300 μl 1 M TEAB (Sigma) to 2.7 ml mass spec grade water and vortex
Recommended to keep solution on ice - Trypsin digestion solution (Trypsin/Lys-C prepared in 100 mM TEAB to 10 µg/ml)
Add 2,000 μl 100 mM TEAB to a single 20 μg vial of Trypsin/Lys-C (Promega) for a final concentration of 10 μg/ml, and gently pipet-mix until completely dissolved. DO NOT VORTEX. Recommended to keep solution on ice - 0.5 M NaCl (5 ml)
Note: This will be an excess of solution, but it is a more manageable to weigh out NaCl for this volume).
Weigh out 146.1 mg NaCl (Sigma) and add mass spec grade water to 5 ml
Vortex until dissolved - 10% trifluoroacetic acid
Add 1 ml of 100% trifluoroacetic acid (Thermo Scientific) to 9 ml mass spec grade water and vortex - A Buffer (3% acetonitrile/0.1% formic acid)
Add 1 ml 100% formic acid (Thermo Scientific) to 30 ml 100% acetonitrile, then add mass spec grade water to 1 L - B buffer (95% acetonitrile/0.1% formic acid)
Add 1 ml 100% formic acid to 950 ml 100% acetonitrile, then add mass spec grade water to 1 L
Acknowledgments
RH and DF were supported by the Clayton Foundation for Research, Carson City NV and the College of Agriculture and Natural Sciences, North Carolina State University. TG and GR were supported by the U.S. Army Combat Capabilities Development Command (CCDC) Chemical Biological Center through an internal basic research grant. The opinions presented here are those of the authors and are not the official policy of the U.S. Army, CCDC, or the U.S. Government. Information in this report is cleared for public release and distribution is unlimited. This protocol is adapted from the publication. Schuchman R, Kilianski A, Piper A, Vancini R, Ribeiro JMC, Sprague TR, Nasar F, Boyd G, Hernandez R, Glaros T. 2018. Comparative Characterization of the Sindbis Virus Proteome from Mammalian and Invertebrate Hosts Identifies nsP2 as a Component of the Virion and Sorting Nexin 5 as a Significant Host Factor for Alphavirus Replication. J Virol 92.
Competing interests
The Authors declare no competing interests.
References
- Brown, D. T. (1986). Replication of Alphaviruses in Mosquito Cells. In: The Togaviridae and Flaviviridae. Schlesinger, S. and Schlesinger, M. J. (Eds). Plenum Press. New York and London. 171-207.
- Chen, X. G., Jiang, X., Gu, J., Xu, M., Wu, Y., Deng, Y., Zhang, C., Bonizzoni, M., Dermauw, W., Vontas, J., Armbruster, P., Huang, X., Yang, Y., Zhang, H., He, W., Peng, H., Liu, Y., Wu, K., Chen, J., Lirakis, M., Topalis, P., Van Leeuwen, T., Hall, A. B., Jiang, X., Thorpe, C., Mueller, R. L., Sun, C., Waterhouse, R. M., Yan, G., Tu, Z. J., Fang, X. and James, A. A. (2015). Genome sequence of the Asian Tiger mosquito, Aedes albopictus, reveals insights into its biology, genetics, and evolution. Proc Natl Acad Sci U S A 112(44): E5907-5915.
- Coombs, K. and Brown, D. T. (1987). Topological organization of Sindbis virus capsid protein in isolated nucleocapsids. Virus Res 7(2): 131-149.
- Giraldo-Calderón, G. I., Emrich, S. J., MacCallum, R. M., Maslen, G., Dialynas, E., Topalis, P., Ho, N., Gesing, S., VectorBase, C., Madey, G., Collins, F. H. and Lawson, D. (2015). VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res 43 (Database issue): D707-713.
- Glaros, T. G., Blancett, C. D., Bell, T. M., Natesan, M. and Ulrich, R. G. (2015). Serum biomarkers of Burkholderia mallei infection elucidated by proteomic imaging of skin and lung abscesses. Clin Proteomics 12(1): 7.
- Hernandez, R., Brown, D. T. and Paredes, A. (2014). Structural differences observed in arboviruses of the alphavirus and flavivirus genera. Adv Virol 2014: 259382.
- Johnson, J. E. and Speir, J. A. (1997). Quasi-equivalent viruses: a paradigm for protein assemblies. J Mol Biol 269(5): 665-675.
- Kall, L., Canterbury, J. D., Weston, J., Noble, W. S. and MacCoss, M. J. (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4(11): 923-925.
- Karim, S., Singh, P. and Ribeiro, J. M. (2011). A deep insight into the sialotranscriptome of the gulf coast tick, Amblyomma maculatum. PLOS One 6: e28525.
- Mi, H., Poudel, S., Muruganujan, A., Casagrande, J. T. and Thomas, P. D. (2016). PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44(D1): D336-342.
- Ribeiro, J. M., Topalis, P. and Louis, C. (2004). AnoXcel: an Anopheles gambiae protein database. Insect Mol Biol 13(5): 449-457.
- Rumble, J. R. (2005). CRC Handbook of Chemistry and Physics. Internet Version 2005, CRC Press, Boca Raton, FL.
- Strauss, J. H. and Strauss, E. G. (1994). The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58(3): 491-562.
- Vancini, R., Wang, G., Ferreira, D., Hernandez, R. and Brown, D. T. (2013). Alphavirus genome delivery occurs directly at the plasma membrane in a time- and temperature-dependent process. Journal of Virology 87(8): 4352-4359.
- Wilkins, M. (2009). Proteomics data mining. Expert Rev Proteomics 6(6): 599-603.
- Wilkins, M. R., Pasquali, C., Appel, R. D., Ou, K., Golaz, O., Sanchez, J. C., Yan, J. X., Gooley, A. A., Hughes, G., Humphery-Smith, I., Williams, K. L. and Hochstrasser, D. F. (1996). From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (N Y) 14(1): 61-65.
- Wiśniewski, J. R., Zougman, A., Nagaraj, N. and Mann, M. (2009). Universal sample preparation method for proteome analysis. Nat Methods 6(5): 359-362.
- Wray, W., Boulikas, T., Wray, V. P. and Hancock, R. (1981). Silver staining of proteins in polyacrylamide gels. Anal Biochem 118(1): 197-203.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hernandez, R., Glaros, T., Rizzo, G. and Ferreira, D. F. (2019). Purification and Proteomic Analysis of Alphavirus Particles from Sindbis Virus Grown in Mammalian and Insect Cells. Bio-protocol 9(10): e3239. DOI: 10.21769/BioProtoc.3239.
Category
Microbiology > Microbe-host interactions > Virus
Microbiology > Microbial proteomics > Whole organism
Molecular Biology > Protein > Detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.