- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Luciferase-expressing Tumor Cell Lines
Published: Vol 8, Iss 8, Apr 20, 2018 DOI: 10.21769/BioProtoc.2817 Views: 20118
Reviewed by: Jia LiShalini Low-NamShweta Garg
Original research article
The authors used this protocol in:

Jan 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Murine tumor models have been critical to advances in our knowledge of tumor physiology and for the development of effective tumor therapies. Essential to these studies is the ability to both track tumor development and quantify tumor burden in vivo. For this purpose, the introduction of genes that confer tumors with bioluminescent properties has been a critical advance for oncologic studies in rodents. Methods of introducing bioluminescent genes, such as firefly luciferase, by viral transduction has allowed for the production of tumor cell lines that can be followed in vivo longitudinally over long periods of time. Here we describe methods for the production of stable luciferase expressing tumor cell lines by lentiviral transduction.
Keywords: LentivirusBackground
Paramount to tracking cells in vivo is the ability to detect them externally by minimally invasive methods. Enzymatic bioluminescence using luciferase derived from the firefly (Photinus pyralis) is a widely used method for image-based cell tracking in vivo. Bioluminescence has been used for a variety of in vivo application including the noninvasive imaging of reporter gene expression (Herschman, 2004), studying circadian rhythms (Southern and Millar, 2005), imaging cerebral strokes (Vandeputte et al., 2014), and for tracking genetically engineered T cells (Costa et al., 2001; Cheadle et al., 2010). Perhaps the field where bioluminescent cell lines have been most applicable is oncology where they have been instrumental for the monitoring tumor growth (Jenkins et al., 2005; Brennan et al., 2016; Byrne et al., 2016) and tumor metastasis (Rosol et al., 2003; Simmons et al., 2015) in mouse models. While some subcutaneously implanted tumors can be detected by palpation and measured with calipers, these methods are not effective for monitoring metastases or tracking tumors that disseminate widely, such as hematological malignancies that commonly grow in the bone marrow, lymph nodes and spleen.
Firefly luciferase oxides luciferin in the presence of molecular oxygen, magnesium and adenosine triphosphate to produce yellow-green light at 560 nm (Wilson and Hastings, 1998; Fraga, 2008). Benefits of luciferase bioluminescence in cell tracking include penetration of tissue for non-invasive monitoring and the re-usability of the enzymatic marker. Another advantage of luciferases is that most cells are not luminescent such that high signal-to-noise ratios can be achieved. A limitation bioluminescence is photon attenuation caused by intervening tissues, such as skin, bone, or hair.
Firefly luciferase is a single polypeptide specified by the luc gene that can be readily cloned into vectors used in gene delivery. Transient expression by plasmid transfection or non-integrating virus transduction limits the time over which cell tracking can be performed. This is especially problematic for oncology studies that may last several months. The ability of retroviruses to integrate into the genome is a key attribute that favors their use in producing stable cell lines. However, some oncoretroviruses, such as the Moloney murine leukemia virus can be limited by transgene silencing over time (Jähner et al., 1982). Further, retroviral vectors require cell division for genomic integration and can be inefficient at transducing highly differentiated cells such as neurons, dendritic cells, or resting lymphocytes.
For the purpose of making luciferase expressing cell lines, lentiviral retroviral vectors derived from human immunodeficiency virus-1 (HIV-1) are highly effective. An advantage of lentiviral vectors over other retroviral vectors, is their ability to integrate into the genome of non-dividing cells. This property makes them suitable gene delivery vehicles for targeting highly differentiated cells, such as neurons, dendritic cells and lymphocytes (Naldini et al., 1996). Lentiviruses also deliver very stable genomic integration and long-term transgene expression, to the extent that they have been used to make transgenic mice following embryo transduction (Lois et al., 2002).
In order to track hematologic tumor cells in an in vivo murine leukemia model, we made the FULGW lentiviral vector that co-expresses firefly luciferase (Luc) and enhanced green fluorescent protein (EGFP) for the purpose of B-cell lymphoma (A20) cell line transduction and stable clone production. The FULGW vector is based on a self-inactivating vector previously described by Miyoshi et al. (1998) that had been engineered to express the GFP reporter gene behind the human ubiquitin-C promoter by Lois et al. (2002), making the FUGW vector. To make FULGW, we replaced the EGFP sequence of FUGW with a Luc-IRES-EGFP sequence from rKat.Luc2.IRES.EGFP, previously developed by Cheadle et al. (2010).
FULGW contains unique elements that enhance gene integration and expression (Figure 1A). It encodes the human immunodeficiency virus-1 (HIV-1) flap element, giving it karyotropic properties that permit efficient genomic integration in non-replicating cells (Zennou et al., 2000). It contains the wood-chuck hepatitis virus posttranscriptional regulatory element (WPRE) that increases gene expression by transcript stabilization (Zufferey et al., 1999). In addition, it includes a 3’ self-inactivating long terminal repeat (3’ si-LTR) that contributes to maintaining it as a replication deficient virus. The 3’ si-LTR was developed by the deletion of a 133 bp region in the U3 region (ΔU3) of the 3’ LTR that renders the 5’ LTR of the integrated provirus transcriptionally inactive (Miyoshi et al., 1998).
Virus production is performed by the co-transfection of HEK-293T cells with the lentiviral plasmid (FULGW) and the two packaging plasmids, pCMV-ΔR8.91 and pCMV-VSVG (Figure 1B). HEK-293T cells are a human embryonic kidney cell line that stably expresses the CMV large T antigen, which greatly increases gene expression by the CMV promoter, generating robust virus production. pCMVΔR8.91 is an HIV-1 Gag and Polymerase (Pol) expression plasmid that was modified from the dR8.9 vector by deletion of four accessory HIV-1 gents, Vif, Vpr, Vpu, and Nef (Zufferey et al., 1997). pCMV-VSVG expresses the pantropic envelop (Env) protein derived from the vesicular stomatitis virus glycoprotein (VSVG) (Stewart et al., 2003). [*Note: Both FULGW and pCMV-ΔR8.91 are large plasmids and best grown in chemically competent recA1-deficient E. coli with high transformation efficiency such as One Shot TOP10 E. coli (Invitrogen) grown at 30 °C for 24-28 h]. Using the FULGW lentiviral vector packaged with these helper plasmids, we have produced multiple types of tumor cell lines on various genetic backgrounds that stably express luciferase and GFP for use in oncologic studies (Table 1).
Figure 1. Production of FULGW lentivirus and transduction of tumor cell lines. A. Diagram of key regions of the FULGW vector including the Luc-IRES-EGFP transgene. Transgene expression is driven by the human ubiquitin-C promoter. CMV (cytomegalovirus promoter), U5 (LTR unique 5’ region), R (LTR repeat region), HIV-1 flap (human immunodeficiency virus-1 flap element), Luc (firefly luciferase), IRES (intra-ribosomal element sequence), EGFP (enhanced green fluorescent protein), WPRE (wood-chuck hepatitis virus posttranscriptional regulatory element), si-LTR (self-inactivating LTR). B. FULGW is packaged and pseudotyped by lipophilic co-transfecting with pCMV-ΔR8.91 and pCMV-VSVG. Virus rich culture supernatant (SN) is collected at 48 and 72 h and virus is concentrated by ultracentrifugation. The concentrated virus is used to transduce tumor cell lines by spin-transduction in the presence of polybrene. Illustrated schematics make use of Motifolio templates (www.motifolio.com/).
Table 1. Luciferase-GFP expressing tumor cell lines produced by FULGW transduction
Materials and Reagents
- Pipette tips (USA Scientific, catalog numbers: 1110-3000 , 1110-1000 , 1111-2021 )
- T75 flask (Corning, Falcon®, catalog number: 353136 )
- 100 mm TC-treated Tissue Culture Dish (Corning, Falcon®, catalog number: 353003 )
- Sterile syringe 0.45 μm filter (VWR, catalog number: 28145-505 )
- Beckman ultra-clear 25 x 89 mm tubes (Beckman Coulter, catalog number: 344060 )
- Centricon Plus-70 unit (Merck, catalog number: UFC710008 )
- 15 ml Falcon tubes (Corning, Falcon®, catalog number: 352099 )
- 1.5 ml Eppendorf tubes (USA Scientific, catalog number: 1615-5500 )
- 12-well plates (Corning, catalog number: 3513 )
- 24-well plates (Corning, Costar®, catalog number: 3526 )
- 96-well plates (Greiner Bio One International, catalog number: 650185 )
- Sterile 500 ml 0.22 μm filter system (Corning, catalog number: 430758 )
- 29 ga. needles attached to 0.5 ml syringe (Terumo Medical Corporation, Elkton, MD, USA)
- BALB/c mice (THE JACKSON LABORATORY, catalog number: 000651 )
- HEK 293T (ATCC, catalog number: CRL-3216 )
- A20 B-cell lymphoma (ATCC, catalog number: TIB-208 )
- pCMV-VSVG plasmid (Addgene, catalog number: 8454 )
- pCMVΔR8.91 plasmid (Lifescience Market, catalog number: PVT2323 )
- pFULGW (Lentiviral luciferase-IRES-GFP plasmid, Available on request)
- Lipofectamine 2000 (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11668027 )
- Opti-MEM (Thermo Fisher Scientific, GibcoTM, catalog number: 11058021 )
- Dulbecco’s phosphate buffered saline (DPBS) (Corning, catalog number: 21-031-CM )
- Trypsin EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 25200056 )
- Polybrene (Sigma-Aldrich, catalog number: TR-1003-G )
- Propidium iodide (Sigma-Aldrich, catalog number: P4864-10ML )
- Bright-GloTM Luciferase Assay system (Promega, catalog number: E2610 )
- D-Luciferin, potassium salt (Gold Bio, catalog number: LUCK-1G )
- Isoflurane; Abbott Laboratories (Abbott Park, Illinois, USA)
- Fetal bovine serum (Corning, catalog number: 35-010-CV )
- DMEM–high glucose (Sigma-Aldrich, catalog number: D6429-500ML )
- L-Glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 25030081 )
- Gelatin, 2% in H2O, tissue culture grade (Sigma-Aldrich, catalog number: G1393 )
- RPMI (Thermo Fisher Scientific, GibcoTM, catalog number: 11875093 )
- Penicillin/streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- D10 growth media (see Recipes)
- 2% gelatin (see Recipes)
- D-Luciferin stock solution (see Recipes)
Equipment
- Pipettes (Mettler-Toledo, Rainin, catalog numbers: 17008653 , 17008650 , 17008649 ; Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 4641070N )
- Tissue culture hood
- Tissue culture incubator (Eppendorf, New Brunswick, model: Galaxy® 170 S )
- Fluorescent inverted microscope with GFP filter (Leica Microsystems, model: Leica DM IL LED )
- FACS-Canto flow cytometer (BD Biosciences)
- Microcentrifuge (Eppendorf, model: 5424 )
- Table-top centrifuge (Eppendorf, model: 5810 )
- Ultracentrifuge (Beckman Coulter, model: L8-80M ) equipped with an SW-28 rotor (Beckman Coulter, model: SW 28 )
- Balance (VWR, catalog number: 10204-990 )
- Xenogen IVIS Imaging System (Perkin Elmer, Hopkinton, MA, USA)
- Tabletop Laboratory Animal Anesthesia System (VetEquip, catalog number: 901806 )
- Autoclave
Software
- Acquisition software (CellQuest, BD Biosciences)
- Analysis software (FlowJo v9.3, TreeStar)
- Living Image Software (Caliper Life Sciences, Hopkinton, MA, USA)
- Graphing software (GraphPad Prism v7.0c, La Jolla, CA, USA)
Procedure
- Lentiviral plasmid transfection and virus production
- Prepare tissue culture plates by coating them with 2% gelatin (see Recipes). Briefly rinse each 10-cm plate with 10 ml of the 2% gelatin solution and let dry in a hood.
Note: The same 10 ml of gelatin solution can be reused to coat several plates. - Plate 293T cells on the gelatin-coated tissue culture plates 24 h before transfection at 6-7.5 x 106 cells per 10 cm plate in 10 ml D10 growth media (see Recipes).
Note: We prepare 4 plates for each lentivirus preparation. - Transfect 293T cells with lentivirus (e.g., pFULGW) and packaging plasmids (pCMV-ΔR8.91, pCMV-VSVG).
Notes:- Lentiviruses are classified as Biosafety Level 2 (BSL-2) organisms due to their ability to infect primary human cells and experiments need to be conducted in appropriate facilities.
- Transfection efficiency can be checked by imaging GFP expression using an inverted fluorescent microscope.
- For each plate, prepare 60 μl of Lipofectamine in 1.5 ml Opti-MEM.
- For each plate, also prepare 13.3 μg FULGW, 10 μg pCMV-ΔR8.91, and 6.7 μg pCMV-VSVG in 1.5 ml Opti-MEM.
- Mix the Lipofectamine and plasmid containing Opti-MEM together and incubate for 25 min at room temperature.
- Remove 2 ml of media from each plate (now 8 ml total).
- Add 3 ml of the Lipofectamine/plasmid mix to each plate in a dropwise manner, evenly distributing the mix over the plate.
- Lentiviruses are classified as Biosafety Level 2 (BSL-2) organisms due to their ability to infect primary human cells and experiments need to be conducted in appropriate facilities.
- Incubate for 6 h at 37 °C, aspirate the media and add 10 ml of fresh D10.
- Harvest SN (the culture media) at 48 h and store at 4 °C. Add another 10 ml fresh D10 media and harvest again at 72 h following transfection. Combine the SNs and clear by centrifugation at 2,000 rpm (805 x g) for 5’ in a table-top centrifuge. Then filter SN through a 0.45 μm filter that has been pre-wetted with 10 ml D10.
- Concentrate the virus.
Note: Use proper containment when working with lentiviruses as per BSL-2 and institutional guidelines.- Ultra-centrifugation method
- Put the SN in Beckman ultra-clear 25 x 89 mm tubes (each holds up to 35 ml) and weigh on a balance to the hundredth of a gram to insure they are of equal weight such that the rotor will be balanced.
- Spin at 25,000 rpm (112,000 x g) for 90 min at 4 °C in an SW-28 rotor.
- Discard supernatant by inverting the tube. Maintain the tube in the inverted position in the tissue culture hood and aspirate the remaining fluid from the walls of the tube to dry it completely without disturbing the viral pellet.
Note: The pellet will be small and can be difficult to visualize. - Add 200 μl of serum-free media directly to the viral pellet and allow it to resuspend for 12 h at 4 °C. Gently pipette to improve resuspension.
- Put the SN in Beckman ultra-clear 25 x 89 mm tubes (each holds up to 35 ml) and weigh on a balance to the hundredth of a gram to insure they are of equal weight such that the rotor will be balanced.
- Filtration method
- Place SN in Centricon Plus-70 unit (Millipore, Bedford, MA). Each unit holds approximately 60 ml.
- Centrifuge at 3,000 rpm (1811 x g) at 15 °C for 2-2.5 h using a table-top centrifuge.
- Discard flow through.
- Invert unit and spin at 1,500 rpm (453 x g) at 15 °C for 3 min to collect the viral concentrate.
- Place SN in Centricon Plus-70 unit (Millipore, Bedford, MA). Each unit holds approximately 60 ml.
- Ultra-centrifugation method
- Aliquot 20 μl of virus per Eppendorf tube and store at -80 °C.
Note: Aliquoting the virus helps to avoid freeze-thaw cycles, each of which will decrease the MOI by about 50%.
- Prepare tissue culture plates by coating them with 2% gelatin (see Recipes). Briefly rinse each 10-cm plate with 10 ml of the 2% gelatin solution and let dry in a hood.
- Titer lentivirus
- One day prior, plate 293T cells in 24-well plates at 250,000 cells/well in 0.5 ml D10.
- Transfect cells by adding the equivalent of 10, 3, 1, 0.3, 0.1, and 0 μl of viral concentrate per well of each prep.
- After 2 days, harvest cells by rinsing with 100 μl of PBS and then adding 100 μl per well of trypsin-EDTA and incubating for 5 min at 37 °C.
- FACS cells and determine % GFP positive.
- Calculate:
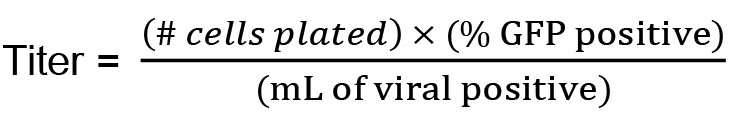
- One day prior, plate 293T cells in 24-well plates at 250,000 cells/well in 0.5 ml D10.
- Cell line transduction
- Mix 5-10 MOI of lentivirus in serum-free media with polybrene (6-8 μg/ml) and use it to replace the media of your cell line in a 12- or 24-well tissue culture plate. Spin transfect at 32 °C for 3-4 h at 1,000 x g (~2,300 rpm on a table-top centrifuge).
- Incubate at 37 °C for another 3 h, then replace the media with fresh culture media.
- Repeat viral transduction the next day if starting with lower MOI.
- Grow for 2 days and check transduction efficiency with FACS or with a fluorescent microscope. GFP expression is heterogeneous in cells 2 days following transduction with FULGW and will decrease over time in culture (Figure 2A).
Notes:- This decrease likely is due to the competition of untransduced clones or transgene loss where viral integration did not occur.
- When testing by FACS, add 0.5 μg/ml of propidium iodide to the FACS solution to exclude dead cells.
- This decrease likely is due to the competition of untransduced clones or transgene loss where viral integration did not occur.
- Mix 5-10 MOI of lentivirus in serum-free media with polybrene (6-8 μg/ml) and use it to replace the media of your cell line in a 12- or 24-well tissue culture plate. Spin transfect at 32 °C for 3-4 h at 1,000 x g (~2,300 rpm on a table-top centrifuge).
- Isolate luciferase expressing clones
- Plate transduced cells into round-bottom 96-well plates with the goal of obtaining one positive cell per well. The most efficient method to accomplish this is to FACS sort individual GFP-expressing cells 2-3 days after transduction into wells containing 100 μl of growth media. Alternatively, single-cell clones can be obtained by methods of limiting dilution. For example, by preparing plates with 100 μl of growth media containing ~5 cells/ml.
Note: Plate multiple plates to ensure the development of sufficient clones. - Allow cell clones to grow for ~2 weeks, giving them another 100 μl of fresh culture media at 1 week. Observe for clone growth by simply looking for wells with media that is becoming yellow, or by holding the plate up to a light and looking for colonies in the bottom of the wells.
- Transfer the clones to 24-well plates and expand over a few days.
- Test for GFP expression by FACS analysis. Clones derived from single cells will have a homogenous, narrow range of GFP expression. Select clones with different levels of expression for further testing (Figure 2B).
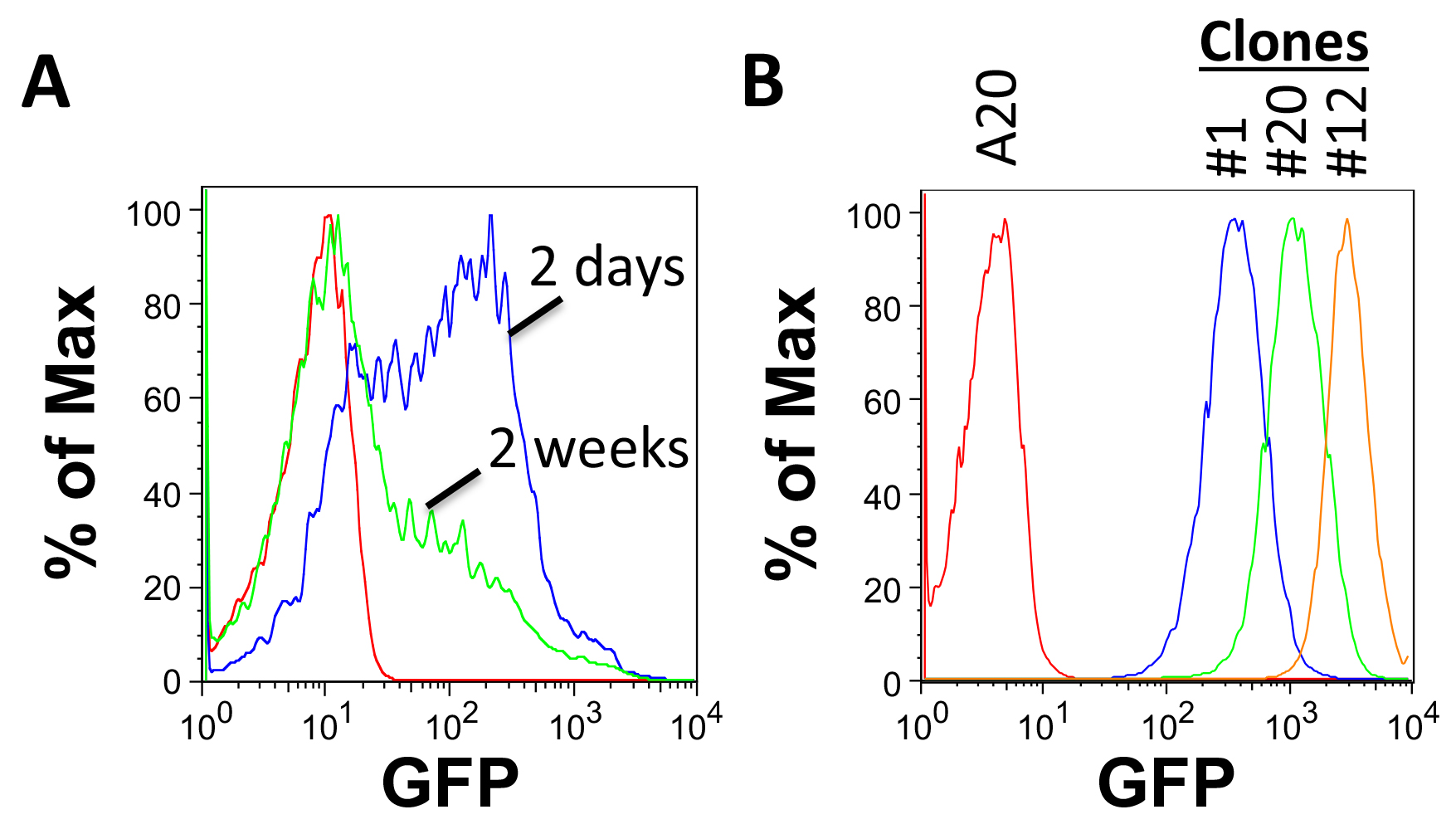
Figure 2. Assessing tumor cell line transduction by FACS. A. GFP expression in A20 cells 2 days and 2 weeks following transduction with FULGW. B. Isolated A20-Luc/GFP clones (#1, 12 and 20) demonstrate stable and homogenous GFP expression. - Test for luciferase activity.
- Place 10,000 cells in round-bottom 96-well tissue culture plates.
- To each well add a volume of Bright-GloTM Reagent equal to the volume of culture medium in the well, and mix. For 96-well plates, typically 100 μl of reagent is added to cells in 100 μl of culture medium.
- Wait at least 2 min to allow cell lysis, then measure luminescence in a luminometer or an IVIS system (Figures 3A and 3B).
- Place 10,000 cells in round-bottom 96-well tissue culture plates.
- Select clones and retest for GFP and luciferase expression after two weeks of culture to insure stable integration of the transgenes.
- Freeze and store multiple aliquots of the cell line in liquid nitrogen for future use.
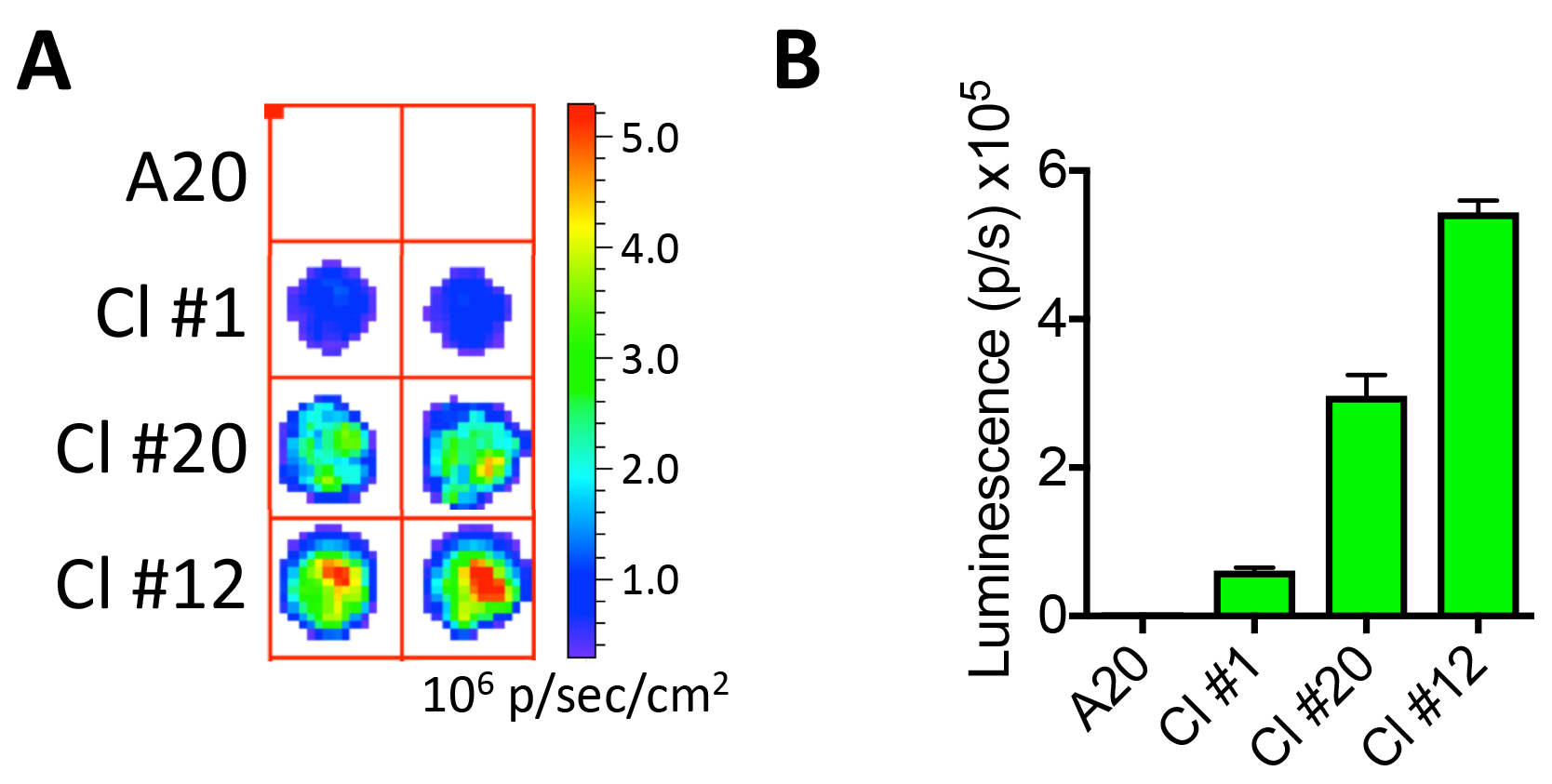
Figure 3. Confirmation of luciferase expression. A. Imaging A20 and A20-Luc/GFP B-cell lymphoma clones #1, 20, and 12 (10,000 cells from each clone are plated in a round-bottom 96-well plate) and tested for luciferase activity using an IVIS imaging system following the addition of D-Luciferin. B. Luminescence for each clone is quantified in photons per second (p/sec).
- Plate transduced cells into round-bottom 96-well plates with the goal of obtaining one positive cell per well. The most efficient method to accomplish this is to FACS sort individual GFP-expressing cells 2-3 days after transduction into wells containing 100 μl of growth media. Alternatively, single-cell clones can be obtained by methods of limiting dilution. For example, by preparing plates with 100 μl of growth media containing ~5 cells/ml.
- Image luciferase cell lines in vivo
- It is important to determine whether cell lines will grow efficiently in vivo.
- Administer 0.5-1 x 106 Luc/GFP tumors cells either subcutaneously or intravenously into mice of the same genetic background (e.g., BALB/c for A20 cells).
Note: It may be necessary to precondition mice with 4 Gy of total body irradiation 4 h prior to tumor administration to permit tumor growth in some strains. - Quantify tumor burden by measuring luciferase activity by IVIS (Figure 4).
- Inject mice with D-Luciferin (150 mg/kg, i.p.) and anesthetize by exposure to 4% isoflurane in an anesthetic chamber.
- Once sedated, transfer the mice onto the pre-warmed stage inside the IVIS imaging system specimen chamber with continuous exposure to 2% isoflurane flowing into the nose-cone to sustain sedation.
- Image mice and measure photon flux approximately 10 min following injection of the substrate.
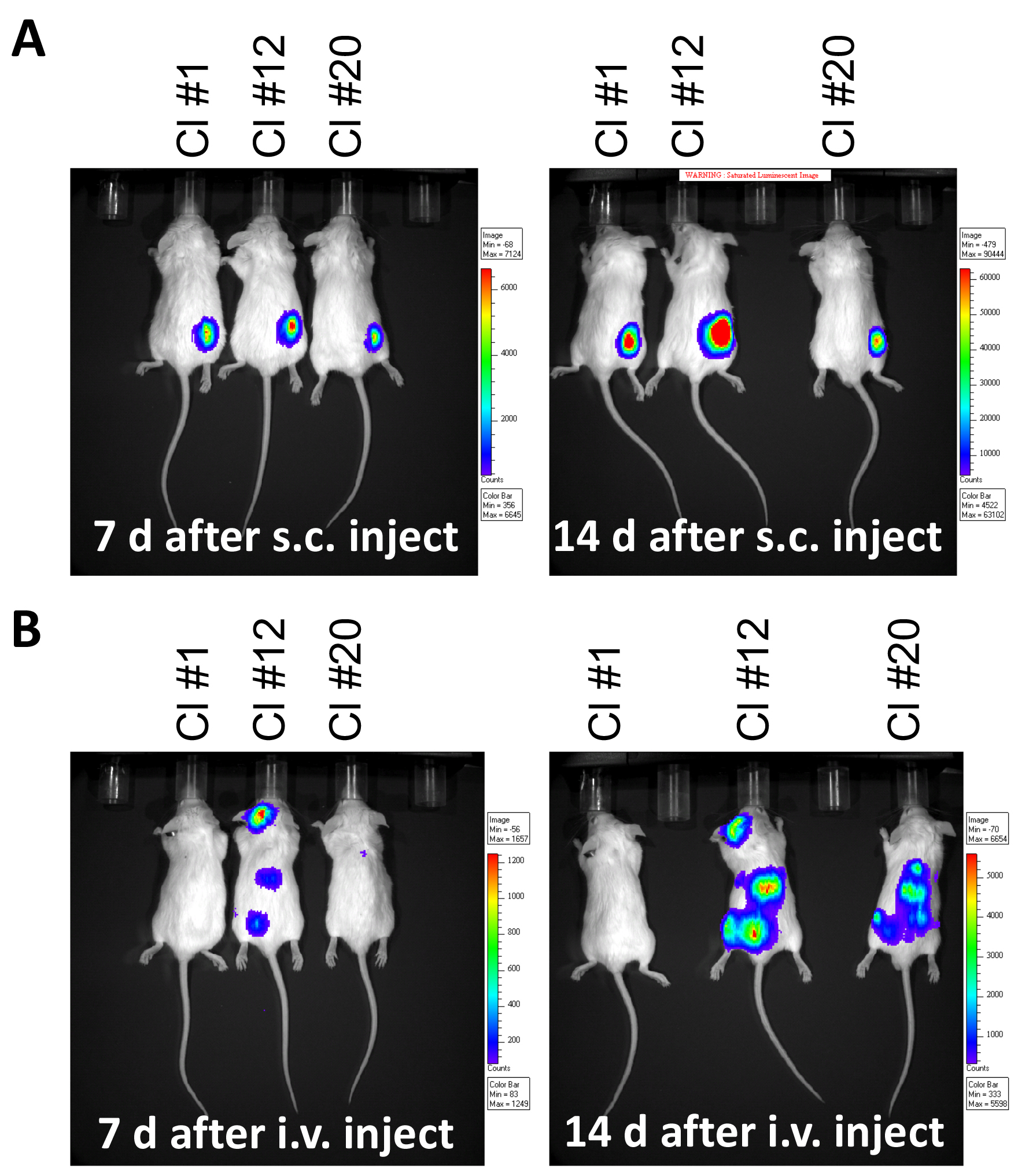
Figure 4. Monitoring tumor burden in mouse leukemia model. Luminescence imaged by IVIS at 7 and 14 days following, A. subcutaneous (s.c.) injection of 1 x 106 A20-gfp/luc clones on the right flank, or B. after intravenous (i.v.) injection of 5 x 105 A20-gfp/luc clones by tail vein injection. - Inject mice with D-Luciferin (150 mg/kg, i.p.) and anesthetize by exposure to 4% isoflurane in an anesthetic chamber.
- Euthanize mice when primary tumors reach ~1 cm3 (estimate from palpation), if animals lose significant weight (> 20%), develop hind-limb paralysis or become moribund, whichever comes first according to the method approved by your Institutional Animal Care and Use Committee (IACUC). For intravenously injected leukemia models, where tumor burden is difficult to determine by palpation, we typically euthanize animals when their measured luminescence reaches 107 p/sec.
- As a control, obtain a baseline measurement of luminescence in an untreated mouse following D-Luciferin injection.
- It is important to determine whether cell lines will grow efficiently in vivo.
Data analysis
IVIS data is collected in regions of interest and can be exported to Excel spreadsheet for analysis. The significance of differences between treatment groups can be determined by unpaired Student’s t-test.
Recipes
- D10 growth media
Add 50 ml of heat-inactivated fetal bovine serum (FBS) to 450 ml DMEM supplemented with glutamine and glucose - 2% gelatin
Dissolve 1 mg gelatin in 500 ml H2O and autoclave at 121 °C, 15 psi for 30 min - D-Luciferin stock solution
Dilute to 15 mg/ml in 1x Dulbecco’s phosphate buffered saline (DPBS) (Life Technologies) and sterile-filtered (0.22 μm)
Acknowledgments
- This work was supported by grants CA136934 (Y.Y.), CA047741 (Y.Y.), AI083000 (Y.Y.) and AI101263 (T.V.B.) from the National Institutes of Health. Conflict of interest: The authors have declared that no conflict of interest exists.
References
- Brennan, T. V., Lin, L., Brandstadter, J. D., Rendell, V. R., Dredge, K., Huang, X. and Yang, Y. (2016). Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J Clin Invest 126(1): 207-219.
- Byrne, F. L., McCarroll, J. A. and Kavallaris, M. (2016). Analyses of tumor burden in vivo and metastasis ex vivo using luciferase-expressing cancer cells in an orthotopic mouse model of neuroblastoma. Methods Mol Biol 1372: 61-77.
- Cheadle, E. J., Hawkins, R. E., Batha, H., O'Neill, A. L., Dovedi, S. J. and Gilham, D. E. (2010). Natural expression of the CD19 antigen impacts the long-term engraftment but not antitumor activity of CD19-specific engineered T cells. J Immunol 184(4): 1885-1896.
- Costa, G. L., Sandora, M. R., Nakajima, A., Nguyen, E. V., Taylor-Edwards, C., Slavin, A. J., Contag, C. H., Fathman, C. G. and Benson, J. M. (2001). Adoptive immunotherapy of experimental autoimmune encephalomyelitis via T cell delivery of the IL-12 p40 subunit. J Immunol 167(4): 2379-2387.
- Fraga, H. (2008). Firefly luminescence: a historical perspective and recent developments. Photochem Photobiol Sci 7(2): 146-158.
- Herschman, H. R. (2004). Noninvasive imaging of reporter gene expression in living subjects. Adv Cancer Res 92: 29-80.
- Jähner, D., Stuhlmann, H., Stewart, C. L., Harbers, K., Lohler, J., Simon, I. and Jaenisch, R. (1982). De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature 298(5875): 623-628.
- Jenkins, D. E., Hornig, Y. S., Oei, Y., Dusich, J. and Purchio, T. (2005). Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res 7(4): R444-454.
- Lois, C., Hong, E. J., Pease, S., Brown, E. J. and Baltimore, D. (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295(5556): 868-872.
- Miyoshi, H., Blomer, U., Takahashi, M., Gage, F. H. and Verma, I. M. (1998). Development of a self-inactivating lentivirus vector. J Virol 72(10): 8150-8157.
- Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F. H., Verma, I. M. and Trono, D. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272(5259): 263-267.
- Rosol, T. J., Tannehill-Gregg, S. H., LeRoy, B. E., Mandl, S. and Contag, C. H. (2003). Animal models of bone metastasis. Cancer 97(3 Suppl): 748-757.
- Simmons, J. K., Hildreth, B. E., 3rd, Supsavhad, W., Elshafae, S. M., Hassan, B. B., Dirksen, W. P., Toribio, R. E. and Rosol, T. J. (2015). Animal models of bone metastasis. Vet Pathol 52(5): 827-841.
- Southern, M. M. and Millar, A. J. (2005). Circadian genetics in the model higher plant, Arabidopsis thaliana. Methods Enzymol 393: 23-35.
- Stewart, S. A., Dykxhoorn, D. M., Palliser, D., Mizuno, H., Yu, E. Y., An, D. S., Sabatini, D. M., Chen, I. S., Hahn, W. C., Sharp, P. A., Weinberg, R. A. and Novina, C. D. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9(4): 493-501.
- Vandeputte, C., Reumers, V., Aelvoet, S. A., Thiry, I., De Swaef, S., Van den Haute, C., Pascual-Brazo, J., Farr, T. D., Vande Velde, G., Hoehn, M., Himmelreich, U., Van Laere, K., Debyser, Z., Gijsbers, R. and Baekelandt, V. (2014). Bioluminescence imaging of stroke-induced endogenous neural stem cell response. Neurobiol Dis 69: 144-155.
- Wilson, T. and Hastings, J. W. (1998). Bioluminescence. Annu Rev Cell Dev Biol 14: 197-230.
- Zennou, V., Petit, C., Guetard, D., Nerhbass, U., Montagnier, L. and Charneau, P. (2000). HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101(2): 173-185.
- Zufferey, R., Donello, J. E., Trono, D. and Hope, T. J. (1999). Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J Virol 73(4): 2886-2892.
- Zufferey, R., Nagy, D., Mandel, R. J., Naldini, L. and Trono, D. (1997). Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15(9): 871-875.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Brennan, T. V., Lin, L., Huang, X. and Yang, Y. (2018). Generation of Luciferase-expressing Tumor Cell Lines. Bio-protocol 8(8): e2817. DOI: 10.21769/BioProtoc.2817.
Category
Cancer Biology > General technique > Tumor formation > Cell isolation and culture
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.