- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Method for CRISPR/Cas9 Mutagenesis in Candida albicans
Published: Vol 8, Iss 8, Apr 20, 2018 DOI: 10.21769/BioProtoc.2814 Views: 12920
Reviewed by: David CisnerosEmmanuel Orta-ZavalzaSneh Lata Singh
Original research article
The authors used this protocol in:

Apr 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Candida albicans is the most prevalent and important human fungal pathogen. The advent of CRISPR as a means of gene editing has greatly facilitated genetic analysis in C. albicans. Here, we describe a detailed step-by-step procedure to construct and analyze C. albicans deletion mutants. This protocol uses plasmids that allow simple ligation of synthetic duplex 23mer guide oligodeoxynucleotides for high copy gRNA expression in C. albicans strains that express codon-optimized Cas9. This protocol allows isolation and characterization of deletion strains within nine days.
Keywords: Candida albicansBackground
C. albicans is a difficult organism to manipulate genetically. Since it normally exists as a diploid that does not readily undergo sexual reproduction, homozygous recessive mutations require sequential modification of each locus. The development and application of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) mutagenesis in C. albicans facilitates genetic manipulations because it allows simultaneous mutation of both alleles (Vyas et al., 2015; Min et al., 2016; Ng and Dean, 2017). CRISPR gene editing involves recruitment of an RNA-guided nuclease to a complementary target site adjacent to an NGG protospacer adjacent motif (PAM) (Jinek et al., 2012; Cong et al., 2013; Mali et al., 2013). CRISPR associated (Cas) nuclease is targeted with high specificity through complementary base pairing between a guide RNA associated with trans-activating CRISPR RNA (tracrRNA), which binds Cas9 (Gasiunas et al., 2012). Since the chromosomal target sequence is only ~20 nucleotides, expression of a single guide RNA (sgRNA) fused to a ~80 nucleotide tracrRNA, along with Cas9, is sufficient for targeted double-strand DNA cleavage. Since chromosomal breaks are lethal, there is a strong selective pressure for double stranded break repair. In C. albicans, co-expression of a donor repair fragment, containing homology to regions flanking the break, allows repair of the break by homologous recombination. Thus, appropriate design of the donor repair fragment allows introduction of sequence deletions, replacements or other chromosomal alterations.
Our previous studies demonstrated that a key factor contributing to high efficiency CRISPR mutagenesis of C. albicans genes relies on optimal gRNA expression (Ng and Dean, 2017). Toward this end, we created gRNA expression vectors that permit high levels of gRNA expression. The basis for this high-level expression is in part due to the presence of a strong, RNA polymerase II promoter (PADH1). This promoter drives the expression of an sgRNA flanked by a 5’ tRNA and a 3’ hepatitis delta virus (HDV) ribozyme RNA. The presence of these 5’ and 3’ flanking RNA sequences serve to promote efficient post-transcriptional processing to produce a mature sgRNA with precise ends. In the presence of an appropriate donor repair fragment, this increased sgRNA expression dramatically improves CRISPR/Cas mutagenesis in C. albicans. In practice, execution of mutagenesis is quite simple. The gRNA expression plasmid cloning relies on the annealing and ligation of two short gRNA encoding oligonucleotides into pre-cut vectors. Mutagenesis involves co-transformation of an sgRNA plasmid and the donor repair fragment into C. albicans strains that express codon-optimized Cas9 nuclease. Described below are detailed protocols for the design, synthesis, and cloning of gRNA oligonucleotides, healing fragment construction, yeast transformations, and mutant verification.
Materials and Reagents
- 1.5 ml microfuge tubes (acceptable as sold by any science distributor)
- Glass or disposable round bottom sterile tubes for growing 2-5 ml cultures of yeast and bacteria (acceptable as sold by any science distributor)
- Plastic Petri dishes (acceptable as sold by any science distributor)
- PCR tubes
- Toothpicks
- Yeast strains (Table 1)
Table 1. Strain list
- Plasmids (Table 2)
Table 2. Plasmids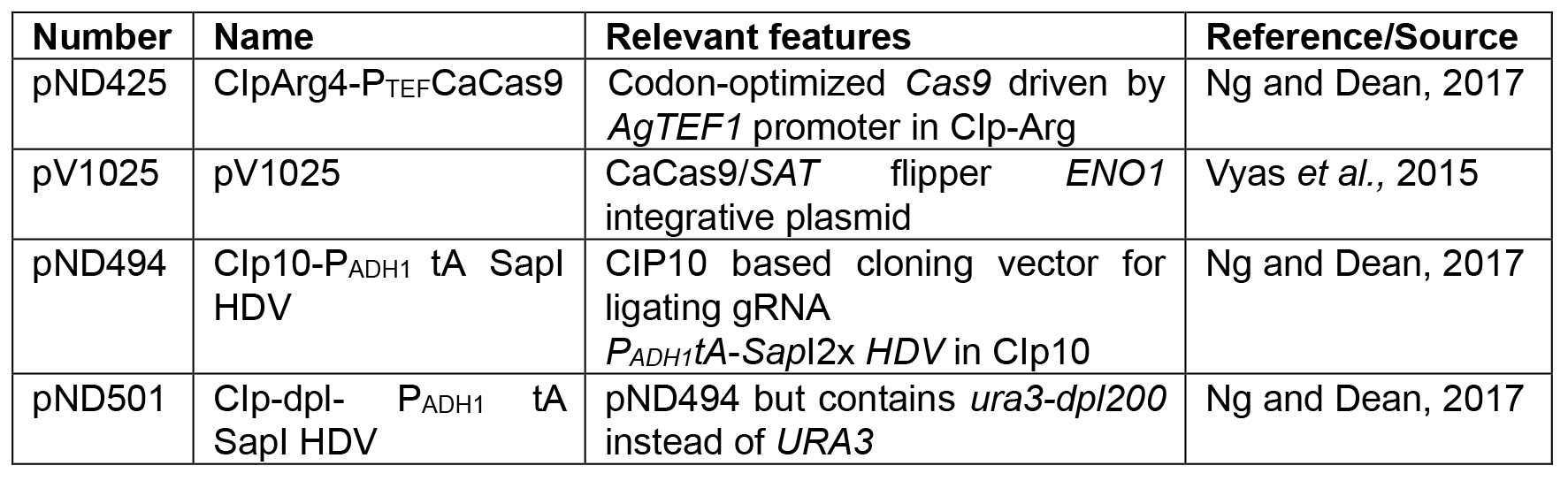
- E. coli competent cells (DH5α) (made as described, e.g., https://www.neb.com/protocols/2012/06/21/making-your-own-chemically-competent-cells or can be purchased from New England Biolabs)
Note: We use CaCl2-treated DH5α but these can be purchased. - Glycerol (any source) (50% in H2O, autoclaved)
- Salmon sperm DNA (Sigma-Aldrich, catalog number: D1626 ; 10 mg/ml, sonicated and boiled)
- SapI (New England Biolabs, catalog number: R0569S )
- ClaI (New England Biolabs, catalog number: R0197S )
- Calf Intestinal Phosphatase (CIP) (New England Biolabs, catalog number: M0290L )
- Custom oligonucleotides (between 18-60 nucleotides in length, Eurofins MWG Operon, Alabama)
- T4 polynucleotide kinase (New England Biolabs, catalog number: M0201S )
- T4 ligase (New England Biolabs, catalog number: M0202L )
- 2x YT (see https://www.elabprotocols.com/protocols/#!protocol=5436 for recipe) (+ 100 µg/ml ampicillin) liquid media and plates (bacterial selection)
- Ampicillin (any source)
- Taq polymerase (any source)
- Polyethylene glycol (PEG) 3350 (Sigma-Aldrich, catalog number: P4338 )
- Lithium acetate (any source)
- NcoI or StuI
- Uridine (any source)
- 0.2% SDS
- Agarose (any source)
- Qiaquick Gel Extraction Kit (QIAGEN, catalog number: 28706 )
- LB
- YPD
- Synthetic dextrose lacking uracil (SD-URA) liquid media and plates (uracil prototrophic yeast selection) (see http://cshprotocols.cshlp.org/content/2015/2/pdb.rec085639.short for recipe)
- SD-complete + 5’ FOA (ura3∆ yeast selection plates) (see http://cshprotocols.cshlp.org/content/2016/6/pdb.rec086637.short for recipe).
Equipment
- Pipettes (capable of accurately pipetting from 1 µl-10 ml) (any source)
- Thermocycler (MJ Research, model: PTC-200 )
- Vortex (any source)
- Micro centrifuge capable of 14 kG (Eppendorf)
- Heating blocks (37 °C, 44 °C)
- Incubators (30 °C, 37 °C)
- DNA gel electrophoresis apparatus
- UV transilluminator (or handheld UV lamp)
- Shaker incubator (or rolling drum) (30 °C, 37 °C)
- Apparatus for media sterilization (autoclave or pressure cooker)
Procedure
The overall procedure entails identification of appropriate target site in the chromosome, design of the gRNA oligonucleotides and their ligation into the gRNA expression plasmid, design and construction of the donor repair fragment, co-transformation of gRNA plasmid and repair fragment into yeast, and finally, screening and verification of mutant strains.
Detailed protocols for each of these steps are described following the timeline outlined below.
Basic protocol/time line for single gene knockout
Day 1-2. Design and order oligonucleotides (see Procedure A)
- 3 pairs of different gRNA targets (if possible, target one PAM in promoter region up to 150 bases upstream the initiating ATG) (~23 bases/oligonucleotide).
- 2 oligonucleotides for donor repair fragment (~60 bases/primer).
- 2 oligonucleotides (forward and reverse) for genotyping.
Day 3-4. Make gRNA plasmid and repair fragment (see Procedure B, C, and D)
- Anneal and fill in donor repair fragment.
- Anneal and ligate gRNA-encoding oligonucleotides into SapI digested and dephosphorylated vector.
- Transform into E. coli competent cells.
Day 4-5. Confirm plasmid inserts (see Procedure B)
- Grow bacterial culture and prepare gRNA plasmid (6-8 h incubation at 37 °C).
- Check gRNA plasmid for loss of ClaI site.
- Digest w/StuI for yeast transformation.
- Put up overnight yeast culture for next day transformation.
Day 6-7. C. albicans transformation (see Procedure E)
- Dilute yeast culture 1:20 for transformation (5 h incubation at 30 °C).
- Transform into C. albicans.
Day 9-10. Genotype mutants (see Procedure F and G)
- Assay Ura+ transformants by PCR
- Streak single colonies to avoid mixture of mutants, recheck by PCR, then stock 2-3 mutant strains.
- Design and order gRNA, knock-out verification oligonucleotides
- Identify a gene or DNA region you wish to target for mutagenesis and find an appropriate gRNA target sequence. The requirement for Cas9 cleavage is the presence of a PAM sequence (5’-NGG-3’) immediately downstream of the ~20 base pair target. This 5’-NGG-3’ can be on either DNA strand. This sequence must be unique in the C. albicans genome to prevent off-target cleavage. A useful web site for identification and ranking of appropriate gRNA is CHOP CHOPv2 (http://chopchop.cbu.uib.no/) (Labun et al., 2016). Generally, we adhere to the ranking of PAM sites obtained using this web site to guide the choice of target site.
- Two complementary oligonucleotides corresponding to the 20 bp double stranded sequence are required. Each oligonucleotide is 23 bases in length and must also contain appropriate sequences at their 5’ end to provide sticky ends for direct ligation of annealed dsDNA into pND494 or pND501 cut with SapI (Figure 1). The choice of which vector to use is based on whether or not subsequent experiments require a uracil prototrophic or auxotrophic strain. If a uracil auxotroph is required, pND501 allows recycling of the Ura- genotype (see Procedure H). The cloning cassette (see Figure 1 below), into which these annealed oligonucleotides are ligated, has been designed to allow precise fusion to both an upstream tRNA sequence and a downstream tracrRNA (Ng and Dean, 2017). This entire tRNA-sgRNA-HDV sequence is driven by the ADH1 promoter in both pND494 and pND501 vectors (Figure 1A). The end result is the expression of an sgRNA flanked with tRNA and ribozymes for precise post-transcriptional processing to produce appropriate 5’ and 3’ ends.
- Figure 1 depicts gRNA expression vector maps (A) and an example of the design of the two complementary gRNA oligonucleotides (B). The top sequence in Figure 1B depicts the ClaI-containing SapI cassette present in the pND494/501 expression vectors. The lower sequence depicts the gRNA-encoding oligonucleotides after annealing and ligation into this cassette. This example shows the ligation of the duplex encoding LEU2 gene-specific gRNA, with the boxed sequences indicating each of the oligonucleotides that were synthesized.
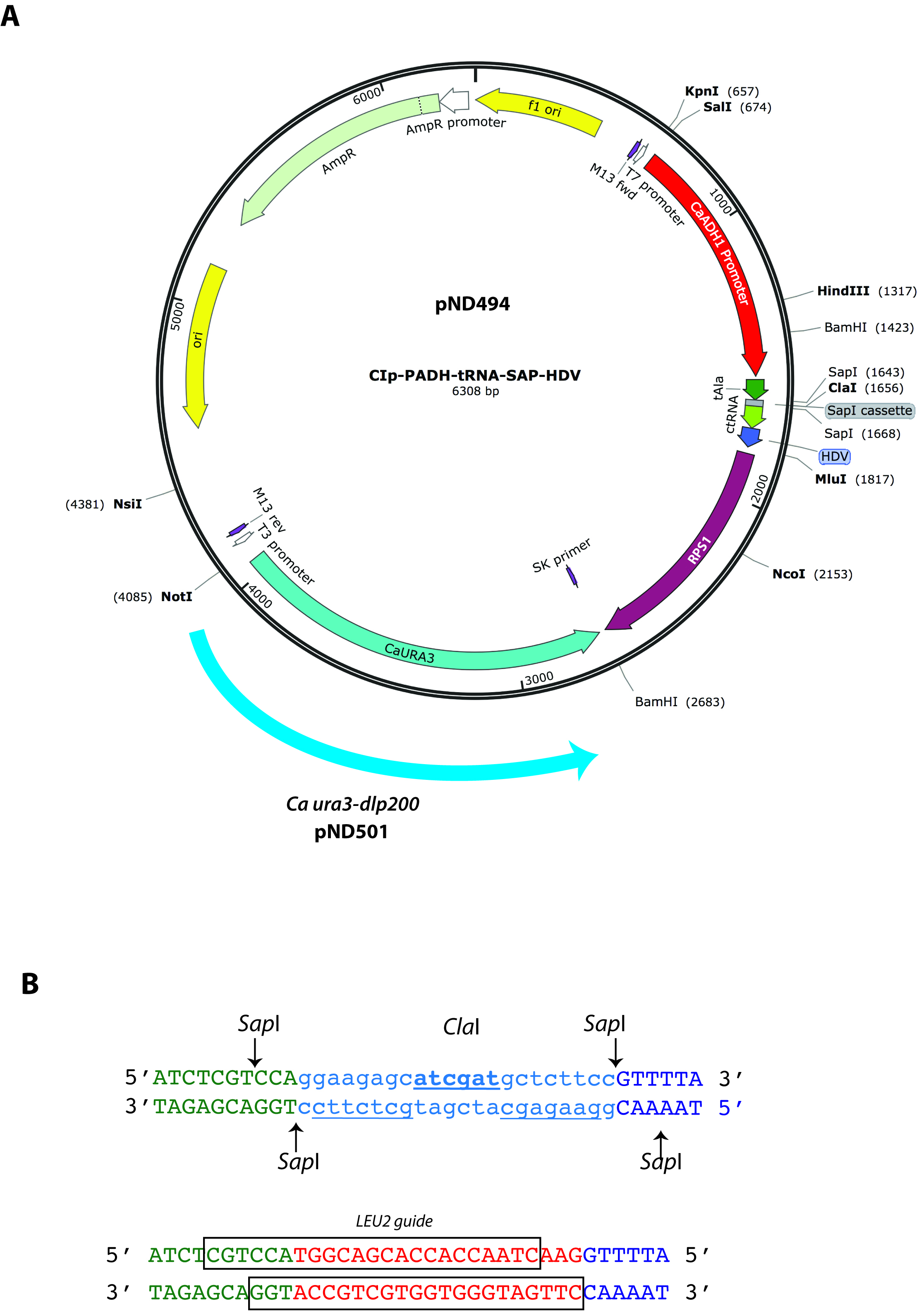
Figure 1. How to design gRNA-encoding oligonucleotides for expression cloning. The pND494 and pND501 cloning vectors (Panel A) contain a SapI cloning cassette designed for direct ligation of annealed gRNA oligonucleotides (Panel B). The DNA sequence of the SapI cloning cassette is indicated in the upper DNA duplex (B). Shown below is an example of the ligation of double-stranded annealed oligonucleotides encoding a LEU2 targeting gRNA (from Ng and Dean, 2017). The boxed in region indicates the sequence of each oligonucleotide. Note that each 23 bases oligonucleotide includes an overhang sequence that results in a sticky end cohesive with that of the vector digested with SapI. Since SapI cleavage is non-palindromic, re-ligation of the vector is not possible thus greatly reducing the background of false-positive E. coli transformants (C4). - When ordering gRNA oligonucleotides, we generally target 3 different PAM sites (which requires 6 different gRNA oligonucleotides) because the efficiency of gRNA-dependent CRISPR mutagenesis varies in a sequence dependent way. Targeting 3 different PAM sites maximizes the probability of obtaining a correct mutant. Experimental evidence suggests that targeting 5’ proximal regions, near the start site of transcription results in higher mutagenesis frequency (Radzisheuskaya et al., 2016). Thus if possible, choose one target within 150 bases upstream (5’) the first initiating ATG methionine codon as one of target sites.
Note: DO NOT INCLUDE PAM SITE in gRNA oligonucleotide. - Design primers for verification of correct chromosomal mutation
Order two ~18-30 bp oligonucleotides, forward and reverse, for checking chromosomal gene deletion, by PCR amplification of genomic DNA. If possible, the product of this PCR amplification should be less than 500 bp. This allows PCR extension time to be less than 30 sec (using Taq polymerase), thereby minimizing the time required for verification.
- Identify a gene or DNA region you wish to target for mutagenesis and find an appropriate gRNA target sequence. The requirement for Cas9 cleavage is the presence of a PAM sequence (5’-NGG-3’) immediately downstream of the ~20 base pair target. This 5’-NGG-3’ can be on either DNA strand. This sequence must be unique in the C. albicans genome to prevent off-target cleavage. A useful web site for identification and ranking of appropriate gRNA is CHOP CHOPv2 (http://chopchop.cbu.uib.no/) (Labun et al., 2016). Generally, we adhere to the ranking of PAM sites obtained using this web site to guide the choice of target site.
- Create an appropriate repair template
A repair template is a double stranded DNA fragment that spans the cut site, has sufficient homology for homologous recombination, and creates appropriate mutation. In addition to introducing the desired mutation, repair templates must lack the PAM site. Otherwise the target break site will be re-introduced into the chromosome after its repair.
The simplest repair template for creating deletion allele uses a pair of 60 bp oligonucleotides with ~47 bp of homology (at 5’ ends) to sequences flanking the chromosomal break site, ~20 bp overlap at 3’ end, and a unique restriction site within the overlap confirmation of correct mutation (e.g., see Vyas et al., 2015; Ng and Dean, 2017). An example that shows repair strategy for deleting the LEU2 gene (from Ng and Dean, 2017) and the sequence of repair oligonucleotides is illustrated in Figure 2.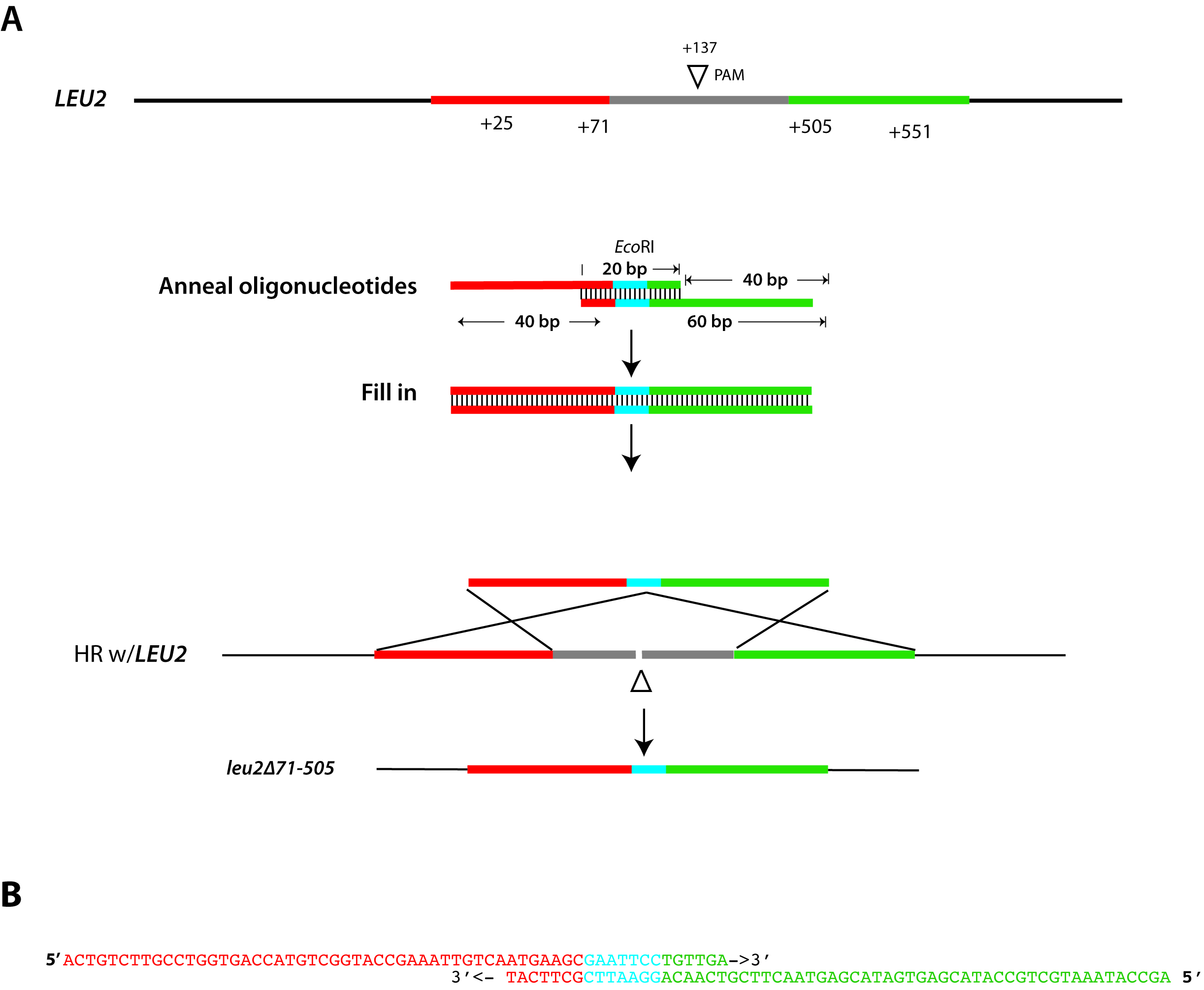
Figure 2. Design of a repair template for gene deletion. Schematic diagram of donor repair fragment synthesis used for deletion of the LEU2 locus, targeting break at the PAM site at position 137 (Panel A) using the oligonucleotides depicted in Figure 1B. Chromosomal sequences homologous to the repair template are colored in red and green. Each 60-mer oligonucleotide ends with a 3’ 20 bp sequence of complementarity, including a restriction site that is absent in LEU2 (EcoRI), shown in blue. When annealed and extended, this repair fragment contains 47 bp of homology to sequence flanking the DSB. In this example, taken from Ng and Dean, 2017, homologous recombination results in a 434 deletion of LEU2 and insertion of a unique EcoRI site, which simplifies genotyping of putative mutants. Panel B shows the sequence of each “repair fragment” oligonucleotide and their alignment. Sequences are color-coded red, blue, and green to indicate the location of each sequence as indicated in Panel A. - Cloning gRNA oligonucleotides into gRNA vector
Both pND494 or pND501 can be used for gRNA expression in any ura3∆ CAS9 C. albicans strain. The only difference in these two vectors is that pND501 has a recyclable URA3 allele (Wilson et al., 2000) if further phenotypic analyses require the loss of URA3 (see below).- Digest 2 μg vector (pND494 or pND501) with SapI
DNA 2 μg 10x buffer 5 μl SapI 1 μl H2O to 50 μl - Incubate at 37 °C for 5-15 min for Fast Digest/High Fidelity enzyme.
- Spin down.
- Add 1 μl calf intestinal phosphatase (CIP) (or Antarctic phosphatase).
- Incubate at 37 °C for 1 h.
- Purify using a QIAquick Gel Column (no need to run it on a gel); elute in 30 μl elution buffer or TE, pH 8.0.
- Incubate at 37 °C for 5-15 min for Fast Digest/High Fidelity enzyme.
- Phosphorylation and annealing sgRNA oligos (from Vyas et al., 2015).
- Add to a PCR tube:
Note: Do a negative control with no oligos.100 μM Oligo 1 (top) 1 μl 100 μM Oligo 2 (bottom) 1 μl 10x T4 ligase buffer 5 μl T4 polynucleotide kinase 1 μl H2O 42 μl - Incubate in a thermocycler:
37 °C for 30 min
95 °C for 5 min
Cool to 16 °C, at the slowest ramp rate your machine can do (ours does 0.1 °C/sec).
- Add to a PCR tube:
- Ligation of annealed sgRNA oligonucleotides
- Assemble in a PCR tube:
10x T4 ligase buffer 1 μl T4 ligase 0.5 μl Annealed Oligo mix 0.5 μl (include a negative control w/no oligos) Vector 20-40 ng H2O to 10 μl - Incubate tubes in a thermocycler or cool/heat blocks:
16 °C for 30 min
65 °C for 10 min
Cool to 25 °C
- Assemble in a PCR tube:
- Transform 5 μl into 50-100 μl competent bacterial DH5α cells.
Plate cells on 2x YT (or LB) + ampicillin plates. Incubate overnight at 37 °C; prepare plasmid DNA using any published protocol. - Screening colonies
This protocol usually results in ~10-20 colonies, with no background from cut vector alone. Identify correct clones by restriction analysis, screening for loss of the ClaI site that is predicted by its replacement in the SapI cassette with gRNA oligonucleotides (note that pND494 contains a single ClaI site, in the cassette, while pND501 contains two ClaI sites). Any potentially correct clone should be verified by DNA sequence analysis. The following primers are useful as for sequencing gRNA ligations especially if these vectors will be used for multiple gene knockouts. These primers (see sequences below) are homologous to the flanking 5’ PADH1 promoter and 3’ HDV ribozyme sequences.
5’ within PADH1 upstream of tRNA (-247 relative to +1)
5’ GCTTATTCAGAATTTTCAGA 3’3’ within HDV (includes the MluI site at 3’ end of HDV)
5’-CGAGATGACGCGTGTCCCATTCGCCAT-3’
- Digest 2 μg vector (pND494 or pND501) with SapI
- Preparation of repair template
To create the repair template using ~60 bp oligonucleotides with ~20 bp overlap at 3’ end, subject mixed oligonucleotides to annealing and DNA replication to generate a product of ~100 bp (see Figure 2).
Per 25 µl reaction:
Incubate tubes in a thermocycler for 30 cycles of 20 sec at 94 °C, 30 sec at 55 °C, and 30 sec at 72 °C.Each oligonucleotide (100 mM stock) 3 µl Taq polymerase 0.5 µl dNTP mix (final 0.2 mM) 2.5 µl 10x buffer 2.5 µl H2O to 25 µl
Transform 20 µl of this reaction per yeast transformation. - C. albicans transformationgRNA vectors are marked with URA3 for selection, and RPS1 to allow integration at the chromosomal RPS1 locus after linearization with StuI. Plasmids are linearized with StuI (or NcoI if gRNA sequence contains a StuI site), transformed into a Cas9-expressing yeast strain (i.e., those in Table 1), and selected on SD (–Ura) plates.
The transformation should include controls lacking gRNA plasmid, as well as those containing just the parental vector (lacking the gRNA sequences). Co-transformation of the donor repair fragment allows for double stranded break repair. Since a break that cannot be repaired is lethal, there is generally at least a two-fold increase in the number of transformants obtained in the presence of repair template compared to its absence.
A modified lithium acetate protocol (Walther and Wendlund, 2003), outlined below, is used for transformation.Transformation (TF) mix (per transformation)50% PEG (sterile) 240 µl 1 M LiOAc (sterile) 32 µl Salmon sperm DNA (10 mg/ml boiled; keep on ice) 5 µl Plasmid DNA (~10 µg, digested with NcoI or StuI) 33 µl Donor repair fragment 25 µl Yeast cell suspension 50 µl Total 360 µl
Yeast transformation protocol (for ~2-3 transformation reactions)- Inoculate fresh colony into YPD + 75 µg uridine/ml. Grow overnight in a shaker at 30 °C.
- Dilute 100 μl from overnight into 2 ml of YPD + uridine, shake at 30 °C for 4-6 h.
- Spin down 1 ml of cells (about 5 OD units), wash with H2O, then wash with 1 ml 0.1 M LiOAc.
- Resuspend in 100 µl of 0.1 M LiOAc (should appear to be thick and concentrated).
- Mix TF mix, 50 µl of yeast cells and 30 µl linearized plasmid DNA (~30 µg) or 15 μl PCR reaction (1-10 μg/μl) so that the total volume is 360 µl.
- Incubate overnight (up to 24 h) at 30 °C.
- Heat shock at 44 °C for 15 min.
- Spread the entire mixture on SD-URA plates. Incubate for 2-3 days at 30 °C.
- Inoculate fresh colony into YPD + 75 µg uridine/ml. Grow overnight in a shaker at 30 °C.
- Mutant verification
To identify the desired mutants, use colony PCR to amplify the flanking region of the wild type gene and mutant alleles. Isolate genomic DNA as follows:- Inoculate a toothpick full of cells from a single colony in ~400 μl of LB media and culture for ~3 h at 37 °C, 220 rpm.
- Spin down cells and resuspend in 30 μl of 0.2% SDS and boil for 4 min.
- After centrifugation of cells, 3 μl of the supernatant (containing genomic DNA) is added to 18.25 μl water, 2.5 μl PCR buffer, 20 μl 2.5 mM dNTP, 1 μl of each primer (10 mM stock) and 0.25 μl Taq polymerase. The PCR conditions are 30 cycles of 0.5 min at 94 °C, 1 min at 55 °C, and 1 min at 72 °C.
- PCR products can be analyzed by agarose gel electrophoresis. It is strongly recommended that prior to this PCR analysis, streak for single colonies to avoid the possibility of mixing colonies of differing genotypes. For an example of these analyses, see Ng and Dean, 2017, Figure 7.
- The design of oligonucleotides used for verification (see Procedure C above) should consider the need to detect a size mobility shift if deletion or alteration produces a smaller PCR product than the wild type allele. As described in Procedure B, susceptibility to restriction digest with a unique enzyme introduced into the donor repair fragment is very useful for mutant verification. In cases where this is not possible, PCR products can be screened directly by DNA sequencing. In either case, genomic sequencing of the deletion alleles should be performed for any mutant strain that will be used for subsequent phenotypic analyses.
- CRISPR-mediated mutation frequency varies depending on the locus and the nature of the mutation, but this procedure generally results in frequencies ranging from 30-95%. Once mutant strains have been obtained and verified, stock 2-3 strains from single colonies in 15% glycerol at -80 °C.
- Inoculate a toothpick full of cells from a single colony in ~400 μl of LB media and culture for ~3 h at 37 °C, 220 rpm.
- Regeneration of ura3 auxotrophic strains
- When using strains transformed with pND501, ura3 mutants that lose the ura3dpl200 allele can be isolated by selection on 5-FOA containing plates (http://cshprotocols.cshlp.org/content/2016/6/pdb.rec086637.short). Prior to 5-FOA selection, streak out the mutant strain on fresh media for single colonies.
- Pick 3-4 colonies and streak them on 5-FOA containing plates. Alternatively, use fresh liquid culture, dilute, and plate out (frequency of looping out is ~1 in 105).
- Dilute overnight culture 1:100, 1:10,000, 1:100,000. Plate 100 µl of each dilution on FOA-containing plate. Screen FOA-resistant colonies on SD-URA plates to confirm the loss of URA3 gene.
- When using strains transformed with pND501, ura3 mutants that lose the ura3dpl200 allele can be isolated by selection on 5-FOA containing plates (http://cshprotocols.cshlp.org/content/2016/6/pdb.rec086637.short). Prior to 5-FOA selection, streak out the mutant strain on fresh media for single colonies.
Data analysis
During yeast transformation, it is important to include the following controls: No plasmid DNA (to ensure that transformants are uracil prototrophs and that SD (–Ura) plates or transformation solutions are not contaminated); no gRNA plasmid (to ensure that transformants arise from gRNA vector uptake), no donor repair fragment. This last control provides an estimate of the number of colonies that are likely to arise from homologous recombination of the repair fragment at the target locus. The fold stimulation of colonies that arise in the presence versus absence of healing fragment may provide an estimate of mutation frequency. Generally, we have found that a high mutation frequency results in a large stimulation of colonies that arise in the presence versus absence of repair fragment. The frequency of mutation and repair by this method is about 30-90%. It should be noted that several other CRISPR-mediated systems for introducing mutations in C. albicans have been described (Vyas et al., 2015; Min et al., 2016; Nguyen et al., 2017), each having its own strength and weakness. It is hoped that as these methods are used more frequently, facile genetic analyses of C. albicans will become the norm.
Acknowledgments
This protocol was adapted primarily from Ng and Dean, 2017. Authors have no conflicts of interest or competing interests. HN was supported in part by a Stony Brook University URECA-Biology Alumni Research award. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Gasiunas, G., Barrangou, R., Horvath, P. and Siksnys, V. (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109(39): E2579-2586.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B. and Valen, E. (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44(W1): W272-276.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Min, K., Ichikawa, Y., Woolford, C. A. and Mitchell, A. P. (2016). Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere 1(3).
- Ng, H. and Dean, N. (2017). Dramatic improvement of CRISPR/Cas9 editing in Candida albicans by increased single guide RNA expression. mSphere 2(2).
- Nguyen, N., Quail, M. M. F. and Hernday, A. D. (2017). An efficient, rapid, and recyclable system for CRISPR-mediated genome editing in Candida albicans. mSphere 2(2).
- Radzisheuskaya, A., Shlyueva, D., Muller, I. and Helin, K. (2016). Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res 44(18): e141.
- Vyas, V. K., Barrasa, M. I. and Fink, G. R. (2015). A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 1(3): e1500248.
- Wilson, R. B., Davis, D., Enloe, B. M. and Mitchell, A. P. (2000). A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast 16(1): 65-70.
- Wilson, R. B., Davis, D. and Mitchell, A. P. (1999). Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol 181(6): 1868-1874.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dean, N. and Ng, H. (2018). Method for CRISPR/Cas9 Mutagenesis in Candida albicans. Bio-protocol 8(8): e2814. DOI: 10.21769/BioProtoc.2814.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > DNA > Chromosome engineering
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.