- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection of mRNA by Whole Mount in situ Hybridization and DNA Extraction for Genotyping of Zebrafish Embryos
Published: Vol 9, Iss 6, Mar 20, 2019 DOI: 10.21769/BioProtoc.3193 Views: 15724
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
In situ hybridization is used to visualize the spatial distribution of gene transcripts in tissues and in embryos, providing important information about disease and development. Current methods involve the use of complementary riboprobes incorporating non-radioactive labels that can be detected by immunohistochemistry and coupled to chromogenic or fluorescent visualization. Although recent fluorescent methods have allowed new capabilities such as single-molecule counting, qualitative chromogenic detection remains important for many applications because of its relative simplicity, low cost and high throughput, and ease of imaging using transmitted light microscopy. A remaining challenge is combining high contrast signals with reliable genotyping after hybridization. Dextran sulfate is commonly added to the hybridization buffer to shorten development times and improve contrast, but this reagent inhibits PCR-based genotyping. This paper describes a modified protocol for in situ hybridization in fixed whole mount zebrafish embryos using digoxigenin (DIG) labeled riboprobes that are detected with alkaline phosphatase conjugated anti-DIG antibodies and nitroblue tetrazolium (NBT)/5-bromo-4-chloro-3-indolyl-phosphate (BCIP) chromogenic substrates. To yield embryos compatible with downstream genotyping after hybridization without sacrificing contrast of the signal, this protocol omits dextran sulfate and utilizes a lower hybridization temperature.
Keywords: In situBackground
In situ hybridization is a technique that enables the detection of RNA in single cells, tissues or in whole embryos (Schulte-Merker et al., 1992; Hauptmann and Gerster, 1994; Schulte-Merker et al., 1994). The technique has been widely employed in developmental biology to characterize the spatial and temporal distributions of target mRNAs in fixed embryos (Thisse et al., 2001; Thisse and Thisse, 2004 and 2005). By allowing the comparison of mRNA distributions in wild-type, mutant and experimentally manipulated embryos, in situ hybridization continues to play a vital role in the investigation of gene function in development.
Hybridization is based on the principle of complementary base-pair binding of a specific probe oligonucleotide to a target mRNA. RNA oligonucleotides (riboprobes) are designed to pair with target mRNA molecules and are synthesized with nucleotides containing haptens, such as digoxigenin (DIG). In in situ hybridization, the riboprobes are incubated with previously fixed and permeabilized bulk tissue or intact embryos at an elevated temperature to favor the formation of hybrids with high sequence complementarity. The stable target-probe hybrids are then detected with a hapten-specific antibody. In chromogenic variations of the technique, the antibody is conjugated to alkaline phosphatase. This enzymatic activity results in the local generation of a colored precipitate upon the addition of a chromogenic substrate. NBT and BCIP are commonly used substrates yielding a purple-blue precipitate. Thus, the localization of target mRNA can be visualized with cellular resolution in the embryo or tissue by a colored signal in transmitted light microscopy.
The technique has been modified to compare the localization of two mRNAs by including different haptens on different probes (Hauptmann and Gerster, 1994) or combined with immunohistochemistry to compare the localization of mRNA and protein. Recent advances allow the fluorescent detection of multiple genes (Choi et al., 2010; Gross-Thebing et al., 2014), the sub-cellular localization of mRNAs (Jambor et al., 2015) and the determination of the number of transcripts in a sample (Wang et al., 2012; Little et al., 2013; Stapel et al., 2016; Trivedi et al., 2018). Nevertheless, qualitative chromogenic detection remains important in the field because of its relative simplicity, low cost, high throughput and ease of imaging.
The signal to noise ratio of the colored signal of an in situ hybridization experiment is a function of the specificity of riboprobe, i.e., the ability of the riboprobe to distinguish between different targets, and the stringency of the hybridization. High stringency requires the base pairing of all bases in the oligonucleotide while lower stringency allows some bases to be unpaired. The design of the riboprobe sets the specificity of hybridization. The reaction environment affects the stringency–annealing of oligonucleotides and the thermal stability of the resulting hybrid is influenced by temperature, pH, the concentration of monovalent cations and the presence of organic solvents in the environment.
Riboprobes have to be designed to be maximally complementary to the target mRNA for high specificity. Additionally, maximally complementary RNA-RNA hybrids have the highest thermal stability as base pair mismatches lower the thermal stability. The length of the riboprobe will affect the hybridization rate and also influence the thermal stability of the resulting hybrid–longer probes have a higher hybridization rate and generate thermally stable hybrids (Wetmur, 1976). Riboprobes of lengths between 300-3,200 base pairs specific to the target can be used with the protocol described here.
The temperature used for hybridization is determined from the melting temperature of the designed oligonucleotide. Hybridization is performed 15-25 °C lower than the calculated melting temperature (Wetmur and Davidson, 1968)–the higher this temperature, the greater the stringency of hybridization. However, prolonged incubation at typical RNA hybridization temperatures will degrade sample morphology. Therefore, organic solvents are added to reduce the thermal stability of the RNA duplexes, with formamide being the most common reagent (McConaughy et al., 1969). Hybridization buffers used in in situ hybridization also contain monovalent sodium cations, from sodium chloride and sodium citrate, which interact electrostatically with the RNA-RNA hybrid to stabilize it. Low salt washes are subsequently performed to reduce background by preferentially reducing the stability of non-specific hybrids.
Typical in situ protocols for the qualitative detection of mRNA in whole mount in zebrafish recommend a hybridization temperature of 70 °C (Thisse and Thisse, 2008) for high stringency. Such high stringency conditions are necessary if a riboprobe is required to discriminate between two similar target sequences. We found that for riboprobes with high specificity, by performing hybridization at lower temperatures (55-60 °C), we could achieve a more rapidly developing, higher contrast stain. We could further reduce the development time and enhance the contrast of the stain by increasing the effective concentration of riboprobe by adding dextran sulfate to the hybridization buffer (Lauter et al., 2011). Additionally, for probes that required prolonged development, the addition of polyvinyl alcohol to the NBT-BCIP staining solution reduces background (Kiyama and Emson, 1991). Making these modifications to the in situ hybridization protocol consistently accelerates the development and enhances the contrast of the stain.
A key part of an in situ experiment is the qualitative analysis of variations in mRNA distributions in mutant or otherwise experimentally manipulated embryos. However, differences in mRNA distributions may be evident before the genotype of the embryo can be ascertained by altered morphology. Further, such differences can be subtle, or variable, and may not fall into convenient Mendelian ratios complicating a straightforward assignment of wild-type and perturbed patterns. For these reasons, it is valuable to have a protocol compatible with reliable genotyping of embryos, or fragments of embryos, after hybridization and photographic documentation. We have observed that the presence of dextran sulfate in the hybridization buffer inhibits genotyping by PCR; therefore it is omitted if the embryos need to be genotyped after in situ hybridization.
The protocol described here describes three linked procedures: (1) the generation of DIG-labeled riboprobes suitable for hybridization; (2) in situ hybridization using a single riboprobe to qualitatively detect an mRNA target of interest in developing zebrafish embryos and imaging of the embryos; and (3) DNA extraction that gives a PCR-ready extract, enabling the presence or absence of indel mutations to be identified in the genomic DNA of individual, previously imaged embryos. We have successfully tested the protocol in labs in two different countries. Combined, these procedures form a protocol sufficiently detailed to enable a relatively inexperienced experimentalist to achieve high quality and reliable analysis of mRNA distribution patterns with a range of experimental perturbations.
Materials and Reagents
- Riboprobe synthesis
- 1.5 ml colorless microfuge tubes (Eppendorf, SafeLock, catalog number: 0030120086)
- PCR tubes (Sarstedt, Multiply®-μstrip Pro 8-strip, catalog number: 72.991.002)
- Restriction digest enzymes (Thermo Scientific, FastDigest) (store at -20 °C)
- 2x Phusion Hot Start II High-Fidelity PCR Master Mix (Thermo Scientific, catalog number: F-565) (store at -20 °C)
- rNTP-DIG (Roche, catalog number: 11277073910) (store at -20 °C)
- RNase Inhibitor (RNaseOUT, Invitrogen, catalog number: 100000840) (store at -20 °C)
- RNA polymerases (1,000 U-20 U/μl) (store at -20 °C)
- Sp6 (Roche, catalog number: 10810274001)
- T3 (Roche, catalog number: 11031163001)
- T7 (Roche, catalog number: 10881767001)
- Clean up kits (store at room temperature)
- Qiaquick PCR purification kit (QIAGEN, catalog number: 28104)
- Promega SV Gel Wizard PCR purification kit (Promega, catalog number: A9281)
- RNeasy Min Elute (QIAGEN, catalog number: 74204)
- Agarose (Sigma-Aldrich, catalog number: A9539) (store at room temperature)
- 1x Tris-Acetate, EDTA (TAE) (store at room temperature, see Recipes)
- Tris Base (Sigma-Aldrich, BioXtra, catalog number: T6791) (store at room temperature)
- Glacial acetic acid (Suprapur®, EMD Millipore, catalog number: 1.00066) (store at room temperature)
- EDTA, 0.5 M (UltraPure, Invitrogen, catalog number: 15575-038) (store at room temperature)
- Hybridization buffer (Hyb) (store at -20 °C, see Recipes)
- Formamide, deionized (Roche, catalog number: 1814320) (store at 4 °C)
- 50 mg/ml Heparin stock (store at 4 °C, see Recipes)
Heparin (Sigma-Aldrich, catalog number: H3400) (store at room temperature) - Torula-RNA (Sigma-Aldrich, catalog number: R6875) (store at -20 °C)
- 20x SSC (store at room temperate, see Recipes)
Sodium chloride (Sigma-Aldrich, BioXtra, catalog number: S7653)
Trisodium citrate dihydrate (Sigma-Aldrich, catalog number: C8532) (store at room temperature) - (Optional) Dextran sulfate sodium salt (Sigma-Aldrich, catalog number: D6001) (store at 4 °C)
- 10% Tween-20 (see Recipes)
Tween-20 (Sigma-Aldrich, catalog number: P9416) (store at room temperature)
- Embryo culture
- 94 mm polystyrene Petri dish (Grenier Bio-One, catalog number: 633161)
- 100x PTU stock (store at room temperature, see Recipes)
N-Phenylthiourea (PTU, Sigma-Aldrich, catalog number: P7629) (store at room temperature)
- In situ hybridization
- 24-well plates (Grenier Bio-One, Cell Star, catalog number: 662160)
- 2 ml colorless microfuge tubes (Eppendorf, SafeLock, catalog number: 0030120094)
- 3 ml plastic non-sterile, macrograduated Pasteur pipettes (HuberLab, catalog number: 15.4051.11)
- 94 mm polystyrene Petri dish (Grenier Bio-One, catalog number: 633161)
- Thin walled plastic container (Microwaveable plastic lunch box–any brand)
- Methanol (Fisher, catalog number: M/4000/15) (store at room temperature)
- Phosphate buffered saline, 0.1% Tween (PBST) (store at room temperature, see Recipes)
- Sodium chloride (Sigma-Aldrich, BioXtra, catalog number: S7653) (store at room temperature)
- Potassium chloride (Sigma-Aldrich, BioXtra, catalog number: P9333) (store at room temperature)
- Sodium phosphate dibasic (Sigma-Aldrich, BioXtra, catalog number: S7907) (store at room temperature)
- Potassium phosphate monobasic (Sigma-Aldrich, catalog number: P9791) (store at room temperature)
- 10% Tween-20 (see Recipes)
- 4% Paraformaldehyde (PFA) in PBST (store at 4 °C) (see Recipes)
- PFA powder (Sigma-Aldrich, catalog number: P6148) or 16% PFA (Alfa Aesar, catalog number: 43368.9M) (store at 4 °C)
- PBST (see Recipes)
- Proteinase K stock (store at -20 °C, see Recipes)
Proteinase K (Merck, catalog number: 1.24568) (store at 4 °C) - Hybridization buffer (Hyb) (store at -20 °C, see Recipes)
- Post-hybridization washes (see Recipes)
- Wash 1
- Wash 2
- Wash 3
- Detection
- AP-conjugated anti-DIG antibody (Anti-Digoxigenin-AP, Fab fragments from sheep) (Roche, catalog number: 11093274910) (store at 4 °C)
- Maleic acid buffer, 0.1% Tween (MABT) (store at room temperature, see Recipes)
- Maleic acid (Sigma-Aldrich, catalog number: M0375) (store at room temperature)
- Sodium chloride (Sigma-Aldrich, BioXtra, catalog number: S7653) (store at room temperature)
- Sodium hydroxide (Fisher Chemical, catalog number: S/4920/60) (store at room temperature)
- 10% Tween-20 (see Recipes)
- 10% Blocking reagent in maleic acid buffer
- Blocking Reagent (Roche, catalog number: 11096176001) (store at room temperature)
- Maleic acid buffer (see Recipes)
- 2% Blocking reagent in MABT (2% Roche block) (see Recipes)
- Staining buffer (see Recipes)
- Tris-HCl (Sigma-Aldrich, BioXtra, catalog number: T6666) (store at room temperature)
- Magnesium chloride hexahydrate (Sigma-Aldrich, BioXtra, catalog number: M2670) (store at room temperature)
- Sodium chloride (Sigma-Aldrich, BioXtra, catalog number: S7653) (store at room temperature)
- Staining solution (see Recipes)
- NBT (Roche catalog number: 11383213001) (aliquot and store at -20 °C)
- BCIP (Roche, catalog number: 11282221001) (aliquot and store at -20 °C)
- Staining buffer (see Recipes)
- (Optional) Polyvinyl alcohol (PVA, Sigma-Aldrich, catalog number: P1763) (store at room temperature)
- Mounting
- 60 mm Petri dish
- A human eyelash glued to a tooth pick/syringe needle or a fine pipette tip (GELoader Tips 0.5-20 μl, 62 mm, Eppendorf, catalog number: 0030001222)
- (Optional) Mold for embryos for photographic documentation in whole mount [the mold negative is made in our laboratory (Herrgen et al., 2009)]
- (Optional) Dissection hook (Picker et al., 2009) made with Tungsten wire (Hamilton, catalog number: 18306)
- 10 μl pipette tip
- Glass slides (Thermo Scientific, Superfrost, catalog number: 12372098)
- Glass coverslip (VWR, Menzel Gläser, #1.5 rectangular 22 x 40 mm, catalog number: 631-1370)
- Silicone grease or petroleum jelly (such as Vaseline)
- Transparent nail varnish (any brand)
- 87% glycerol in distilled water (store at room temperature, see Recipes)
Glycerol (Fisher, catalog number: G/0650/08) (store at room temperature) - Genomic DNA extraction (store at room temperature)
- 50x base solution (see Recipes)
- Potassium hydroxide (Fisher Chemical, catalog number: P/5640/60)
- EDTA, 0.5 M (UltraPure, Invitrogen, catalog number: 15575-038)
- 1x base solution (see Recipes)
- 50x neutralization solution (see Recipes)
Tris-HCl (Sigma-Aldrich, catalog number: T6666) - 1x neutralization solution (see Recipes)
- 50x base solution (see Recipes)
Equipment
- Stock solution preparation
- Magnetic stirrer (Heidolph Instruments, model: MR Hei-Standard, catalog number: 505-20000-00)
- pH meter (Mettler Toledo, model: Seven compact and InLab Expert Pro-ISM)
- Riboprobe synthesis
- PCR machine (Eppendorf, model: Mastercycler pro S)
- Thermoblock (Eppendorf, model: ThermoMixer C and SmartBlock 2.0 ml)
- Gel electrophoresis (Bio-Rad, model: PowerPac Basic Power Supply; Wide Mini-Sub Cell GT Cell)
- NanoDrop spectrophotometer (Thermo Fisher, model: NanoDropTM 2000, catalog number: ND 2000)
- In situ hybridization
- Forceps (Dumont, model: 5-Inox-E)
- Stereomicroscope (Olympus, model: SZ61)
- Shaker (Heidolph, model: Duomax1030, catalog number: 543-32205-00)
- (Optional) Vacuum set-up (Integra, Vacusafe aspiration system)
- Water bath (GFL, model: 1003)
- (Optional) Well plate stand (Diversified Biotech, catalog number: WPST-1000)
- Documentation
- RGB Camera (Olympus, model: DP22)
- Ring light (Photonic Optics, model: LED 40 ring light)
- Genomic DNA extraction
- Micro knife (Fine Science Tools, catalog number: 10315-12)
Software
- Fiji (Schindelin et al., 2012, https://fiji.sc/)
- Snapgene (GSL Biotech LLC)
Procedure
- Riboprobe synthesis
Note: DNA templates for riboprobes can be either cDNA cloned into a plasmid backbone downstream of an RNA polymerase promoter or PCR product amplified from cDNA or genomic DNA. See Note 1.- Template preparation
Preparing a template from a plasmid- Linearize the plasmid by digestion with the appropriate restriction enzyme at 37 °C. Choose an enzyme such that the template is linearized immediately downstream of the cDNA insert.
Set the reaction up as:
Make up to 100 μl with distilled water/nuclease-free water.Plasmid template 5 μg Restriction enzyme 5 μl 10x restriction buffer 10 μl - Purify the linearized plasmid with a commercial clean up kit. We recommend the Qiaquick PCR purification kit. Purify the plasmid as per the manufacturer’s instructions and elute the plasmid in 30 μl of elution buffer.
- Dilute 1 μl of the eluent in 4 μl of distilled water. Use 1 μl to assess concentration on a spectrophotometer and 4 μl to check the linearization with agarose gel electrophoresis. A single band corresponding to the linearized plasmid should be visible on the gel. Proceed with riboprobe synthesis if a single band corresponding to the fully linearized plasmid is visualized.
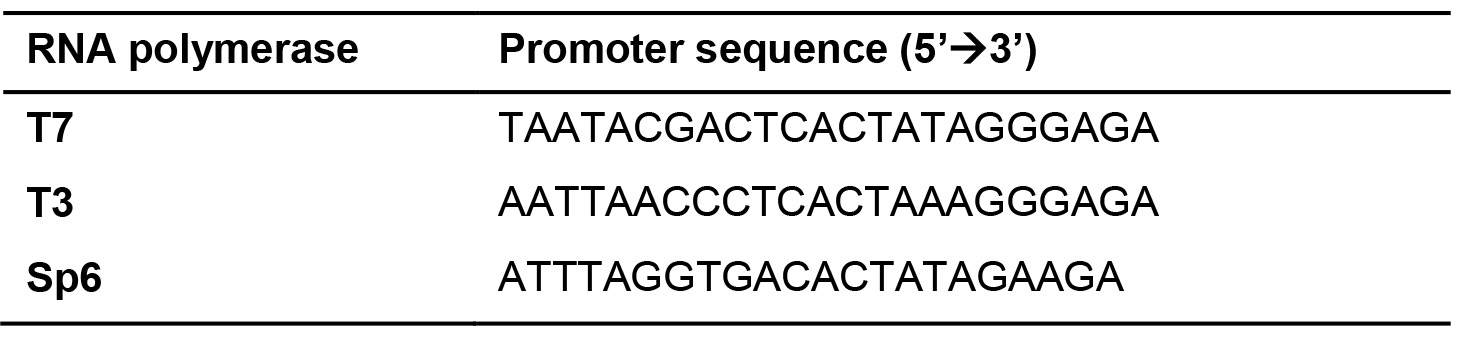
- Design primers to amplify the desired target from cDNA or genomic DNA. To make a riboprobe that is complementary to the target mRNA, add the RNA polymerase (T7/T3/Sp6) promoter sequence to the reverse primer as: 5’-RNA polymerase promoter sequence + template specific sequence-3’.
- Carry out the PCR with the optimal cycling conditions. We recommend using a high fidelity DNA polymerase like Phusion.
- Clean up the PCR product. We recommend the Promega SV Gel Wizard PCR purification kit. Elute in 30 μl of the kit elution buffer. Dilute 1 μl of the eluent in 4 μl of distilled water. Use 1 μl of this dilution to assess concentration on a NanoDrop and 4 μl to assess product quality by gel electrophoresis. Proceed with riboprobe synthesis if a single band of the appropriate length is obtained. It is not necessary to sequence the PCR product to confirm sequence accuracy of the template (see Note 6).
- Linearize the plasmid by digestion with the appropriate restriction enzyme at 37 °C. Choose an enzyme such that the template is linearized immediately downstream of the cDNA insert.
- In vitro transcription
- Thaw the reaction components (except the enzymes) and place on ice.
- Set up each 20 μl transcription reaction as:
Adjust the volume to 20 μl with RNase-free water, if needed.DNA Template 1-1.5 μg of linearized plasmid
or 100-500 ng of PCR product10x Transcription buffer 2 μl rNTP-DIG 2 μl RNase Inhibitor 0.5 μl RNA polymerase 2 μl - Transcribe for 2-4 h at 37 °C. The yield of the reaction is equivalent when the transcription time is between 2 h and 4 h.
- Purify the riboprobe from the reaction with a commercial kit like QIAGEN RNeasy Min Elute. Follow the manufacturer’s instructions and elute the riboprobe in 30 μl of RNase-free water. Dilute 1 μl of the probe in 4 μl of water. Use 1 μl to assess the concentration on a spectrophotometer and 4 μl to assess the quality of the transcription by agarose gel electrophoresis. The typical A280/A260 ratio of the synthesized probe after clean up is between 1.85 and 2.00. To resolve the RNA riboprobe on a 1% agarose gel in 1x TAE, run it at 140 V for 10-15 min. The riboprobe should resolve as a distinct band. Occasionally, a longer band is also obtained–this is typically an electrophoresis artifact arising due to RNA secondary structure, if a high-quality PCR product or completely linearized plasmid template has been used.
- Store the remainder of the synthesized riboprobe at -20 °C. Dilute the riboprobe in 5 ml of Hyb to perform in situ hybridization. See Note 2.
- Test the probe by performing in situ hybridization and assessing the pattern of hybridization obtained.
- Template preparation
- In situ hybridization
Notes:- This protocol has been optimized for embryos up to 48 h post fertilization (hpf) and is carried out in 24-well plates. Use 800 μl for washes and 500 μl for incubations. When exchanging media, the embryos being stained should not be allowed to dry out and should always remain covered with liquid. To ensure that they do not get stuck to the bottom or on to the sides of the wells, pipette gently and check after each solution change that embryos are covered by liquid.
- All incubation steps, unless specified, are performed at room temperature on a shaker.
- Fixation and dehydration of embryos (see Note 3).
Fixing embryos younger than 24 hpf
- Transfer embryos into 2 ml microfuge tubes (for long-term storage) or into wells of a 24-well plate (for immediate use for in situ) with a 3 ml plastic Pasteur pipette. Up to 100 embryos can be stored in a single tube and up to 30 embryos can be processed per well in a 24-well plate.
- Remove as much embryo medium as possible. If the embryos are in their chorions, all the liquid can be removed. Add 4% PFA in PBST (fix)–1 ml per tube, 800 μl per well. Incubate on a shaker for 5 min at room temperature. This step serves to remove any excess embryo medium.
- Exchange the fix with fresh fix–up to 2 ml per tube, 800 μl-1 ml per well. Incubate at room temperature for 1 to 4 h or overnight at 4 °C, with shaking.
- Remove the fix and add PBST–up to 2 ml per tube, 800 μl-1 ml per well.
- Transfer embryos into PBST in a 35 mm Petri dish with a 3 ml plastic Pasteur pipette and use forceps to dechorionate the embryos under a stereo microscope (Video 1). Transfer embryos back into the well plate or into a fresh tube/vial. The pipette should be rinsed by pipetting up and down in PBST, and the PBST should be exchanged between batches of embryos to prevent cross-contamination.
Fixing embryos older than 24 hpf
- From 24 hpf onwards, add PTU to the embryo medium to a final concentration of 0.003% to inhibit pigment development in the embryos.
- Dechorionate embryos in embryo medium in a 35 mm Petri dish under a stereomicroscope (Video 2) and transfer them with a 3 ml plastic Pasteur pipette to the well plate or microfuge tube vial–30 for a well, 100 for a tube. The embryo medium should be exchanged between batches of embryos to prevent cross-contamination.
- Remove as much embryo medium as possible. Add 4% PFA in PBST (fix)–1 ml per tube, 800 μl per well. Incubate on a shaker for 5 min at room temperature. This step serves to remove any excess embryo medium.
- Exchange with fresh fix–up to 2 ml per tube or vial, 800 μl to 1 ml per well in the well plate. Incubate with shaking at room temperature for 1 to 4 h or overnight at 4 °C.
- Briefly rinse the embryos by removing fix and adding PBST–up to 2 ml per tube, 800 μl-1 ml per well.
- Exchange the PBST and wash the embryos in PBST 3 times, for a duration of 5 min each time, on a shaker at room temperature.
Dehydration and permeabilization of embryos- Dehydrate the embryos in a graded methanol series: add 800 μl of 30% methanol in PBST, then replace with 800 μl of 50% methanol in PBST and then with 800 μl of 70% methanol in PBST. Incubate on a shaker between each replacement for 5 min.
- Incubate the embryos in 800 μl of absolute methanol for 5 min on a shaker.
- Replace the methanol and incubate embryos at -20 °C overnight to permeabilize the embryos. Use 1.1 ml of methanol per well or 2 ml per microfuge tube.
The protocol can be stopped here if necessary as embryos can be stored at -20 °C in methanol indefinitely (see Note 3). Video 1. Dechorionation of fixed somitogenesis stage embryos Video 2. Dechorionation of live 36 hours post fertilization (hpf) embryos
Video 1. Dechorionation of fixed somitogenesis stage embryos Video 2. Dechorionation of live 36 hours post fertilization (hpf) embryos
- Enzymatic embryo permeabilization
- Rehydrate embryos in a graded methanol series: add 800 μl of 70% methanol in PBST, then replace with 800 μl of 50% methanol in PBST and then with 800 μl of 30% methanol in PBST. Incubate on a shaker for 5 min at each step.
- Perform two 5-min washes with PBST.
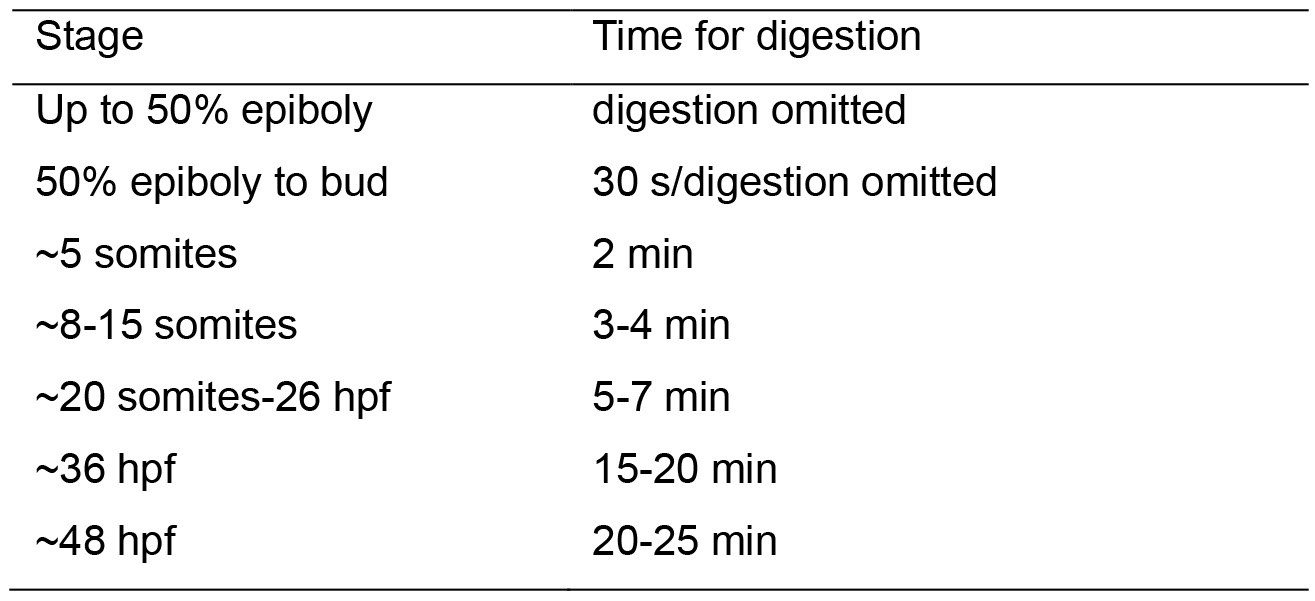
- Digest embryos in Proteinase K (PKA). Dilute the PKA stock 1:200 for embryos younger than 24 hpf and 1:100 for embryos older than 24 hpf in PBST. Incubate in 500 μl without shaking at room temperature. Digestion time is embryonic stage dependent; the timings listed below can be used as a guideline. Stages indicated here are as per Kimmel et al. (1995).
- Stop the reaction by making a rapid exchange with PBST followed by a 20-min re-fixation with 500 μl of fix at room temperature on a shaker.
- Remove the fix; perform one rapid wash with PBST followed by two 5-min PBST washes.
- Hybridization
Note: The temperature used for hybridization is probe-dependent, but a good starting temperature is 60 °C (see Note 4). Perform hybridization steps in a water bath set to the hybridization temperature. Use a thin-walled plastic container lined with moistened tissue to incubate the well plate in the water bath. The container and hybridization solutions need to be pre-warmed to the hybridization temperature in the water bath before use.
- Pre-hybridize in 500 μl of Hyb at the hybridization temperature for a minimum of 30 min. Embryos can be pre-hybridized for up to 3 h at the hybridization temperature.
- Remove the Hyb and replace it with 500 μl of labeled riboprobe diluted in Hyb. Hybridize overnight at the hybridization temperature. See Note 5.
- Post-hybridization washes
Note: These washes are also performed in a water bath at the hybridization temperature. Prepare the wash solutions fresh and warm them in the water bath before use. Timings indicated in this step are critical–do not wash for shorter or longer than the time recommended.- Remove and recycle probe. Store it at -20 °C. See Note 2.
- Wash embryos 2 times, for 30 min each time in Wash 1.
- Wash embryos for 15 min in Wash 2.
- Wash embryos 2 times, for 30 min each time in Wash 3.
- Detection
- Make a quick rinse with MABT to remove any remaining Wash 3 from the embryos.
- Add 500 μl of 2% blocking reagent and incubate without shaking in the dark at room temperature for at least 30 min.
- Dilute AP-conjugated anti-DIG antibody 1:2,000 in 2% blocking reagent. Remove the blocking reagent and add 500 μl of the diluted antibody in blocking reagent to the embryos. Incubate for 2 h, without shaking, at room temperature in the dark.
- Remove antibody solution and make 2 quick washes with MABT.
- Wash in 10-20 min intervals over 1 to 2 h at room temperature; at least 6 exchanges of MABT should be made. Alternatively, embryos can be washed at 4 °C overnight in MABT, followed by three 5-min washes the next day.
- Equilibrate embryos in freshly prepared staining buffer twice for 5 min each.
- Add 500 μl of the staining solution to the embryos. Stain in the dark at room temperature without shaking. See Note 5.
- Control the staining reaction by eye under a stereomicroscope. Place the well plate on a white background and illuminate the plate from above. Monitor the development of the embryos in the first 5 min after the staining solution has been added and then in 10-15 min intervals till the stain saturates or the desired contrast is achieved.
- Stop the staining reaction by performing three 5-min washes in PBST.
- Incubate embryos in methanol at room temperature till the yolk is cleared and appears white, or overnight at 4 °C or -20 °C.
Note: See Figures 2, 3B and 4B for stains we have made with this protocol. See Note 6 for notes on troubleshooting.
- Mounting and photography
- Remove the methanol from the embryos by making two 5-min washes in PBST.
- Re-fix in 4% PFA in PBST for at least 15 min at room temperature.
- Wash embryos twice each for 5 min in PBST.
- Add 87% glycerol to the embryos and leave to equilibrate at 4 °C. When the embryos sink to the bottom of the well, they are completely equilibrated. This takes ~12 h. Embryos can be subsequently stored in 87% glycerol either at 4 °C or -20 °C.
- Photograph the embryos:
- Photographing whole mount preparations:
Photograph the embryos in 87% glycerol in a 35 mm Petri dish or an imaging mold (see Note 7) with an RGB camera coupled to a stereomicroscope. Illuminate the embryos from above using a ring light. Place a white sheet of paper below the dish to provide a white background for the image. An eyelash, tungsten wire or fine pipette tip can be used to manipulate the embryos. - Photographing flat mount preparations:
See also the JoVE video protocol for de-yolking and flat mounting (Cheng et al., 2014).1)Transfer the embryos to be flat mounted onto a glass slide with a drop of 87% glycerol. De-yolk the embryos with an eyelash or sharpened tungsten wire (see Note 8) in the drop. Move the embryo to the edge of the drop so that a minimal volume of glycerol covers it. Remove the remaining yolk cells with the eyelash. Periodically rinse the loosened yolk cells off the embryo in the glycerol drop. Move the embryo to a fresh drop of glycerol after de-yolking.2)Place the embryonic axis, dorsal side up, on a clean glass slide with the wire or the eyelash. Place approximately 15-20 μl total volume of 87% glycerol in small drops next to the embryos. Up to 10 de-yolked embryos can be flat mounted on a single slide.3)Make 4 small grease pillars with a 10 μl pipette tip to hold the corners of a glass coverslip in place. Carefully lower the coverslip onto the slide (Figure 1).4)Seal the 4 edges of the coverslip with transparent nail varnish.5)Photograph the embryos with transmitted light on a stereoscope with an RGB camera.
Figure 1. Preparing a flat mount. A. Placing drops of glycerol next to de-yolked embryos (circled) and gently lowering a coverslip on the slide that will be held in place by grease pillars at the corners. B. The finished slide with flat mounted embryos (circled) and coverslip edges sealed with transparent nail varnish.
- Photographing whole mount preparations:
- Genotyping
Note: Genomic DNA can be extracted from intact embryos or from tissue fragments after in situ hybridization. Fragments of tissue are taken before de-yolking and flat mounting the embryos. Perform the following steps in PCR tubes–1 embryo per tube.- Select the embryos to be genotyped and rinse them together in a small Petri dish or singly in PCR tubes with PBST to remove the glycerol. A few rapid exchanges followed by three 5-min washes on a shaker at room temperature are typically sufficient.
- Transfer the whole embryo with forceps into a PCR tube. If DNA is being extracted from tissue fragments, transfer the embryo onto a glass slide into a 15-20 μl drop of PBST. Cut one-third to one-half of the embryo that is not stained with a micro knife. Transfer the piece to a PCR tube. Ensure that no excess liquid is transferred. Add 87% glycerol to the tissue on the glass slide to continue with de-yolking and flat mounting.
- Add 15 μl of 1x base solution to the tube and incubate at 95 °C for 30 min.
- Bring to room temperature and add 15 μl of 1x neutralization solution.
Note: This solution is PCR ready (see Note 9).
Data analysis
- Whole mount mRNA in situ hybridization coupled to chromogenic detection is a qualitative assay for gene expression in the embryo. In the whole mount format, the distribution of the mRNA of interest in specific tissue(s) at a developmental stage of interest can be assessed. Performing mRNA in situ hybridization for a target in embryos fixed at different developmental stages can help the investigator characterize temporal changes in the mRNA expression of the gene of interest over the course of development.
In situ hybridization can also be utilized to assess the effect of the loss or knock down of gene function on the expression of genes of interest. For example, in situ hybridization has been used in our laboratory to assay the wave patterns of cyclic genes arising from their transcriptional oscillations in zebrafish embryos during somitogenesis and to demonstrate that loss of function mutant cyclic gene alleles lead to the loss of these wave patterns (Figure 1, Lleras Forero et al., 2018). Working from a set of microscope images of embryos, in situ hybridization can also be used as a semi-quantitative endpoint assay to measure the spatio-temporal effects of an experimental perturbation, for example to investigate the upregulation or downregulation of Wnt signaling (Figures 3, 5 and 6, Bajard et al., 2014). mRNA in situ hybridization has also been used to perform a comparison between the spatial distribution of endogenous cyclic gene expression and a transgenic reporter (Figure S1, Soroldoni et al., 2014). However, care must be taken in interpreting results that depend on the intensity of the chromogenic signal, either qualitatively, or after quantifying the intensity of the signal with a tool such as plot profile in Fiji (https://fiji.sc/), as the intensity of the stain is initially development time-dependent, but the enzymatic reaction can later saturate. - Designing a genotyping strategy from the extracted genomic DNA will depend entirely on the nature of the specific alleles in the experiment. For some alleles, base transitions will lead to the gain or loss of a site for restriction digestion, allowing for a restriction digestion-based analysis of the PCR amplicon obtained from the genomic DNA using primers specific for the gene of interest. When such a strategy is not possible, the PCR amplicon can be sequenced. Analysis of genotyping sequence data generated can be done with open source or commercially available software (Snapgene, GSL Biotech LLC). For example, we detected base-pair transitions causative for a TALEN induced her1 mutation in a her7hu2124 background initially by sequencing (Figure S1, Lleras Forero et al., 2018). The mutation led to the loss of a restriction site and embryos were subsequently genotyped by the resistance of the her1 mutant allele to restriction digestion.
Notes
- Riboprobe design
In general, using long probes provides a good stain. Long probes yield hybrids with high thermal stability and provide greater signal amplification. Therefore, a good general approach is to design a riboprobe that is complementary to the entire coding sequence of the mRNA. However, such an approach might not be suitable in all cases. For example, to distinguish between mRNAs of paralogs, designing a shorter riboprobe with high complementarity to the paralog of interest is the better approach. In our hands, probes of approximately 300-3,200 bp lengths have reliably yielded a good stain with this protocol (Figure 2).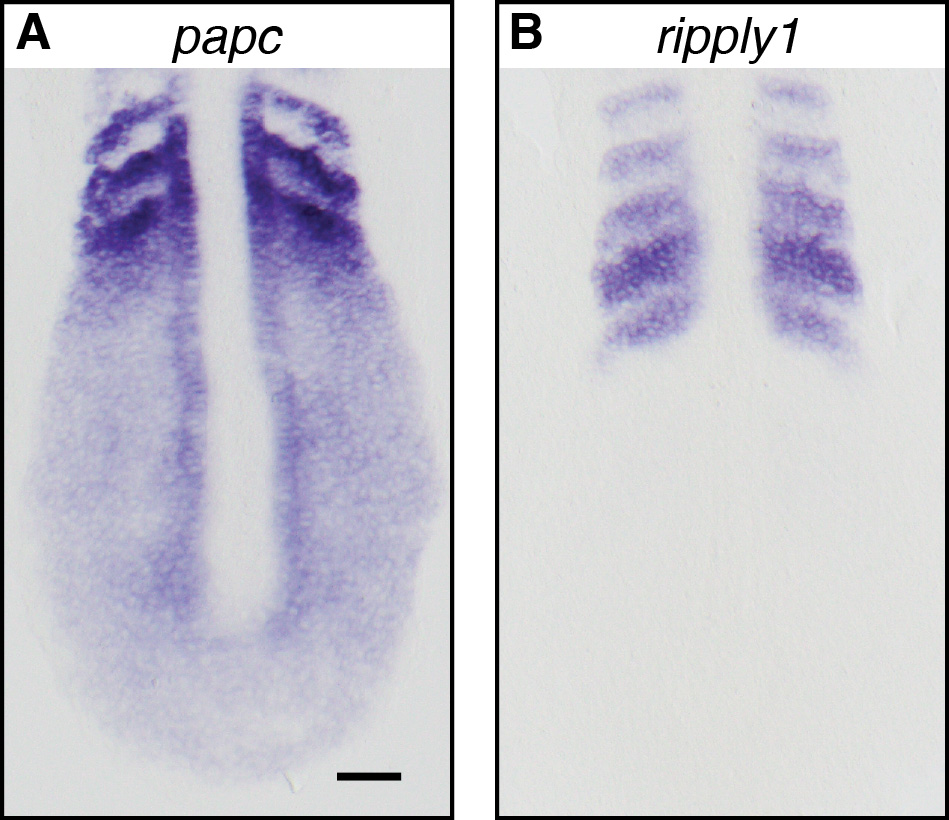
Figure 2. Representative results of the in situ hybridization protocol using riboprobes of lengths ~300 and 3,200 base pairs. A. Stain for paraxial protocadherin (papc/pcdh8) using a riboprobe of 3,137 base pair length, antisense to the coding region of papc mRNA (Yamamoto et al., 1998). B. Stain for ripply1 using a 325 base pair riboprobe, antisense to the coding region of ripply1 mRNA (Lleras et al., 2018). Hybridization has been performed at 60 °C on 10-somite stage wild-type embryos. Flat mounted preparations are shown here oriented with the posterior towards the bottom of the panels. Scale bar is 50 μm and applies to both panels. - Probe concentration
The final working concentration of the probe should be optimized experimentally. In general, concentrations of 0.3-1 ng/μl produce a good in situ stain; however, working concentrations should be titrated every time a new probe is synthesized. The probe can be recycled and reused many times. Note down the initial volume, as the probe will dilute over time and fail to produce a high contrast stain. - Embryo fixation and storage
Embryos can be fixed and dehydrated ahead of time. The quality of the fix will affect the signal to noise ratio of the stain. Freshly prepared fix gives the best results, but fix up to a week old can be used. Embryos should not be incubated in fix for more than 4 h at room temperature or 1 week at 4 °C. Fixed and dehydrated embryos can be stored in methanol at -20 °C indefinitely. - Hybridization temperature
The hybridization temperature is probe-dependent and has to be experimentally determined for each probe. The choice of temperature also sets the stringency of the protocol–stringency can be increased by increasing the hybridization temperature and decreased by lowering it. When using probes that need to discriminate between targets with high sequence similarity, the stringency of the protocol needs to be raised to favor the formation of the desired RNA hybrids as a 1% base pair mismatch decreases the thermal stability of RNA-RNA duplexes by roughly 1 °C (Wetmur et al., 1976).
In our hands, a good starting temperature has been 60 °C for probes designed to detect the coding sequence of the target mRNA (Figures 2 and 3). For short probes and intronic probes, 55 °C has been used as the starting temperature. The hybridization temperature has then been optimized by changing the temperature in 2 °C steps.
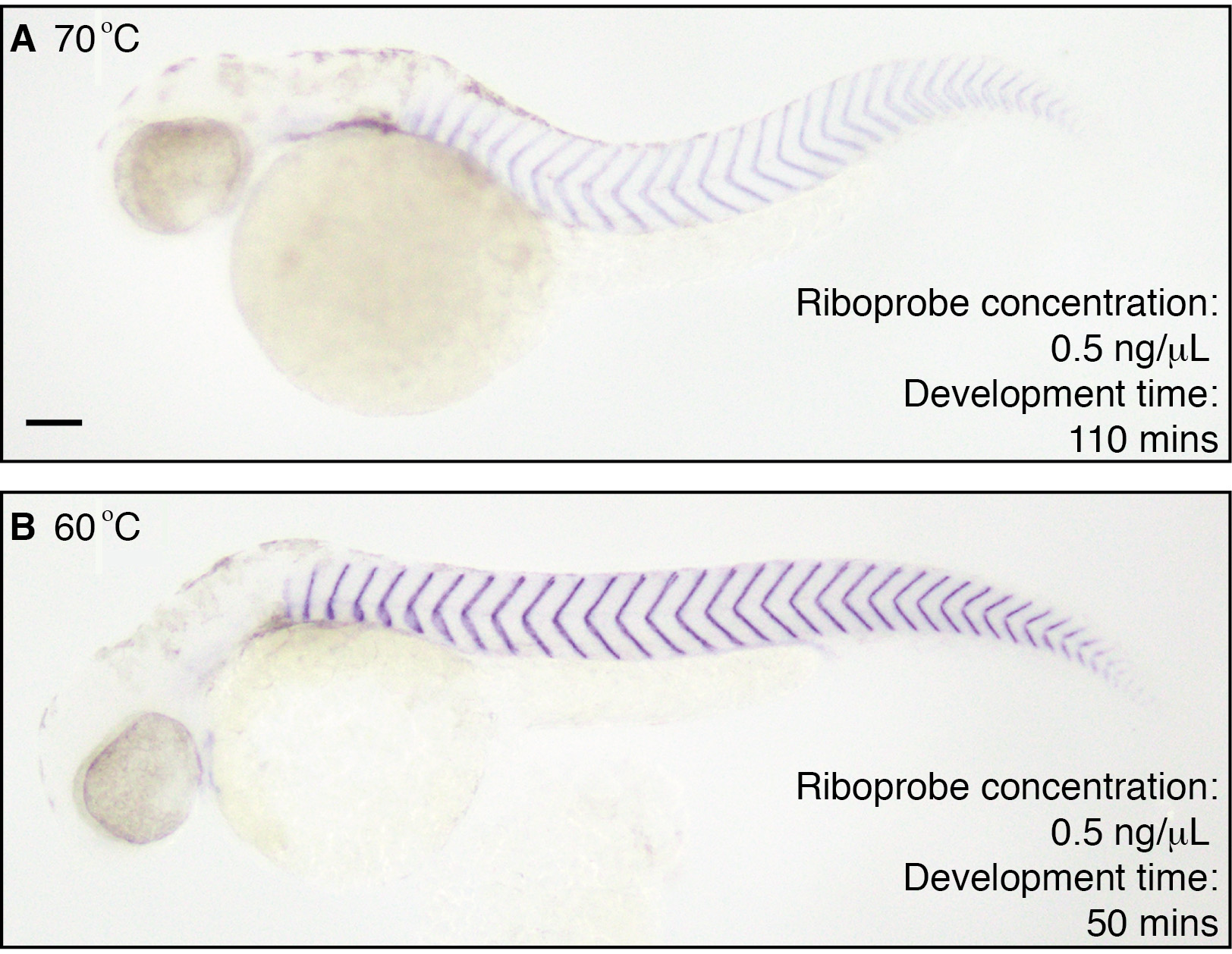
Figure 3. Hybridization at 60 °C is sufficiently stringent to achieve a high contrast final stain with a specific probe. In situ hybridization to detect xirp2a (Deniziak et al., 2007) performed at 70 °C and 60 °C in 36 hpf wild-type embryos. A. Stain after 110 min of development when probe is hybridized at 70 °C. B. Stain after 50 min of development when hybridized at 60 °C. Riboprobe concentration used is 0.5 ng/μl in hybridization buffer. Scale bar is 100 μm and applies to both panels. - Improving contrast of the stain
Adding dextran sulfate to the hybridization buffer will yield high contrast stains with a relatively short development time, compared to stains produced without dextran sulfate (Figure 4). If long development times are required to detect the target mRNA, adding 10% PVA to the staining solution will decrease the rate at which background staining will develop.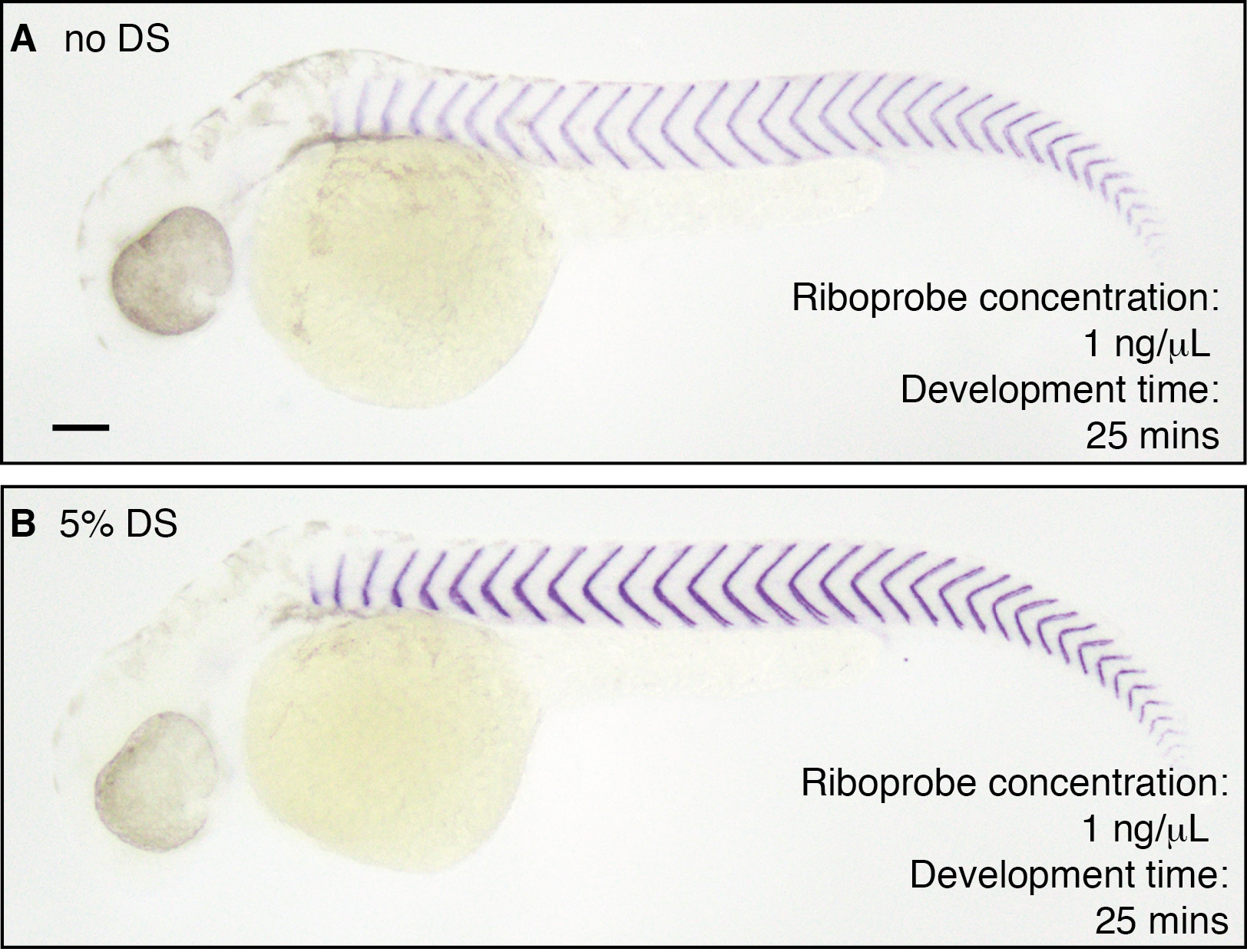
Figure 4. Addition of dextran sulfate to the hybridization buffer enhances the contrast of the final stain. In situ hybridization to detect xirp2a performed at 60 °C in 36 hpf wild-type embryos. A. Stain after 25 min of development when probe is hybridized in hybridization buffer B. Stain after 25 mins of development when probe is hybridized in hybridization buffer with 5% dextran sulfate (DS). Riboprobe concentration used is 1 ng/μl. Scale bar is 100 μm and applies to both panels. - Troubleshooting
No signal from an antisense riboprobe
In vitro transcription has not been successful: The integrity of the plasmid could have been compromised. Re-prep the plasmid or generate a template by PCR.
Sub-optimal signal to noise ratio
- Poor quality probe: Control for probe quality by performing agarose gel electrophoresis of templates and synthesized riboprobes. They should resolve as distinct bands. Templates can be sequenced to assess sequence accuracy.
- Probe concentration is too high: Diluting the probe can reduce the background.
- Background due to non-specific hybridization: Increase the stringency of the protocol by raising the hybridization temperature.
- Background due to prolonged development: Reduce the hybridization temperature to lower the stringency of the protocol, add 10% PVA to the staining solution.
Poor signal intensity- Probe is too dilute: Synthesize new probe, increase the concentration of the probe.
- Probe design: Increase the length of the probe for better signal amplification.
- Hybridization is too stringent: Decrease the hybridization temperature.
- Add 5% dextran sulfate to the hybridization buffer.
- Mold for photography of whole mount preparations
An imaging dish for photographing whole mount specimens can be made by casting a mold for the embryos in 1-1.5% agarose in distilled water using a polydimethylsiloxane (PDMS) negative. This negative is cast from a positive made from Perspex plastic machined with the conical depressions of the desired diameter and depth (Figure 4B, the positive and negative for the imaging mold are made by our laboratory and are described in Herrgen et al., 2009). Add 87% glycerol to the dish. Transfer the stained embryos into the mold and orient them using the tip of a tungsten wire, an eyelash or a fine pipette tip such that their yolks fit in the conical depressions under a stereomicroscope (Figure 5).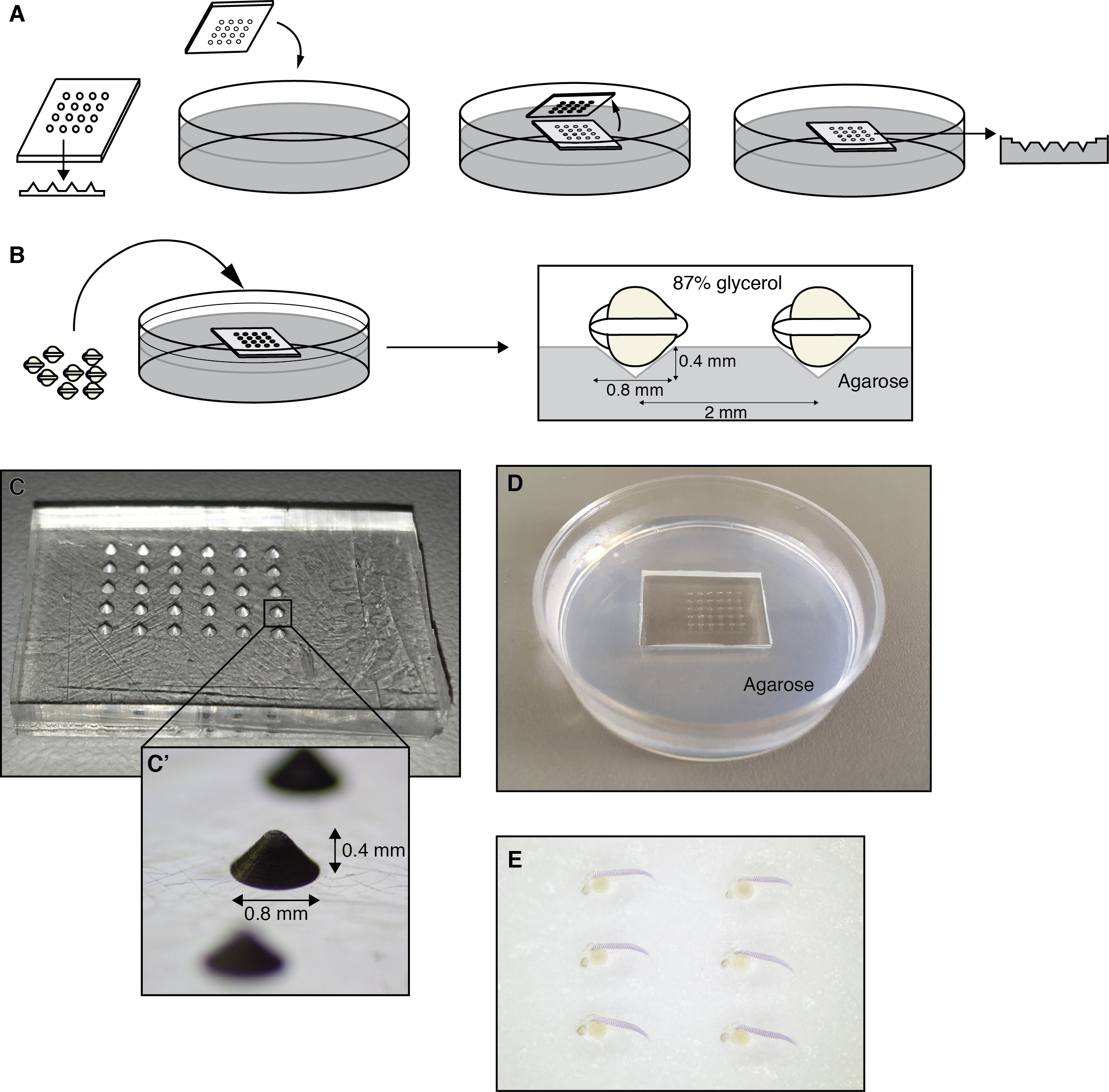
Figure 5. Preparing to document the whole mount in situ hybridization preparations. A PDMS negative with an array of raised cones of 0.8 mm diameter and 0.4 mm height is used to cast the agarose mold with conical depressions. The yolk of the embryos fits into the depressions and embryos can be oriented laterally such that they lie flat on the surface of the agarose. A. Casting a mold to orient embryos in 1%-1.5% agarose. B. Embryos are oriented in the conical cavities of the mold in 87% glycerol. C-C’. Polydimethylsiloxane (PDMS) negative used to cast the mold. D. Agarose mold for the embryos. E. Embryos oriented for photography. - De-yolking the embryo
- We recommend referring to the video protocol for de-yolking and flat mounting (Cheng et al., 2014).
- A sharpened tungsten wire or “hook” can be used instead of an eyelash for de-yolking embryos. To sharpen and shape the wire, fire-polish the end of the tungsten wire by holding the last 0.5 to 1 cm length of the wire in a Bunsen burner flame until it glows orange for 30-60 s. With a pair of forceps shape the flamed wire into a hook in 87% glycerol. Detailed instructions to make the tungsten hook and holder are provided in Picker et al., 2009.
- Using the embryo extract for PCR
A dilution series ranging from no dilution up to 1:5 in distilled water should be trialed to find the optimal volume of genomic DNA extract that is necessary for PCR for a given embryonic stage. Extracts from 8 to 10-somite stage whole embryos and embryo fragments typically do not need any further dilution and 1-2 μl can be used per 10 μl PCR reaction. Extracts from tissue of 24-36 hpf embryos typically need to be diluted 1:4.
Recipes
Note: This protocol does not require DEPC treated water or solutions or specific RNase free conditions. Distilled or ultrapure water, autoclaved glassware and single use polypropylene consumables such as microfuge tubes, centrifuge tubes, plastic Pasteur pipettes micropipette tips are sufficient.
- 1x TAE
40 mM Tris-acetate
20 mM Acetate
1 mM EDTA
pH 8.3
Dilute 10x TAE 1:10 in ultrapure water
Store at room temperature
To make 1 L of 10x TAE (0.4 M Tris-acetate, 0.2 M Acetate and 0.1 M EDTA):
48.5 g Tris base
11.4 ml glacial acetic acid
20 ml 0.5 M EDTA, pH 8.0
Make up to 1 L with ultrapure water
Autoclave - 100x PTU stock (0.3% [weight by volume] in distilled water) (100 ml)
Dissolve 0.3 g N-Phenylthiourea (PTU) in 100 ml of distilled water with heating and stirring (store in the dark at room temperature) - 10% Tween-20 (dilute Tween-20 1:10 in distilled water) (100 ml)
10 ml Tween-20
Make up to 100 ml with distilled water
Stir till a homogeneous solution is achieved
Store at room temperature - PBST
137 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4 in distilled water, pH 7.4
0.1% Tween-20
To make 1 L PBST:
8 g NaCl
0.2 g KCl
1.44 g Na2HPO4
0.24 g KH2PO4
Make up to 1 L with distilled water after adjusting pH and autoclave
Add 10 ml of 10% Tween-20
Store at room temperature - 4% PFA in PBST (4% weight by volume in PBST) (100 ml)
- Pre-warm 100 ml PBS to 68 °C in a water bath
Add 4 g PFA
Incubate at 68 °C with periodic agitation for 50 – 60 mins
Add 1 ml of 10% Tween-20 when the solution has cooled to room temperature
Store for up to a week at 4 °C
Note: The PFA should dissolve in 50 min to 1 h
OR - Dilute 16% PFA 1:4 in PBST. Store for up to a week at 4 °C
- Pre-warm 100 ml PBS to 68 °C in a water bath
- Proteinase K stock
1 mg/ml in distilled water
Store as aliquots at -20 °C - 20x SSC
3 M NaCl
300 mM tri-sodium citrate
pH 7.0-7.2
To make 1 L 20x SSC:
175.2 g NaCl
88.2 g tri-sodium citrate
Make up to 1 L with distilled water after adjusting pH
Autoclave
Store at room temperature - 50 mg/ml Heparin stock
Dissolve in Heparin in distilled water to a final concentration of 50 mg/ml
Store at 4 °C - Hybridization buffer (Hyb)
50% Formamide
5x SSC
50 μg/ml Heparin
0.5 mg/ml Torula-RNA
0.1% Tween-20
Optional: 5% weight by volume dextran sulfate
To make 50 ml Hyb:
25 ml formamide
12.5 ml 20x SSC
50 μl Heparin stock
25 mg Torula-RNA
2.5 g dextran sulfate
Vortex well till a homogenous is achieved
Add 500 μl 10% Tween-20
Make up to 50 ml with distilled water
Store at -20 °C
Note: Dextran sulfate is omitted from hyb used for pre-hybridization. - Post hybridization washes
Wash 1: 50% Formamide, 2x SSC, 0.1% Tween-20 in distilled water
To make 50 ml:
25 ml Formamide
5 ml 20x SSC
500 μl 10% Tween-20
Make up to 50 ml with distilled water
Wash 2: 2x SSC, 0.1% Tween-20 in distilled water
To make 50 ml:
5 ml 20x SSC
500 μl 10% Tween-20
Make up to 50 ml with distilled water
Wash 3: 0.2x SSC, 0.1% Tween-20 in distilled water
To make 50 ml:
500 μl 20x SSC
500 μl 10% Tween-20
Make up to 50 ml with distilled water
Note: Make the appropriate volume of wash solution fresh. - MABT
100 mM Maleic Acid
150 mM NaCl in distilled water
pH 7.5
0.1% Tween-20
To make 1 L:
8.77 g maleic acid
11.6 g NaCl
Adjust pH with NaOH pellets
Make up to 1 L with distilled water
Autoclave
Add 10 ml 10% Tween-20
Store at room temperature - 10% blocking reagent (Roche block)
Prepare 10% blocking reagent in MAB as per the manufacturer’s instructions
Store at -20 °C as aliquots - 2% Blocking reagent in MABT (2% Roche block)
Dilute 10% blocking reagent (Roche block) 1:5 in MABT
Make fresh - Staining buffer
100 mM Tris-HCl, pH 9.5
100 mM NaCl
50 mM MgCl2 in distilled water
To make 50 ml:
5 ml 1 M Tris-HCl, pH 9.5
5 ml 1 M NaCl
2.5 ml 1 M MgCl2
500 μl 10% Tween-20
Note: Make fresh. - Staining solution
4.5 μl of NBT and 3.5 μl of BCIP per 1 ml of the staining buffer
Vortex to mix
Protect from light
Note: Prepare the appropriate volume fresh.
Optional: add PVA to a final concentration of 10%, weight by volume
To make 10 ml of staining solution with 10% PVA:
Add 1 g PVA to 10 ml of staining buffer (without Tween-20)
Bring into solution with stirring and heating
Cool to room temperature
Add 45 μl NBT and 35 μl of BCIP
Vortex to mix
Protect from light
Note: 10 ml is the minimum volume that can be made. - 87% glycerol in distilled water (weight by weight)
To make 100 g:
87 g glycerol in 13 g distilled water
Stir till a homogenous solution is achieved
Store at room temperature - 50x base solution
1.25 M KOH
10 mM EDTA in distilled water
To make 200 ml:
14 g KOH
4 ml 0.5 M EDTA
Add distilled water to a final volume of 200 ml
Store at room temperature - 1x base solution
Dilute 50x base solution 1:50 in distilled water
Note: Prepare the appropriate volume fresh. - 50x neutralization solution
2 M Tris-HCl in distilled water
To make 200 ml:
63 g Tris-HCl
Add distilled water to a final volume of 200 ml
Store at room temperature - 1x neutralization solution
Dilute 50x neutralization solution 1:50 in distilled water
Note: Prepare the appropriate volume fresh.
Acknowledgments
The protocol presented here is based on G. Hauptmann’s work (PMID: 11252185), which was implemented and continuously improved by D. Soroldoni. We acknowledge A. Faro and G. Gestri of the Wilson laboratory at UCL, UK who shared the genomic DNA extraction protocol with us. We also thank O. Venzin of the Oates lab for constructive feedback on the manuscript.
Funding: Institutional support from the Swiss Federal Institute of Technology in Lausanne (EPFL) (to ACO and RN); The Francis Crick Institute (receiving its core funding from Cancer Research UK, the Medical Research Council, and Wellcome) (to ACO and RN); Wellcome (WT098025MA to ACO); the Medical Research Council (MC_UP_1202/3 to ACO and RN).
Competing interests
The authors declare no competing interests.
References
- Bajard, L., Morelli, L. G., Ares, S., Pecreaux, J., Julicher, F. and Oates, A. C. (2014). Wnt-regulated dynamics of positional information in zebrafish somitogenesis. Development 141(6): 1381-1391.
- Cheng, C. N., Li, Y., Marra, A. N., Verdun, V. and Wingert, R. A. (2014). Flat mount preparation for observation and analysis of zebrafish embryo specimens stained by whole mount in situ hybridization. J Vis Exp(89). doi: 10.3791/51604.
- Choi, H. M., Chang, J. Y., Trinh le, A., Padilla, J. E., Fraser, S. E. and Pierce, N. A. (2010). Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat Biotechnol 28(11): 1208-1212.
- Deniziak, M., Thisse, C., Rederstorff, M., Hindelang, C., Thisse, B. and Lescure, A. (2007). Loss of selenoprotein N function causes disruption of muscle architecture in the zebrafish embryo. Exp Cell Res 313(1): 156-167.
- Gross-Thebing, T., Paksa, A. and Raz, E. (2014). Simultaneous high-resolution detection of multiple transcripts combined with localization of proteins in whole-mount embryos. BMC Biol 12: 55.
- Hauptmann, G. and Gerster, T. (1994). Two-color whole-mount in situ hybridization to vertebrate and Drosophila embryos. Trends Genet 10(8): 266.
- Herrgen, L., Schroter, C., Bajard, L. and Oates, A. C. (2009). Multiple embryo time-lapse imaging of zebrafish development. Methods Mol Biol 546: 243-254.
- Jambor, H., Surendranath, V., Kalinka, A. T., Mejstrik, P., Saalfeld, S. and Tomancak, P. (2015). Systematic imaging reveals features and changing localization of mRNAs in Drosophila development. Elife 4: e05003.
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B. and Schilling, T. F. (1995). Stages of embryonic development of the zebrafish. Dev Dyn 203(3): 253-310.
- Kiyama, H. and Emson, P. C. (1991). An in situ hybridization histochemistry method for the use of alkaline phosphatase-labeled oligonucleotide probes in small intestine. J Histochem Cytochem 39(10): 1377-1384.
- Lauter, G., Soll, I. and Hauptmann, G. (2011). Two-color fluorescent in situ hybridization in the embryonic zebrafish brain using differential detection systems. BMC Dev Biol 11: 43.
- Little, S. C., Tikhonov, M. and Gregor, T. (2013). Precise developmental gene expression arises from globally stochastic transcriptional activity. Cell 154(4): 789-800.
- Lleras Forero, L., Narayanan, R., Huitema, L. F., VanBergen, M., Apschner, A., Peterson-Maduro, J., Logister, I., Valentin, G., Morelli, L. G., Oates, A. C. and Schulte-Merker, S. (2018). Segmentation of the zebrafish axial skeleton relies on notochord sheath cells and not on the segmentation clock. Elife 7: e33843.
- McConaughy, B. L., Laird, C. D. and McCarthy, B. J. (1969). Nucleic acid reassociation in formamide. Biochemistry 8(8): 3289-3295.
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C. and Brand, M. (2009). Tissue micromanipulation in zebrafish embryos. Methods Mol Biol 546: 153-172.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Schulte-Merker, S., Ho, R. K., Herrmann, B. G. and Nusslein-Volhard, C. (1992). The protein product of the zebrafish homologue of the mouse T gene is expressed in nuclei of the germ ring and the notochord of the early embryo. Development 116(4): 1021-1032.
- Schulte-Merker, S., van Eeden, F. J., Halpern, M. E., Kimmel, C. B. and Nüsslein-Volhard, C. (1994). No tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development 120(4): 1009-1015.
- Soroldoni, D., Jorg, D. J., Morelli, L. G., Richmond, D. L., Schindelin, J., Julicher, F. and Oates, A. C. (2014). Genetic oscillations. A Doppler effect in embryonic pattern formation. Science 345(6193): 222-225.
- Stapel, L. C., Lombardot, B., Broaddus, C., Kainmueller, D., Jug, F., Myers, E. W. and Vastenhouw, N. L. (2016). Automated detection and quantification of single RNAs at cellular resolution in zebrafish embryos. Development 143(3): 540-546.
- Thisse, B. and Thisse, C. (2004). Fast release clones: a high throughput expression analysis. ZFIN Direct Data Submission ZFIN ID: ZDB-PUB-040907-1.
- Thisse, B., Pflumio, S., Fürthauer, M., Loppin, B., Heyer, V., Degrave, A., Woehl, R., Lux, A., Steffan, T., Charbonnier, X.Q. and Thisse, C. (2001). Expression of the zebrafish genome during embryogenesis (NIH R01 RR15402). ZFIN Direct Data submission ZFIN ID: ZDB-PUB-010810-1.
- Thisse, C. and Thisse, B. (2005). High throughput expression analysis of ZF-models consortium clones. ZFIN Direct Data submission ZFIN ID: ZDB-PUB-051025-1.
- Thisse, C. and Thisse, B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc 3(1): 59-69.
- Trivedi, V., Choi, H. M. T., Fraser, S. E. and Pierce, N. A. (2018). Multidimensional quantitative analysis of mRNA expression within intact vertebrate embryos. Development 145(1): dev156869.
- Wang, F., Flanagan, J., Su, N., Wang, L. C., Bui, S., Nielson, A., Wu, X., Vo, H. T., Ma, X. J. and Luo, Y. (2012). RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14(1): 22-29.
- Wetmur, J. G. (1976). Hybridization and renaturation kinetics of nucleic acids. Annu Rev Biophys Bioeng 5: 337-361.
- Wetmur, J. G. and Davidson, N. (1968). Kinetics of renaturation of DNA. J Mol Biol 31(3): 349-370.
- Yamamoto, A., Amacher, S. L., Kim, S. H., Geissert, D., Kimmel, C. B. and De Robertis, E. M. (1998). Zebrafish paraxial protocadherin is a downstream target of spadetail involved in morphogenesis of gastrula mesoderm. Development 125(17): 3389-3397.
Article Information
Copyright
![]() Narayanan and Oates. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Narayanan and Oates. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Narayanan, R. and Oates, A. C. (2019). Detection of mRNA by Whole Mount in situ Hybridization and DNA Extraction for Genotyping of Zebrafish Embryos. Bio-protocol 9(6): e3193. DOI: 10.21769/BioProtoc.3193.
- Lleras Forero, L., Narayanan, R., Huitema, L. F., VanBergen, M., Apschner, A., Peterson-Maduro, J., Logister, I., Valentin, G., Morelli, L. G., Oates, A. C. and Schulte-Merker, S. (2018). Segmentation of the zebrafish axial skeleton relies on notochord sheath cells and not on the segmentation clock. Elife 7: e33843.
Category
Developmental Biology > Morphogenesis > Organogenesis
Cell Biology > Tissue analysis > Tissue staining
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.