- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Production, Titration and Imaging of Zika Virus in Mammalian Cells


(*contributed equally to this work) Published: Vol 8, Iss 24, Dec 20, 2018 DOI: 10.21769/BioProtoc.3115 Views: 11813
Reviewed by: David PaulVinay PanwarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Since the outbreak of Zika virus (ZIKV) in Latin America and the US in 2016, this flavivirus has emerged as a major threat for public health. Indeed, it is now clear that ZIKV is vertically transmitted from the infected mother to the fetus and this may lead to severe neurological development defects including (but not restricted to) neonate microcephaly. Although ZIKV has been identified in the late 1940s, very little was known about its epidemiology, symptoms and molecular biology before its reemergence 60 years later. Recently, tremendous efforts have been made to develop molecular clones and tools as well as cell culture and animal models to better understand ZIKV fundamental biology and pathogenesis and to develop so-far-unavailable antiviral drugs and vaccines. This bio-protocol describes basic experimental procedures to produce ZIKV stocks and to quantify their concentration in infectious virus particles as well as to image and study this pathogen within infected cells using confocal microscopy-based imaging.
Keywords: Zika virusBackground
Zika virus (ZIKV), a mosquito-borne Flavivirus within the Flaviviridae virus family, was first isolated from a sentinel Rhesus monkey in 1947 in the Ziika Forest in Uganda and is closely related to dengue virus (DENV) (World Health Organization, 2018). Since then, it became famous in the last decade for its outbreaks in some Pacific islands (2007 and 2013) and in the Americas (2015). Symptomatic patients (25%-20% of the cases) may usually show rather mild clinical manifestations such as fevers, rashes, conjunctivitis, muscle/joint pains and/or headaches. However, since ZIKV re-emergence, severe neurological symptoms including (but not restricted to) Guillain-Barre syndrome in adults and congenitally transmitted newborn microcephaly were identified in infected individuals (Lazear and Diamond, 2016; Grubaugh et al., 2018). Unfortunately, there are currently no available treatments or vaccines against Zika virus, and this is partly due to our poor understanding of its biology.
In terms of phylogeny, there are 2 distinct lineages of Zika virus (Haddow et al., 2012). The so-called “historical” African lineage including the prototype strain MR766 and the “contemporary” Asian lineage derived from the African lineage after decades of mutations. For instance, the Asian strain H/PF/2013 is responsible for the outbreak in French Polynesia in 2013. Similarly, the virus strain responsible for the Brazilian outbreak also phylogenetically derives from the Asian lineage. Indeed, experiments in mouse and mosquito infection models support the idea that the Asian strain has acquired several key mutations which led to microcephaly in infected newborns in Brazil (Yuan et al., 2017) and to enhanced viral infectivity in the arthropod transmission vector Aedes aegypti (Liu et al., 2017).
Upon entry in the host cell, the viral genome, a positive single-stranded RNA, is translated into a single polyprotein at the endoplasmic reticulum (ER), which is subsequently cleaved into 3 structural proteins and 7 non-structural proteins by host and viral proteases. After replication, the genome is encapsidated into neosynthesized virions which are subsequently released from the cell (Neufeldt et al., 2018). Like all other tested flaviviruses, ZIKV remodels the endomembranes of the infected cell to generate a cytoplasmic environment which is favorable to ZIKV life cycle (Cortese et al., 2017). These membranous compartments, called viral replication factories are all ER-derived and can be sub-divided into three classes of ultrastructures (Cortese et al., 2017): 1. vesicle packets, which are ER invaginations believed to be the site of viral RNA replication, 2. virus bags in which assembled immature viruses accumulate in regular arrays, and 3. convoluted membranes whose roles remain unclear although it was proposed for DENV that they modulate host processes for the benefit of replication (Chatel-Chaix et al., 2016).
Before 2016, little was known about the general biology of ZIKV, and most of our knowledge relied on a direct transposition of fundamental discoveries about other flaviviruses closely related to this pathogen like DENV. However, it remains unclear what makes ZIKV so unique in terms of neuropathogenesis as compared to the other members of the Flavivirus genus. Hence, since ZIKV re-emergence and the recent outbreaks, the scientific community around the world has begun to decipher the mysteries surrounding this virus and to develop tools such as molecular clones as well as cell culture and animal models (Schwarz et al., 2016; Shan et al., 2016; Xie et al., 2016; Morrison and Diamond, 2017; Mutso et al., 2017; Munster et al., 2018). Knowing how to culture ZIKV and to measure its infectivity constitutes key methods to perform any descriptive or functional studies about this virus both in cellulo and in vivo. Moreover, given that Asian and African lineages may differ in terms of symptom severity in infected mouse fetuses (Cugola et al., 2016), it is sometimes relevant to compare model strains of these two lineages. In this bio-protocol, we describe basic cell culture methods to produce ZIKV stocks from Asian and African lineages and to quantify their concentration in infectious virus particles (more generally referred to as “titer”) as well as to image this pathogen inside infected cells using immunofluorescence-based microscopy.
Materials and Reagents
- Materials
- 1.5 ml microtubes (Ultident Scientific, catalog number: 87-B150-C)
- 10 cm cell culture dishes (Falcon, Fisher Scientific, catalog number: 08-772-E)
- 15 cm cell culture dishes (Falcon, Fisher Scientific, catalog number: 08-772-6)
- 24-well polystyrene culture plates (Corning Life Sciences, Fisher Scientific, catalog number: 08-772-1)
- MCE 0.45 μm filters, 30 mm diameter (Ultident Scientific, catalog number: 229753)
- 20 ml sterile syringes (Ultident Scientific, catalog number: BD-302830)
- 15 ml sterile conical tubes (Falcon, Fisher Scientific, catalog number: 352096)
- Coverslips No. 1 (diameter: 12 mm, thickness: 0.13 to 0.17 mm), sterilized by autoclaving (Fisher Scientific, catalog number: 12-545-80)
- Microscope glass slides, frosted clear glass 26 mm x 76 mm, 1-1.2 mm thick (Ultident Scientific, catalog number: 170-8105-W)
- Absorbent paper
- Parafilm
- Aluminum foil
- Viruses
- ZIKV MR766 (African lineage) (EVAg, catalog number: 001v-EVA143) (under material and transfer agreement. Passage history: P5. Store original desiccated stocks at -80 °C)
- ZIKV H/PF/2013 (Asian lineage) (EVAg, catalog number: 001v-EVA1545) (under material and transfer agreement. Passage history: P6. Store original desiccated stocks at -80 °C)
- Cell lines
- Vero E6 monkey epithelial cells (a kind gift from Drs. Tom Hobman and Anil Kumar, University of Alberta) (ATCC, catalog number: CRL-1586)
- Huh7 hepatocarcinoma cells (a kind gift from Dr. Patrick Labonté, INRS) (Creative Bioarray, catalog number: CSC-C9441L)
Alternatively, this cell line is typically available from most laboratories working on hepatitis C virus or DENV.
- Reagents
- UltraPure distilled water (Life Technologies, catalog number: 10977-015)
- Dulbecco's modified Eagle medium (DMEM) (Life Technologies, catalog number: 111965-092)
- Fetal bovine serum (FBS) performance (Wisent, catalog number: 098150)
- Penicillin-streptomycin (Life Technologies, catalog number: 15140-122)
- MEM non essential amino acids solution (100x) (Life Technologies, catalog number: 11140-050)
- Phosphate buffered saline (PBS) (Life Technologies, catalog number: 14190-144)
- 1 M HEPES buffer, pH range: 7.2-7.5 (Life Technologies, catalog number: 15630-080)
- 0.25% Trypsin-EDTA (Life Technologies, catalog number: 25200-072)
- Minimum Essential Medium (MEM) with L-Glutamine (Life Technologies, catalog number: 11095-080)
- Carboxymethylcellulose (CMC) sodium salt, medium viscosity (Sigma-Aldrich, catalog number: 21902-100G )
- 37% formaldehyde (Fisher Scientific, catalog number: BP531-500)
- Crystal violet (Fisher Scientific, catalog number: C581-100)
- 95% ethanol (Commercial Alcohols, catalog number: 1011C)
- Triton X-100 (Mallinktrot, catalog number: 3555)
- 4% paraformaldehyde (Sigma-Aldrich, catalog number: P6148-1KG)
- Normal goat serum (Thermo-Fisher, catalog number: 01-6201)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A-9647)
- Sodium azide (Fisher Scientific, catalog number: BP922I-500)
- Rabbit polyclonal anti-ZIKV NS4B (GeneTex, catalog number: GTX133311; dilution 1:200)
- Mouse monoclonal anti-dsRNA, clone J2 (Scicons, catalog number: 10010200; dilution 1:400)
- Rabbit polyclonal anti-DENV NS4B (GeneTex, catalog number: GTX124250; dilution 1:1,000)
- Mouse monoclonal anti-DENV NS3, clone GT2811 (GeneTex, catalog number: GTX629477; dilution 1:100)
- Mouse monoclononal panflaviviral anti-E, clone 4G2 (Sigma-Aldrich, catalog number: MAB10216, dilution 1:200)
- Goat anti-Rabbit AlexaFluor 488 (Life Technologies, catalog number: A11034)
- Goat anti-Mouse AlexaFluor 568 (Life Technologies, catalog number: A11004)
- 4',6-Diamidino-2-Phenylindole (DAPI) (Life Technologies, catalog number: D1306)
- Fluoromount G (Southern Biotech, catalog number: 0100-01)
- Complete DMEM (see Recipes)
- Plaquing medium (MEM-CMC) (see Recipes)
- Formaldehyde fixative (see Recipes)
- Paraformaldehyde fixative (see Recipes)
- Crystal violet staining solution (see Recipes)
- Triton X-100 permeabilization solution (see Recipes)
- BSA Blocking solution (see Recipes)
Equipment
- -80 °C freezer
- Vortexer
- Pipette
- Beaker
- CO2 incubator
- Hemacytometer
- Magnetic stirrer
- BSL2 cell culture cabinet
- 2D rocking platform shaker
- Zeiss LSM780 confocal microscope
- Chemical hood
- Tweezers
- 4 °C refrigerator
- Autoclavable glass bottle
- Autoclave
Procedure
IMPORTANT NOTE: All experiments with live viruses (i.e., all pre-fixation steps) should be performed inside a biosafety level 2 (BSL2) or a BSL2+ tissue culture laboratory according to the country and institution regulations and required permits regarding Zika virus handling and storage.
- Virus stock production
- Virus preparation
- Resuspend with 200 μl ultraPure sterile water the desiccated virus stocks by pipetting up and down, at room temperature in a BSL2 cell culture cabinet.
- Aliquot 50 μl of virus in sterile microtubes and store at -80 °C.
- Virus amplification
Day -1: Preparation of cells for infection- Prepare cells from 15 cm culture stock dishes of Vero E6 cells showing 80%-100% confluence. PBS, complete DMEM and trypsin-EDTA should be pre-warmed at 37 °C. It is worth mentioning that we never use Vero E6 cells “older” than 50 passages since this may affect cell permissiveness to ZIKV and yields of virus production.
- Remove the culture medium.
- Wash the cells with 10 ml PBS.
- Remove PBS and add 7 ml trypsin-EDTA.
- Incubate for 2 min at 37 °C in the CO2 incubator.
- Remove trypsin-EDTA.
- Thoroughly tap the cell culture dish in order to detach the cells.
- Resuspend the cells in 10 ml complete DMEM (see Recipes section for composition).
- Transfer the cells into a 15 ml sterile tube.
- Using a hemacytometer, determine the number of cells per ml.
- Seed 2 x 106 Vero E6 cells in 8 ml of complete DMEM in 10 cm cell culture dishes and incubate overnight in the 37 °C incubator.
Day 0: Infection- In a 15 ml sterile tube, dilute 50 μl of virus stock in 6 ml of complete DMEM.
- Remove by aspiration the medium of Vero E6 cells. Cells should show a confluence of approximately 70%-80%).
- Incubate for 2 h at 37 °C.
- After incubation, remove the virus inoculum.
- Add 8 ml of pre-warmed complete DMEM.
- Incubate for 3 days at 37 °C.
Days 3, 4, 5, 6 and 7 post-infection: virus harvest- At each day, harvest the supernatant with a sterile 20 ml syringe directly from the cell culture dish. Place an MCE 0.45 μm filter at the tip of the syringe.
- Filter the supernatant into a sterile 15 ml tube.
- Add 8 ml of complete DMEM in the cell culture dish and place it back into the incubator.
- Add 80 μl HEPES buffer 1 M, pH 7.2-7.5 (final concentration of 10 mM) in the filtered supernatant. This will prevent acidification of virus stocks following freeze/thawing and contribute maintaining optimal viral infectivity.
- Aliquot virus supernatants into sterile 1.5 ml microtubes (1 ml per tube).
- Store aliquots at -80 °C.
- Proceed with viral titration of each harvest using plaque assays.
- Virus preparation
- Viral titration using plaque assays
Day -1: Preparation of cells for infection
Notes:- Cells should be prepared from 15 cm dishes of Vero E6 with 80%-100% confluence.
- One confluent 15-cm dish of cells is generally enough to prepare five 24-well plates corresponding to 10 samples to titer. PBS, trypsin-EDTA and culture media should be pre-warmed at 37 °C.
- Remove by aspiration the medium from the culture dishes.
- Wash the cells with 10 ml of PBS.
- Remove PBS and add 7 ml trypsin-EDTA.
- Incubate for 2 min in the incubator.
- Remove the trypsin by aspiration.
- Thoroughly tap the cell culture dish in order to detach the cells.
- Resuspend the cell in 10 ml complete DMEM.
- Transfer the cells into a sterile 15 ml tube.
- Determine cell concentration with a hemacytometer.
- Dilute the cells with complete DMEM to a final concentration of 4 x 105 Vero E6 cells/ml.
- Seed 24-well plates with 500 μl diluted cells per well. One 24-well plate allows the titration of two virus samples.
- Incubate overnight at 37 °C.
Day 0: Infection
Note: The day of infection, the cells should show a confluence of 90%-100%.- Prepare 6 microtubes per sample to titer and identify them (10-1 to 10-6). Add 450 μl complete DMEM in each microtube.
- Dilute 10 times the sample in series:
- Add 50 μl of virus sample in the first microtube (dilution 10-1). Vortex the tube for 5 s.
- Change the tip to avoid any cross-contamination between samples. Pipet 50 μl of the 10-1 dilution and transfer into the second tube (dilution 10-2). Vortex the tube for 5 s.
- Repeat these steps for the other dilutions (10-3 to 10-6).
- Remove medium from rows A and B of one 24-well plate (Figure 1).
- Pipet up-and-down twice the 10-6 dilution and add 200 μl with the same tip on the side of the last wells of rows A and B according to the plate layout shown in Figure 1. Thus, infection is performed in duplicates.
- Proceed similarly with other sample dilutions (10-5 to 10-1 dilutions) according to the plate layout (Figure 1).
IMPORTANT NOTE: It is critical to change used tips between each dilution to avoid cross-contamination between wells. Moreover, always start dispensing the samples into the plate starting with the most diluted samples. To avoid that cells dry, always wait to have processed one sample before aspirating the medium for another dilution series (e.g., rows C and D).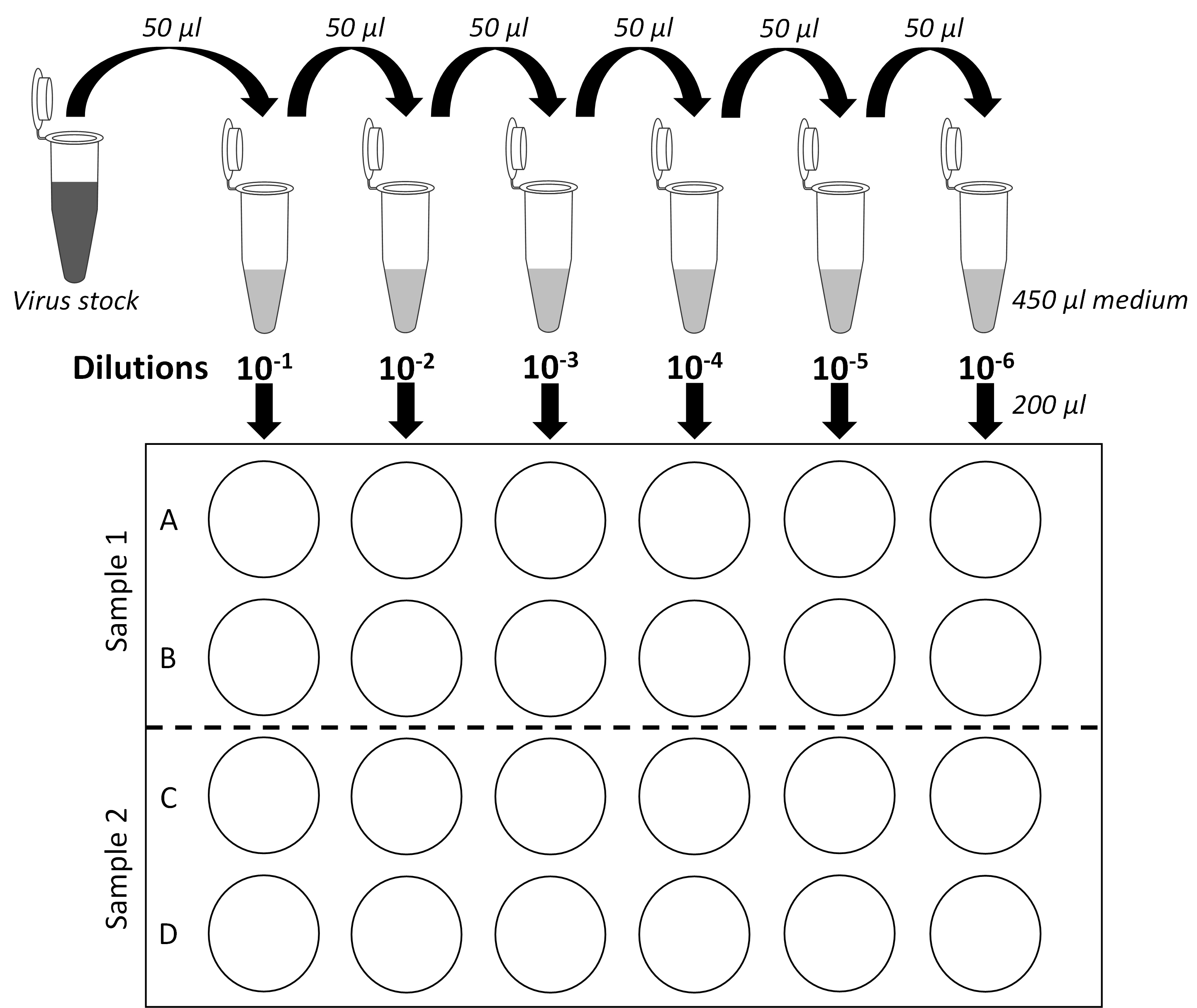
Figure 1. Serial dilution plan and plate layout for plaque assays. Schematic representation of the virus stock 10-fold dilutions and of 24-well plates used for plaque assays. Two samples per plate can be titered. Each dilution is assessed in biological duplicates. - Once a 24-well plate has been entirely processed, put it back to the incubator and proceed with the next plate and two new samples to titer.
- Incubate all plates with gentle agitation at 37 °C for at least one hour using a 2D rocking platform shaker. Incubation period can be extended up to several hours without impacting titers.
- After this incubation, take the first plate and aspirate the supernatant of the row A and B starting by the 10-6 dilution to avoid cross-contamination.
- Add on the side of wells, starting from the highest dilutions, 1 ml of MEM-CMC (see Recipes) with a 10 ml pipette. Discard the pipette. It is important to change the pipette between each sample series to avoid contamination of the stock bottle of MEM-CMC with virus. Especially, contamination with fast-growing viruses may result in future experiments in the appearance of very large plaques in the wells which will render impossible to count smaller expected plaques.
- Repeat for rows C and D and then for all plates.
- Incubate for 5 days at 37 °C.
IMPORTANT NOTE: Do not move the plates during the 5-day incubation since this will result in the formation of “comet-shaped” plaques which might render them difficult to count.
- Add 1 ml 10% formaldehyde per well.
- Incubate for at least 2 h at room temperature in the BSL2 laboratory. Incubation period can be extended up to several days.
- Discard the liquid by inversion in a large beaker in a chemical hood. Formaldehyde-containing liquid wastes should be handled according to the chemical safety regulations of the research institution.
- Wash plates by vigorously rinsing them with tap water to remove all the methylcellulose. Remove the excess of water by taping the plates on an absorbent paper.
- Add 500 μl crystal violet-containing staining solution (see Recipes) on cell monolayers.
- Incubate for 15 min at room temperature. Incubation period can be extended up to several hours.
- Remove crystal violet solution by inversion in a reusable plastic container. Collected crystal violet solution can be transferred back to the stock bottle and be reused for future staining.
- Wash the plates extensively with tap water to remove the excess of staining solution.
- Dry the plates on absorbent paper to remove excess of water.
- Count the number of plaques in the appropriate dilution and determine infectious titers (see Figure 2 and Data analysis).
- Typical infection experiments for imaging
Day -1: Preparation of cells for infection- Using tweezers which have been cleaned with 70% ethanol, transfer sterile coverslips in each well of a 24-well culture dish.
- Seed Huh7 cells (or any cell line of interest to be tested): 20,000 cells in 500 µl complete DMEM. Vigorously shake the plate to homogenously disperse the cells throughout the well. Of note, we never use Huh7 cells “older” than 50 passages since we have noticed a significant change of morphology and growth properties beyond this passage.
- Incubate overnight at 37 °C with 5% CO2.
Day 0: Viral infection- For infection, appropriate multiplicity of infection (MOI) and virus dilution must be chosen according to stock infectious titers (see Procedure B and Data analysis). For immunofluorescence purposes, MOI of 1 is typically used. MOI of 1 means that on average, each target cell is inoculated with 1 infectious virus particle.
- In this case (20,000 cells) with an MOI of 1, 20,000 viruses must be diluted in a final volume of 500 µl DMEM in each well. Prepare a master mix for all the wells.
- Remove culture medium and add gently on the side of each well 500 µl of the ZIKV/DMEM master mix.
- Incubate for at least 2 h at 37 °C with 5% CO2 and gentle agitation.
- Remove virus inoculum and add 1 ml complete DMEM per well.
- Incubate for 48 or 72 h at 37 °C with 5% CO2.
Day 2 or 3: Fixation- Remove media.
- Wash the cells twice with cold PBS.
- Add in each well 500 µl 4% paraformaldehyde/PBS solution.
- Incubate for 20 min at room temperature with gentle shaking.
- Wash once with 500 µl PBS and add 1 ml PBS.
- Seal the plate with parafilm to avoid that cells dry because of PBS evaporation. Store fixed cells at 4 °C protected from light with an aluminum foil. The cells can be stored at 4 °C for a few months if no staining is immediately planned.
- Immunofluorescence staining for confocal microscopy
- Remove PBS.
- Add 500 µl PBS/Triton X-100 permeabilization solution (see Recipes) and incubate for 15 min at room temperature with gentle shaking.
- Wash the cells once with PBS.
- During permeabilization, freshly prepare a premix of blocking solution (see Recipes). Plan 300 µl of blocking solution per coverslip to be labeled.
- Add 300 µl complete blocking solution per well.
- Incubate for 1 h at room temperature with gentle shaking.
- Quickly wash the cells three times with 500 µl PBS.
- Prepare the primary antibodies
- Dilute rabbit anti-ZIKV NS4B and mouse anti-dsRNA in PBS/5% BSA/0.05% sodium azide (i.e., blocking solution without normal goat serum) with dilution of 1/200 and 1/400, respectively. Plan 30 µl antibody solution per coverslip.
- Alternatively, antibodies against dengue virus NS4B and NS3 may also be used to detect ZIKV since there is a cross-reaction between these two flaviviruses for the antibody epitopes. Using panflaviviral antibodies against E protein is also an option to be considered (see antibody reference and dilutions in the Materials and Reagents).
- To avoid coverslips to dry and maintain a sufficient level of humidity, prepare an airtight container with wet pieces of paper at the bottom.
- On a plastic support inside the box, place a piece of parafilm cleaned with 70% ethanol.
- For each coverslip to label, add 30 µl of primary antibody as drops.
- With tweezers, remove the excess of liquid on a piece of absorbent paper and put each coverslip on the antibody drop, with the cells facing parafilm.
- Incubate for at least 2 h at room temperature, protected from light.
- Take back coverslips and transfer them in a sterile 24-well plate which contains 500 µl PBS per well. Cells must face the top of the plate.
- Wash the coverslips three times for 5 min with 500 µl PBS.
- During the last wash, prepare the secondary fluorescently labeled antibodies at a dilution of 1:1,000 in PBS/5% BSA/0.05% sodium azide. Plan 300 µl per coverslip.
- Cover the coverslips with 300 µl of the secondary antibody solution. Alternatively, in order to spare reagents, coverslips may be incubated on an antibody drop exactly as the primary antibody incubation.
- Incubate for 1 h at room temperature protected from light on a 2D rocking shaker.
- Quickly wash once and then, three times for at least 10 min with gentle agitation with 500 µl PBS.
- Dilute 10,000 fold the DAPI stock into PBS. Plan 500 µl per coverslip.
- Remove PBS from the plate and add 500 µl DAPI/PBS per well.
- Incubate for 10 min with gentle agitation at room temperature protected from light.
- Quickly wash three times each with 500 µl PBS.
- Wash once with 500 µl sterile water.
- Clean a microscope glass slide with 70% ethanol. Add a 5 µl drop of Fluoromount G per coverslip. Depending on the microscope used for imaging, each slide can accommodate 3-4 coverslips.
- Remove the excess of water on coverslip to-be-mounted by touching its side with a piece of absorbent paper. Place the coverslip on a Fluoromount G drop on the glass slide, with cells facing the drop.
- Keep slides protected from light at 4 °C overnight before imaging.
- Image the cells with a confocal or epifluorescence microscope in order to determine the % of infection, i.e., the % of NS4B- or dsRNA-positive cells.
Data analysis
- Infectious titer determination for virus stocks using plaque assays
- Count the number of plaques in the well where they can be discriminated. Select the well duplicate showing the highest number of individual plaques. See Figure 2 for a typical example.
- Calculate the infectious viral titers according to the following formula:
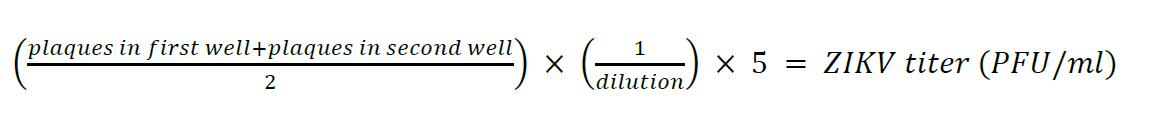
- Values are multiplied by 5 because the virus inoculum is 200 µl and the titers are normalized to 1 ml of the corresponding ZIKV stock.
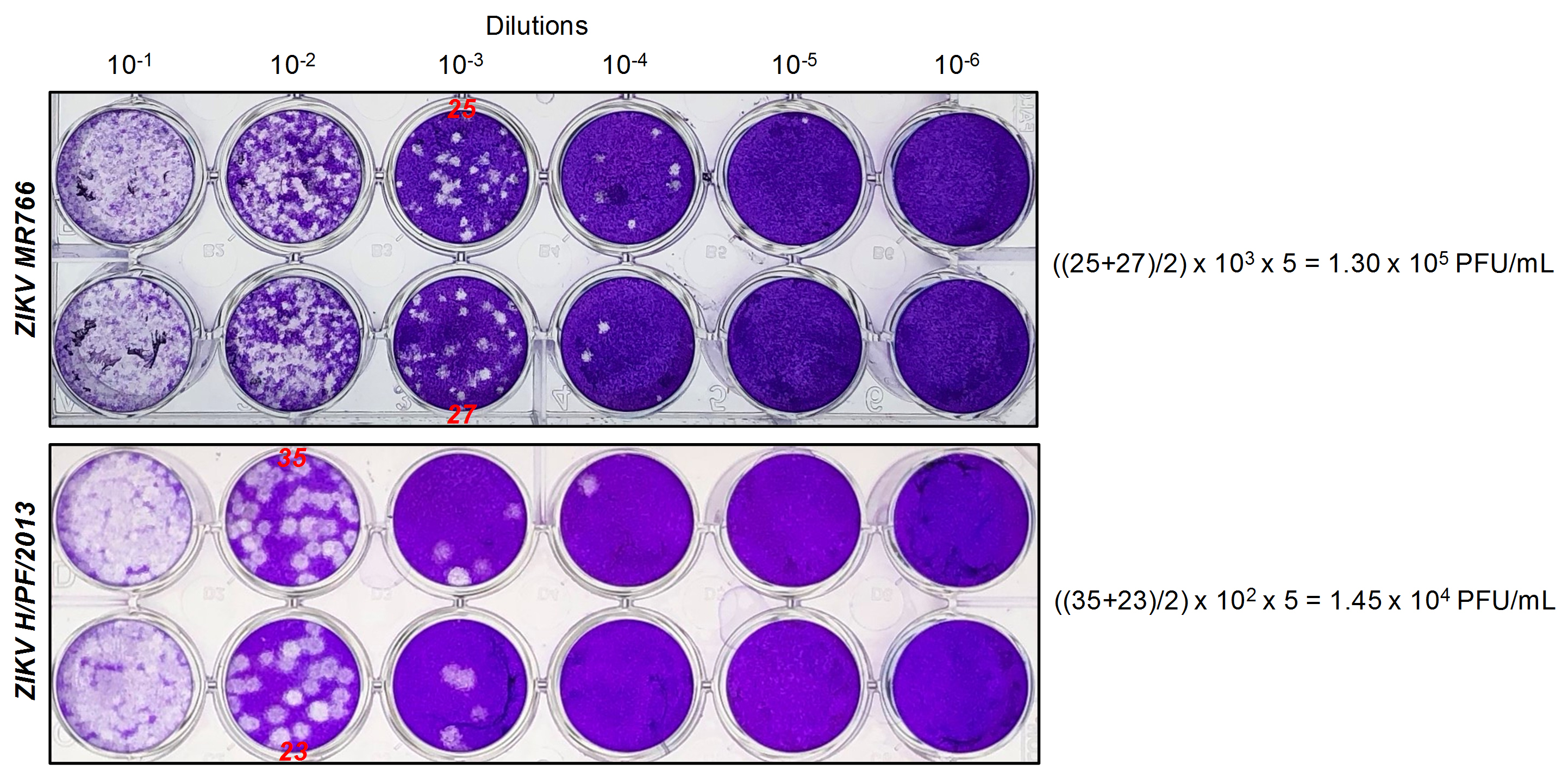
Figure 2. Typical plaque assay for ZIKV MR766 and H/PF/2013. Vero E6 cells were infected with 10-fold serial dilutions of ZIKV MR766 or H/PF/2013 stocks (Day 3 harvests). Five days post-infection, cells were fixed and stained with crystal violet. In this specific example, plaques can be counted in the 10-3 and 10-4 dilution well of ZIKV MR766 and H/PF/2013 (indicated in red), respectively. According to the formula described above, infectious titers are 1.3 x 105 PFU/ml for MR766 and 1.45 x 104 PFU/ml for H/PF/2013.
Typically, starting from ~104-105 PFU/ml depending on the experiment to ~107 PFU/ml (Figure 3), infectious titers for both strains logarithmically increase during the five first days of amplification and then reach a plateau. We never collect viruses at 1 and 2 days post-infection because titers are too low to perform subsequent experiments. In our hands, infectious titers over 5 x 107 PFU/ml were never achieved, even if the amplification period is extended beyond 7 days. It is worth mentioning that plaque morphology, and the profiles of amplification kinetics may change if high passages of viruses as well as of cells are used for virus stock production. This should be carefully considered and addressed if relevant.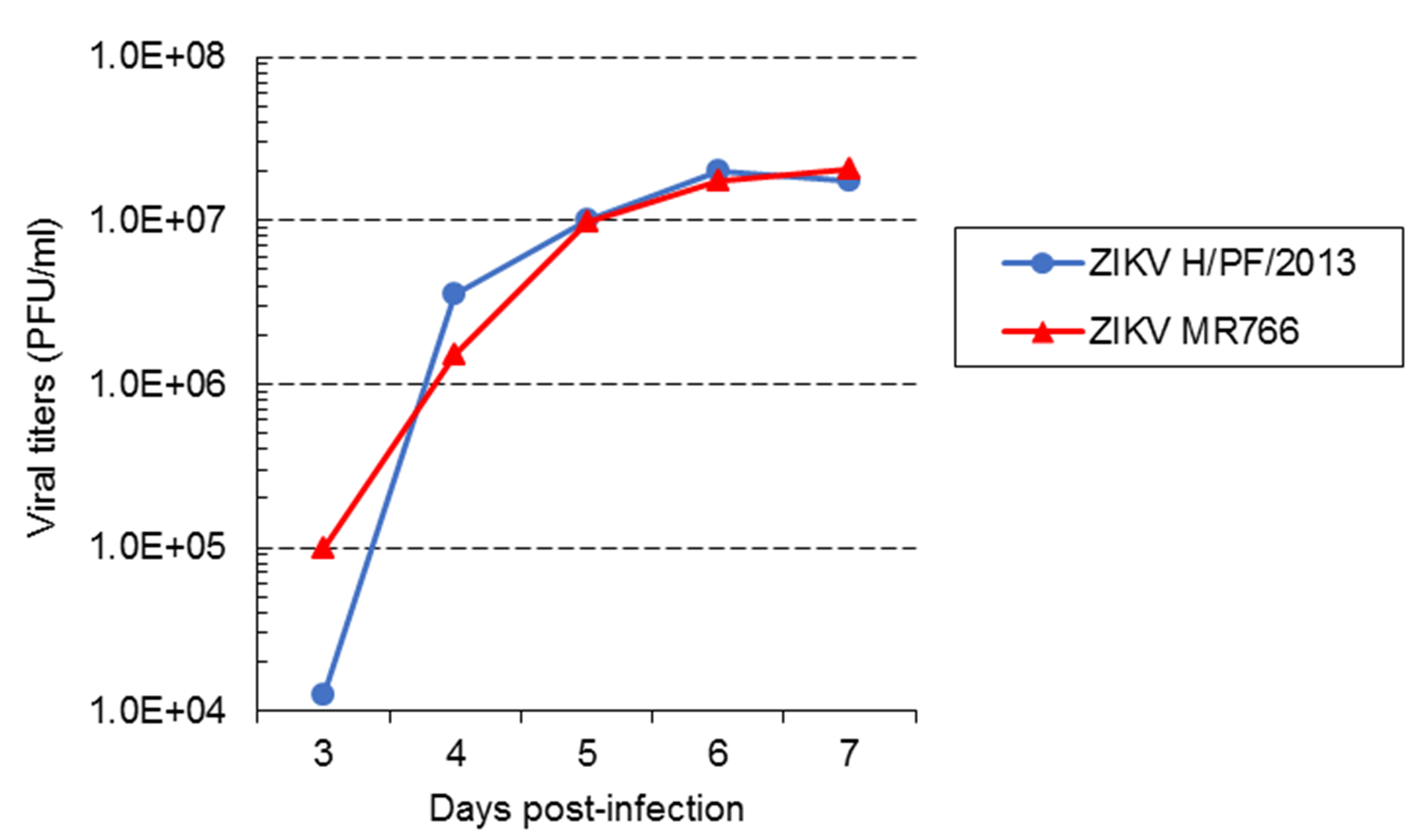
Figure 3. Virus amplification kinetics of ZIKV strains MR 766 and H/PF/2013 in Vero E6 cells. Vero E6 cells were infected with ZIKV MR766 or H/PF/2013 strains at an MOI of 0.01. Virus supernatants were collected at 3, 4, 5, 6 and 7 days post-infection. Infectious virus titers were determined using plaque assays in Vero E6 cells. - Imaging
Observe slides at a confocal microscope and set up the detection parameters with the “uninfected” condition coverslip. Any signal observed in this condition is non-specific. For NS4B antibody, it may be diffuse while it generally appears for anti-dsRNA as small dots with a very weak signal throughout the cell. Although anti-dsRNA is not supposed to recognize RNA helixes smaller than 40 base pairs (which are presumably inexistent or rare in cells), it most likely detects cellular highly structured RNAs with low affinity, explaining the observed background in uninfected cells. If non-specific signals are too high, detergent concentration in permeabilization buffer and/or primary antibody dilution may be increased. The ZIKV-specific signal for dsRNA (corresponding to the ZIKV RNA replication intermediate), typically shows a perinuclear accumulation of small intense puncta which presumably correspond to replication complexes within ER-derived vesicle packets (Chatel-Chaix et al., 2016; Cortese et al., 2017) (Figure 4). It was recently shown that dsRNA is localized in the vicinity of the microtubule organization center (MTOC) surrounded by a "cage-like” structure constituted of rearranged cytoskeleton (Cortese et al., 2017). NS4B labeling for both ZIKV strains shows a diffuse distribution, which colocalizes with dsRNA signal (Figure 4), consistent with the fact that DENV NS4B was shown to be a component of vesicle packets as shown by immunogold labeling followed by electron microscopy (Welsch et al., 2009). In addition, ZIKV H/PF/2013 NS4B also accumulates at the periphery of dsRNA clusters within large and intense punctated structure (diameter of ~1-5 µm) which by analogy with DENV NS4B most likely correspond to convoluted membranes, another replication factory sub-structure (Chatel-Chaix et al., 2016).
To estimate the infection rate, count at least a hundred cells in five different fields of the coverslip and count how many are infected, i.e., NS4B- and dsRNA-positive.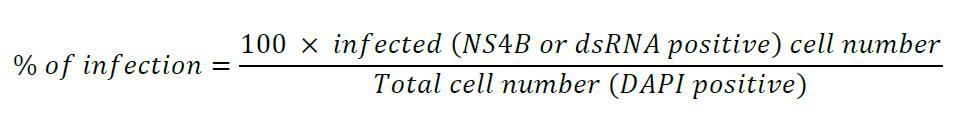
A 100% infection rate is reproducibly achieved within 48 h with ZIKV MR766 in Huh7 cells at an MOI of 1. At 72 h post-infection, cells show obvious signs of cytopathic effect. In contrast, with the same infection conditions, ZIKV H/PF/2013 is generally detected in only 10% of the cells (in infected clusters of 10-20 cells) with no signs of cell death at 48 h post-infection. This infection rate can be improved by fixing the cells one day later. Moreover, we regularly achieve ~100% infection with this strain in Huh7 cells when we increase the MOI to 50. No obvious signs of cytopathic effects were noticed in Huh7 cells in these conditions.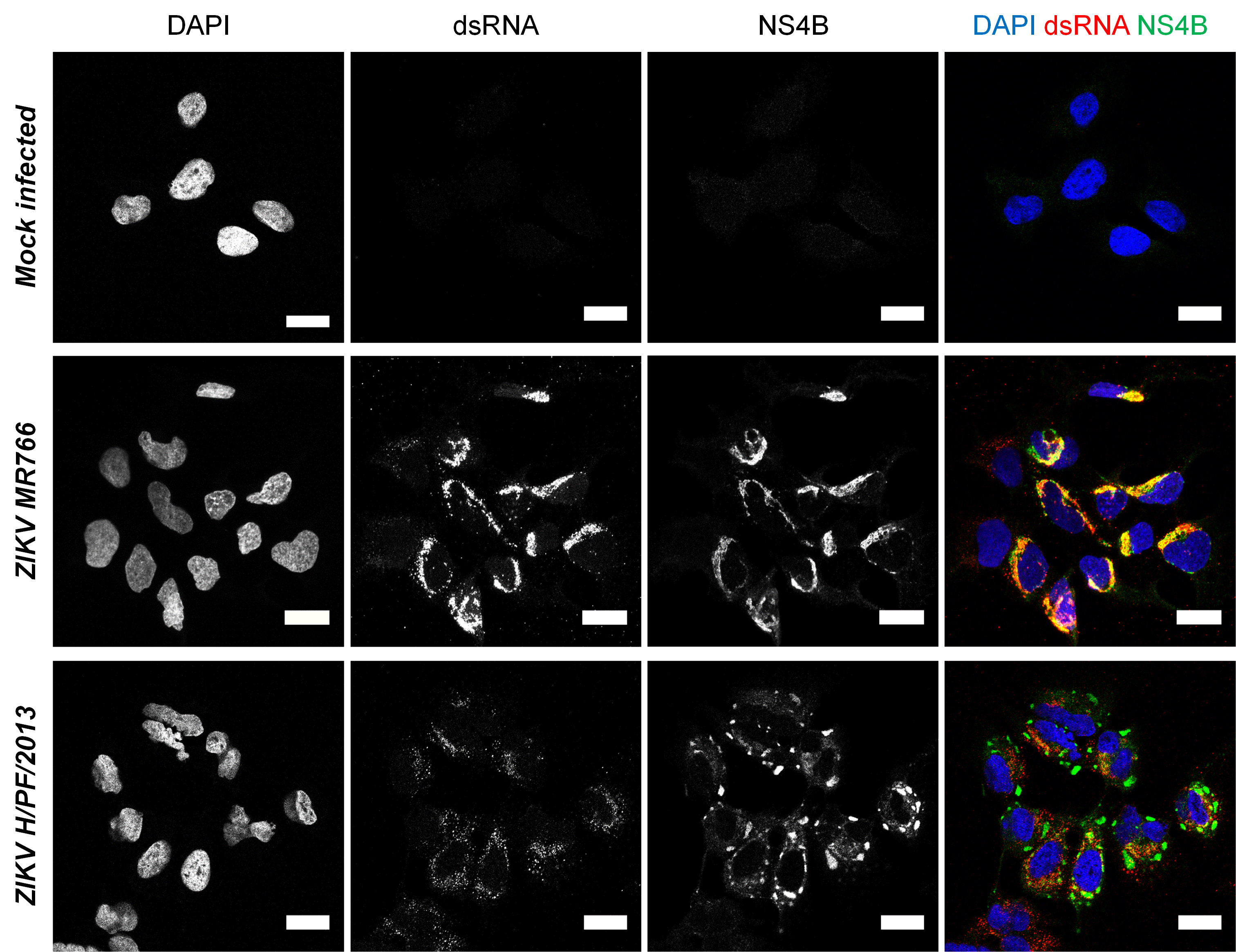
Figure 4. Imaging of Zika virus infection in Huh7 cells. Huh7 cells were infected with ZIKV MR766 or H/PF/2013 strains at an MOI of 1 or left uninfected. Forty-eight hours post-infection, cells were fixed, permeabilized and labeled with anti-dsRNA and anti-ZIKV NS4B antibodies. Nuclei were visualized with DAPI staining. Cells were observed with a Zeiss LSM780 confocal microscope. For ZIKV H/PF/2013, an infected cluster is shown. Scale bars: 20 μm. - Relevant remarks
It is important to note that the infection procedure may be adapted for other end-point readouts such as Western blotting, qPCR or RNA interference combined to any functional assay. For those purposes, the number of cells and volume may be up- or down-scaled according to dish or plate format. Cell density for microscopy experiments is kept rather low so that cells are well individualized for imaging. Hence, it is worth mentioning that more cells may be used as compared to microscopy experiments so that sufficient biological material is available. Typically, those ratios/formats may be used:- 10 cm dish: 1,000,000 Huh7 cells. Infection in 5 ml
- 6-well plate: 200,000 Huh7 cells. Infection in 1 ml
- 12-well plate: 100,000 Huh7 cells. Infection in 500 µl
- 24-well plate: 50,000 Huh7 cells. Infection in 250-300 µl
- 96-well plate: 10,000 Huh7 cells. Infection in 50 µl
Finally, any cell line or primary cells may theoretically be permissive to ZIKV infection. To test this, we typically perform replication kinetics experiments in which we infect cells with ZIKV at an MOI of 1. Cell supernatants are collected and filtered 1, 2, 3 and 4 days post-infection similarly to ZIKV stock harvest. Putative supernatant-associated infectivity is then assessed using plaques assays. The % of infection may be determined using the protocol described in this manuscript. Of note, it should be always kept in mind that the infectious titers determined using plaque assays are relative to Vero E6 cells permissiveness to ZIKV. For many possible reasons that are intrinsic to a given tested cell line (e.g., virus receptor availability/expression, high induction of antiviral responses…) and/or virus strain, the infection rate may be low even with an MOI of 1. To circumvent this limitation, the MOI may be increased up to 10-50 and the infection procedure optimized (e.g., time of infection, presence of serum in the inoculum…) in future experiment to eventually reach values close to 100%.
Recipes
- Complete DMEM
500 ml DMEM
50 ml FBS Performance
5 ml penicillin/streptomycin
5 ml non essential amino acids solution
Store at 4 °C - Plaquing medium (MEM-CMC)
- Put 7.5 g CMC in an autoclavable glass bottle which contains a magnetic bar
- Autoclave the bottle to sterilize the powder and stirrer (121 °C, 30 min, 20 bars; drying for 25 min)
c.Add into the bottle 500 ml MEM within the sterile environment of a cell culture cabinet. Close the bottle without touching the bottlenecks with the hands. Mix by inversion. Keep at 4 °C the original MEM bottle for further use (see below)
d.Incubate the MEM/CMC bottle at 4 °C above a magnetic stirring plate until complete solubilization of CMC. This generally takes between two and three days.
e.Transfer the reconstituted medium from the glass bottle into the original MEM plastic bottle inside the sterile environment of a cell culture cabinet. This will avoid introducing into the BSL2 area any glass-made material
f.Store at 4 °C. The final concentration of CMC is 1.5%
- Formaldehyde fixative
270 ml distilled water
100 ml 37% formaldehyde (10% final concentration)
Store at room temperature - Paraformaldehyde fixative
1 L 1x PBS
40 g paraformaldehyde (4% final concentration)
Facilitate dissolving the paraformaldehyde by heating (~60-70 °C) the preparation inside a chemical hood for ~30 min
Prepare 12 ml aliquots in 15 ml tubes and store at -20 °C - Crystal violet staining solution
5 g crystal violet
Dissolve in 52.6 ml of 95% ethanol
Add 445 ml of double-distilled water
Final concentrations of crystal violet and ethanol are 1% and 10%, respectively
Store at room temperature - Triton X-100 permeabilization solution
500 ml PBS
1 ml Triton X-100 (final concentration 0.2%)
Store at room temperature - BSA Blocking solution
- Dissolve 10 g BSA in 200 ml PBS (5% [w/v] final concentration)
- Add 1 ml 10% (w/v) sodium azide solution (NaN3; final concentration: 0.05%)
- Store at 4 °C
- OPTIONAL: The solution can be sterilized by filtration in order to avoid contamination and to maintain the quality of the solution over several months.
- The day of immunofluorescence staining, supplement the solution with normal goat serum to a final concentration of 10% (v/v).
Acknowledgments
LCC is receiving a research scholar (Junior 2) salary support from Fonds de la Recherche du Québec-Santé (FRQS). LCC’s research is supported by grants from Natural Sciences and Engineering Research Council of Canada (NSERC; RGPIN-2016-05584), the Canadian Institutes of Health Research (CIHR; PJT153020; ICS154142), Fonds de la Recherche du Québec-Nature et Technologies (FRQNT; 2018-NC-205593), Armand-Frappier Foundation and Institut National de la Recherche Scientifique. This protocol is mostly adapted from the experimental procedures used in our previous work (Chatel-Chaix et al., 2016) in which we have optimized the production and detection of ZIKV. We thank the European Virus Archive goes Global (EVAg) and Dr. Xavier de Lamballerie (Emergence des Pathologies Virales, Aix-Marseille University) for providing ZIKV MR766 and H/PF/2013 original stocks. We are grateful to Drs Patrick Labonté (Institut National de la Recherche Scientifique), Tom Hobman (University of Alberta) and Anil Kumar (University of Alberta) for generously providing Huh7 and Vero E6 cells.
Competing interests
The authors declare that they do not have any conflicts of interests or competing interests.
References
- Chatel-Chaix, L., Cortese, M., Romero-Brey, I., Bender, S., Neufeldt, C. J., Fischl, W., Scaturro, P., Schieber, N., Schwab, Y., Fischer, B., Ruggieri, A. and Bartenschlager, R. (2016). Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe 20(3): 342-356.
- Cortese, M., Goellner, S., Acosta, E. G., Neufeldt, C. J., Oleksiuk, O., Lampe, M., Haselmann, U., Funaya, C., Schieber, N., Ronchi, P., Schorb, M., Pruunsild, P., Schwab, Y., Chatel-Chaix, L., Ruggieri, A. and Bartenschlager, R. (2017). Ultrastructural characterization of Zika virus replication factories. Cell Rep 18(9): 2113-2123.
- Cugola, F. R., Fernandes, I. R., Russo, F. B., Freitas, B. C., Dias, J. L., Guimaraes, K. P., Benazzato, C., Almeida, N., Pignatari, G. C., Romero, S., Polonio, C. M., Cunha, I., Freitas, C. L., Brandao, W. N., Rossato, C., Andrade, D. G., Faria Dde, P., Garcez, A. T., Buchpigel, C. A., Braconi, C. T., Mendes, E., Sall, A. A., Zanotto, P. M., Peron, J. P., Muotri, A. R. and Beltrao-Braga, P. C. (2016). The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534(7606): 267-271.
- Grubaugh, N. D., Faria, N. R., Andersen, K. G. and Pybus, O. G. (2018). Genomic insights into Zika virus emergence and spread. Cell 172(6): 1160-1162.
- Haddow, A. D., Schuh, A. J., Yasuda, C. Y., Kasper, M. R., Heang, V., Huy, R., Guzman, H., Tesh, R. B. and Weaver, S. C. (2012). Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis 6(2): e1477.
- Lazear, H. M. and Diamond, M. S. (2016). Zika Virus: new clinical syndromes and its emergence in the Western Hemisphere. J Virol 90(10): 4864-4875.
- Liu, Y., Liu, J., Du, S., Shan, C., Nie, K., Zhang, R., Li, X. F., Zhang, R., Wang, T., Qin, C. F., Wang, P., Shi, P. Y. and Cheng, G. (2017). Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 545(7655): 482-486.
- Morrison, T. E. and Diamond, M. S. (2017). Animal models of Zika virus infection, pathogenesis, and immunity. J Virol 91(8): e00009-17.
- Munster, M., Plaszczyca, A., Cortese, M., Neufeldt, C. J., Goellner, S., Long, G. and Bartenschlager, R. (2018). A reverse genetics system for Zika virus based on a simple molecular cloning strategy. Viruses 10(7): E368.
- Mutso, M., Saul, S., Rausalu, K., Susova, O., Zusinaite, E., Mahalingam, S. and Merits, A. (2017). Reverse genetic system, genetically stable reporter viruses and packaged subgenomic replicon based on a Brazilian Zika virus isolate. J Gen Virol 98(11): 2712-2724.
- Neufeldt, C. J., Cortese, M., Acosta, E. G. and Bartenschlager, R. (2018). Rewiring cellular networks by members of the Flaviviridae family. Nat Rev Microbiol 16(3): 125-142.
- Schwarz, M. C., Sourisseau, M., Espino, M. M., Gray, E. S., Chambers, M. T., Tortorella, D. and Evans, M. J. (2016). Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. mSphere 1(5): e00246-16.
- Shan, C., Xie, X., Muruato, A. E., Rossi, S. L., Roundy, C. M., Azar, S. R., Yang, Y., Tesh, R. B., Bourne, N., Barrett, A. D., Vasilakis, N., Weaver, S. C. and Shi, P. Y. (2016). An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe 19(6): 891-900.
- Welsch, S., Miller, S., Romero-Brey, I., Merz, A., Bleck, C. K., Walther, P., Fuller, S. D., Antony, C., Krijnse-Locker, J. and Bartenschlager, R. (2009). Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5(4): 365-375.
- World Health Organization. Fact sheets-Zika virus. 2018; Available from: http://www.who.int/news-room/fact-sheets/detail/zika-virus.
- Xie, X., Zou, J., Shan, C., Yang, Y., Kum, D. B., Dallmeier, K., Neyts, J. and Shi, P. Y. (2016). Zika virus replicons for drug discovery. EBioMedicine 12: 156-160.
- Yuan, L., Huang, X. Y., Liu, Z. Y., Zhang, F., Zhu, X. L., Yu, J. Y., Ji, X., Xu, Y. P., Li, G., Li, C., Wang, H. J., Deng, Y. Q., Wu, M., Cheng, M. L., Ye, Q., Xie, D. Y., Li, X. F., Wang, X., Shi, W., Hu, B., Shi, P. Y., Xu, Z. and Qin, C. F. (2017). A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 358(6365): 933-936.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Freppel, W., Mazeaud, C. and Chatel-Chaix, L. (2018). Production, Titration and Imaging of Zika Virus in Mammalian Cells. Bio-protocol 8(24): e3115. DOI: 10.21769/BioProtoc.3115.
Category
Microbiology > Microbial cell biology > Virus propagation
Cell Biology > Cell isolation and culture > Virus isolation
Molecular Biology > Protein > Detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.