- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Imaging Cytokine Concentration Fields Using PlaneView Imaging Devices

Published: Vol 8, Iss 7, Apr 5, 2018 DOI: 10.21769/BioProtoc.2788 Views: 7435
Reviewed by: Ivan ZanoniMarco Di GioiaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
We describe here a method to visualize concentration fields of cytokines around cytokine-secreting cells. The main challenge is that physiological cytokine concentrations can be very low, in the pico-molar range. Since it is currently impossible to measure such concentrations directly, we rely on cell’s response to the cytokines–the phosphorylation of a transcription factor–that can be visualized through antibody staining. Our devices aim at mimicking conditions in dense tissues, such as lymph nodes. A small number of secreting cells is deposited on a polylysine-coated glass and covered by multiple layers of cytokine-consuming. The cells are left to communicate for 1 h, after which the top layers are removed and the bottom layer of cells is antibody labeled for the response to cytokines. Then a cross-section of cytokine fields can be visualized by standard fluorescence microscopy. This manuscript summarized our method to quantify the extent of cytokine-mediated cell-to-cell communications in dense collection of cells in vitro.
Keywords: Cytokine concentrationBackground
The mammalian immune system has evolved to identify and limit the spread of potential pathogens while minimizing collateral tissue damage caused by the immune system itself. To achieve this, immune cells rely on a network of cytokine mediators that enable cell-to-cell communications and broadcast information about the magnitude and nature of the pathogenic insult. Vast arrays of different cytokines bind strongly to their cognate receptors, often with characteristic binding affinities in the nano- or pico-molar range. Immunological niches are generated via cytokine communications. For example, in both the bone marrow and the thymus, secretion of Interleukin-7 (IL-7) by stromal cells supports the survival of proliferating B and T cell progenitors, respectively (Tokoyoda et al., 2004; Alves et al., 2009). The size of the cytokine niche controls the number of maturing progenitors, thereby keeping the blood cell compartments in equilibrium (Böyum, 1968; Weist et al., 2015).
We aim to collect information about the spatial and temporal dynamics of cytokines and how these two parameters influence the immune response. This is an area of immunology that is currently under-studied. Many assays test the effects of cytokines in tissue-culture dishes, where media is well-mixed, leading to homogeneous fields of growth and differentiation factors. The intricate and highly specialized architecture of the secondary lymphoid organs sets up niches where cells sense stimuli such as pathogen components and cytokines, proliferate, mature, differentiate, and die. Cytokine concentration gradients are formed within these niches such that some cells have greater or lesser access to cytokines than others (Liu et al., 2015). Measuring how far cytokines spread from their source, and the gradients they form, is key to unravelling the mechanism of the phenotypic heterogeneity of immune cells in differentiation, proliferation, and death (Feinerman et al., 2010; Busse et al., 2010; Höfer et al., 2012; Müller et al., 2012; Thurley et al., 2015).
Due to the typically low concentrations (pM range) of free cytokines in vivo, direct measurement of cytokine fields is difficult at best and maybe impossible. However, due to their high sensitivity to cytokine and graded, concentration-dependent response, the signaling levels of cells in response to cytokines can itself be used as a bio-sensor for cytokine concentrations (Oyler-Yaniv et al., 2017).
In this protocol, we describe how to directly image the signaling response generated around a cytokine producer in vitro, in conditions that mimic in vivo conditions: high cell density and no convection. Our method is general and can be applied to any cell type and any diffusible stimulus, and only depends on the existence of a specific antibody to target the downstream signaling molecule of interest and/or of live cell reporters.
Materials and Reagents
- Pipette tips (USA Scientific, catalog numbers: 1111-1806 , 1111-3800 )
- CELLSTAR Filter Cap Cell Culture Flasks T75 (Greiner Bio One International, catalog number: 658175 )
- 15 ml tube
- Glass slides (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 4951PLUS4 )
- Coverslips (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 25X25-1 )
- Silicone rubber compound (PDMS) (Momentive, catalog number: RTV615 )
- PVP-treated PCTE Membranes, 13 mm diameter, 400 nm pore (Sterlitech, catalog number: PCT0413100 )
- B16-F10 melanoma cells (ATCC, catalog number: CRL-6475 )
- Mouse CD4 (L3T4) MicroBeads (Miltenyi Biotec, catalog number: 130-049-201 )
- Phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, catalog number: P1585-1MG )
- Ionomycin calcium salt (Sigma-Aldrich, catalog number: I0634-1MG )
- Ficoll-paque plus (GE Healthcare, catalog number: 17144003 )
- Recombinant mouse IL-2 (Thermo Fisher Scientific, eBioscienceTM, catalog number: 14-8021-64 )
- Recombinant human IL-2 (gift from Dr. Kendall A. Smith, Cornell University)
- Trypsin/EDTA solution (Thermo Fisher Scientific, GibcoTM, catalog number: R001100 )
- Phosphate buffered saline (Sigma-Aldrich, catalog number: P4417 )
- Glycine (Sigma-Aldrich, catalog number: 50046 )
- Ovalbumin peptide SIINFEKL (Sigma-Aldrich, catalog number: S7951-1MG )
- Cell Trace Far-Red (DDAO-SE) (Thermo Fisher Scientific, InvitrogenTM, catalog number: C34564 )
- Poly-L-lysine solution (Sigma-Aldrich, catalog number: P8920-100ML )
- Paraformaldehyde solution, 4% in PBS (Alfa Aesar, Affymetrix, catalog number: J19943 )
- Methanol (Sigma-Aldrich, catalog number: MX0490-4 )
- Fluoromount Aqueous Mounting Medium (Sigma-Aldrich, catalog number: F4680-25ML )
- Triton 100-X (MP Biomedicals, catalog number: 0230022101-1l )
- α-CD4, Alexa700, Pacific Blue (BD Bioscience clone RM4-5, BD, catalog numbers: 557956 , 558107 )
- α-IL-2Rα, PE (Miltenyi Biotec clone 7D4, Miltenyi Biotec, catalog number: 130-102-593 )
- Primary antibody rabbit α-phospho-STAT5 (pY694) (Cell Signaling clone C71E5, Cell Signaling Technology, catalog number: 9314S )
- Primary antibody rabbit α-phospho-STAT1 (pY701) (Cell Signaling clone 58D6, Cell Signaling Technology, catalog numbers: 9167L )
- Secondary polyclonal antibody α-rabbit IgG, Alexa 488 (Jackson ImmunoResearch, catalog number: 711-176-152 )
- RPMI 1640 media with L-glutamine (Biological Industries, catalog number: 01-100-1A )
- Heat-inactivated fetal bovine serum (Biological Industries, catalog number: 04-127-1A )
- HEPES buffer (Biological Industries, catalog number: 03-025-1B )
- Non-essential amino acids (Biological Industries, catalog number: 01-340-1B )
- Sodium pyruvate (Biological Industries, catalog number: 03-042-1B )
- Penicillin-streptomycin solution (Biological Industries, catalog number: 03-031-5B )
- β-Mercaptoethanol (Sigma-Aldrich, catalog number: M3148-25ML )
- Complete RPMI (see Recipes)
Equipment
- Pipettes
- Heraeus centrifuge with microplate swinging rotor (Thermo Fisher Scientific, Thermo ScientificTM, model: HeraeusTM BiofugeTM StratosTM )
- Zeiss Axiovert 200M microscope (ZEISS, model: Axiovert 200M )
Software
- MATLAB, Mathworks Inc.
- LabVIEW, National Instruments
Procedure
The protocols described here were developed for imaging of cytokine signaling in dense clusters of cytokine consuming cells that are simulated in vivo conditions in secondary lymphoid organs or other solid tissue (Oyler-Yaniv et al., 2017).
- Cell culture
- OT-I and C57BL/6 primary cells are harvested from the lymph nodes and spleen, and mechanically separated into single-cell suspension. CD4+ T-cells are isolated using Mouse CD4 (L3T4) MicroBeads. Primary cells and B16-F10 melanoma cells (ATCC CRL-6475) are maintained in complete RPMI (Recipe 1) throughout the procedure. C57BL/6 T cells are activated using 10 ng/ml PMA and 500 ng/ml Ionomycin and cultured in 30 ml of media, in a T75 flask for 3 days.
- Dead cells are removed using Ficoll-paque plus density gradient media following standard protocols (Böyum, 1968) and subsequently cultured at 106 cells/ml in RPMI supplemented with 2 nM recombinant human IL-2 for an additional day. IL-2 secretion experiments are performed on day 4 of culture.
- B16-F10 cells are maintained in 30 ml of complete RPMI (Recipe 1) in a T75 flask and passaged every 3 days using trypsin/EDTA solution and recultured to 10% confluency (roughly 106 cells at the start of each passage). Cells are never used beyond passage 7.
Note: B16-F10 melanoma cells express the IFNγ receptor but do not produce IFNγ. They also express high levels of MHC-I and therefore serve as antigen presenting cells for TCR recognition.
- OT-I and C57BL/6 primary cells are harvested from the lymph nodes and spleen, and mechanically separated into single-cell suspension. CD4+ T-cells are isolated using Mouse CD4 (L3T4) MicroBeads. Primary cells and B16-F10 melanoma cells (ATCC CRL-6475) are maintained in complete RPMI (Recipe 1) throughout the procedure. C57BL/6 T cells are activated using 10 ng/ml PMA and 500 ng/ml Ionomycin and cultured in 30 ml of media, in a T75 flask for 3 days.
- Preparation of cells for imaging
- To remove receptor-bound cytokines, wash 5 x 107 cultured T-cells (consumer cells) in 10 ml PBS, then expose 0.1 M glycine in PBS (pH 4.0) for 1 min on ice, followed by 3 washes each with 10 ml RPMI, and rest for 1 h at in a tube containing RPMI in a 37 °C incubator.
- To generate CD4 depleted, naïve splenocytes (inert cells), harvest lymph nodes and spleen from a C57BL/6 mouse. Separate tissue into single-cell suspension. Deplete the cell suspension of CD4+ cells using Mouse CD4 (L3T4) MicroBeads.
- Mix cultured consumer cells with inert cells at different ratios.
- Re-administrate the previously activated T-cells cultures with 5 ng/ml PMA and 500 ng/ml Ionomycin to generate IL-2 producing cells. This reactivation leads to a rapid and extensive production of IL-2 by the T-cells. These cells are labeled with DDAO-SE by using the manufacturer’s protocol at 1 µM for subsequent identification. Only the cytokine producers are labeled at this stage!
- Pulse 107 B16-F10 cells with 1 nM of the SIINFEKL peptide in 10 ml of RPMI for 1 h under constant rotation in a 15 ml tube, in an incubator. OT-I Cells are labeled with DDAO-SE by using the manufacturer’s protocol at 1 µM for subsequent identification.
- To remove receptor-bound cytokines, wash 5 x 107 cultured T-cells (consumer cells) in 10 ml PBS, then expose 0.1 M glycine in PBS (pH 4.0) for 1 min on ice, followed by 3 washes each with 10 ml RPMI, and rest for 1 h at in a tube containing RPMI in a 37 °C incubator.
- General description: Fabrication of imaging devices
- Coat glass slides with poly-L-lysine (PLL) by submerging them in 0.01% PLL, diluted in H2O, for 40 min at 37 °C. Wash slides by submerging them in a large volume of H2O and allow them to dry at room temperature for 1 h. Place a small (6 mm) hollow cylinder made of PDMS on the slide, the PDMS rapidly attaches to the slide creating a small well.
- To create a tightly packed cell pellet, cell suspensions are added into the PDMS well, the slide is put inside a pipette box cover and centrifuged for 1 min at 800 x g to allow cells to stick to the glass slide (Figure 1A). As more cells accumulate on the device, they form a 3 dimensional layered structure. Depending on cell dimensions, each layer will contain ~1-2 x 105 individual cells. Moreover, different layers composed of different cell preparations can be added sequentially, creating a stratified structure that can mimic specific in vivo morphologies.
- After cell deposition, the PDMS well is removed and a 13 mm diameter semipermeable hydrophilic membrane is dipped in media and carefully placed on top of the cells. This membrane protects the cells from moving due to changing reagents, and prevents convection flows from distorting the concentration fields. The cells and membrane are then covered by a larger PDMS well (12 mm diameter) to allow for the confinement of reagents around the cells. At the end of the experiment, the cells are fixed for 20 min in 200 μl 37 °C 4% PFA and permeabilized by using 200 μl ice cold 90% MeOH for 10 min. After incubation and fixation, cells that are in contact with the glass slide remain permanently bound to it, preserving their spatial distribution. The PDMS well and the membrane are then removed using forceps, the slides are stained using standard immunofluorescence protocols, and finally are mounted on a coverslip using Fluoromount (Figure 1B). The process is illustrated in Figure 2.

Figure 1. Preparation of slides for imaging. A. PlaneView device after centrifugation; B. Cell patch after staining and mounting.
Figure 2. Graphical protocol of device preparation. Preparation of cells for imaging is done by first coating a glass slide with poly-L-lysine (1). Then, a cell suspension containing a small fraction of cytokine producing cells is deposited in a monolayer by centrifugation (2, 3). 10 layers of cells containing no producers is deposited on top to form a 3 dimensional structure (4, 5). The cells are covered with a semipermeable membrane (6), incubated for 1 h (7), and fixed in situ (8). Cells are then permeabilized and stained (9, 10).
- Coat glass slides with poly-L-lysine (PLL) by submerging them in 0.01% PLL, diluted in H2O, for 40 min at 37 °C. Wash slides by submerging them in a large volume of H2O and allow them to dry at room temperature for 1 h. Place a small (6 mm) hollow cylinder made of PDMS on the slide, the PDMS rapidly attaches to the slide creating a small well.
- In vitro imaging of cytokine concentration fields
- For measuring IL-2 concentration fields (Figure 3), 2 x 105 IL-2 consuming T cells, or a combination of 10% consuming T cells (2 x 104) and 90% inert cells (1.8 x 105) are mixed with 0.1% IL-2 producing T cells (200 cells), each in a total volume of 20 μl, and deposited in a monolayer. Then, 10 more layers (2 x 106) of cells containing no producers and either a 1:0 or a 1:9 ratio of consumers to inert cells, respectfully, each in a volume of 180 μl, are added on top, forming a three dimensional strata with the producing cells dispersed on the bottom (Figure 2). A semipermeable membrane is placed on the cells to preserve their positions during further processing.
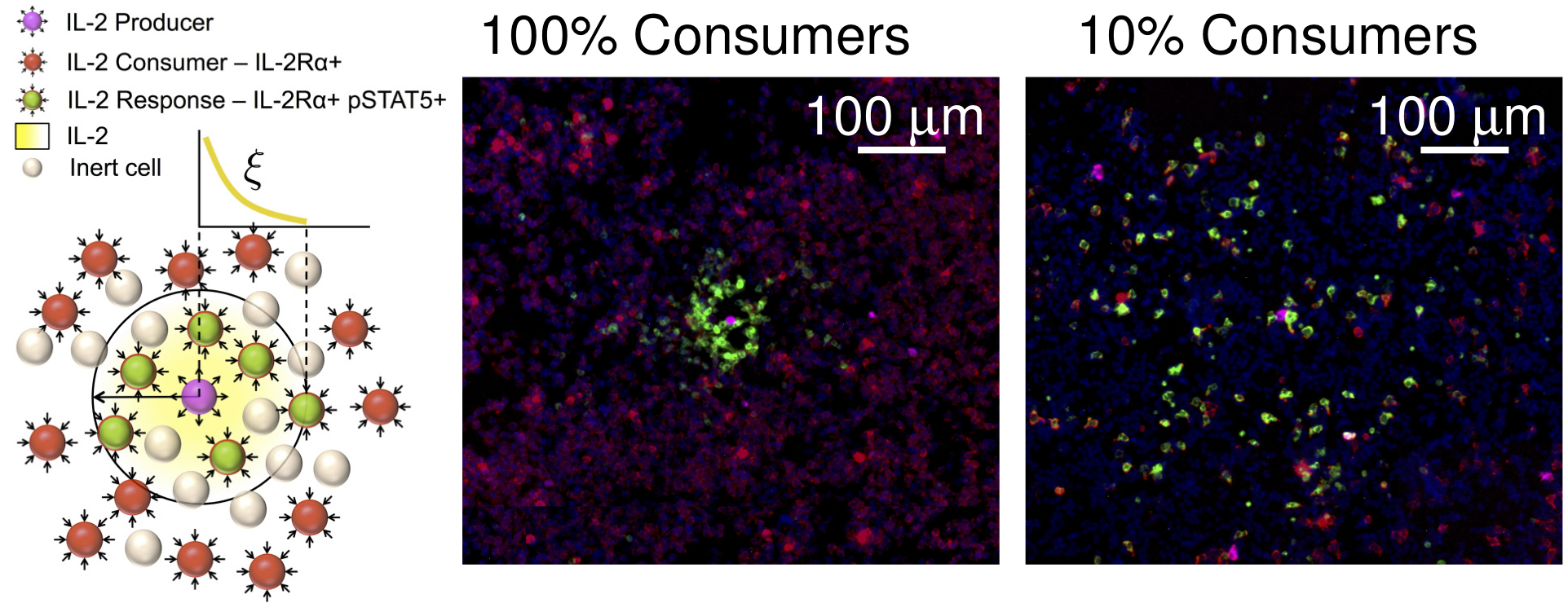
Figure 3. pSTAT5 distribution around IL-2 producers. Immunofluorescence staining of cell preparations containing either 100% IL-2Rα+ consuming cells or 10% consuming cells and 90% IL-2Rα- inert cells, and a small number (< 0.01%) of IL-2 producing T cells in a PlaneView imaging device. Blue: DAPI, Red: IL-2Rα, Green: pSTAT5, Magenta: DDAO-SE. - For measuring IFNγ concentration fields (Figure 4), 105 SIINFEKL pulsed B16-F10 melanoma cells are mixed with 0.2% OT-I T cells (200 cells) and deposited in a monolayer. Then, 10 more layers of B16-F10 cells (106) are added on top. A semipermeable membrane is placed on the cells to preserve their positions during further processing.
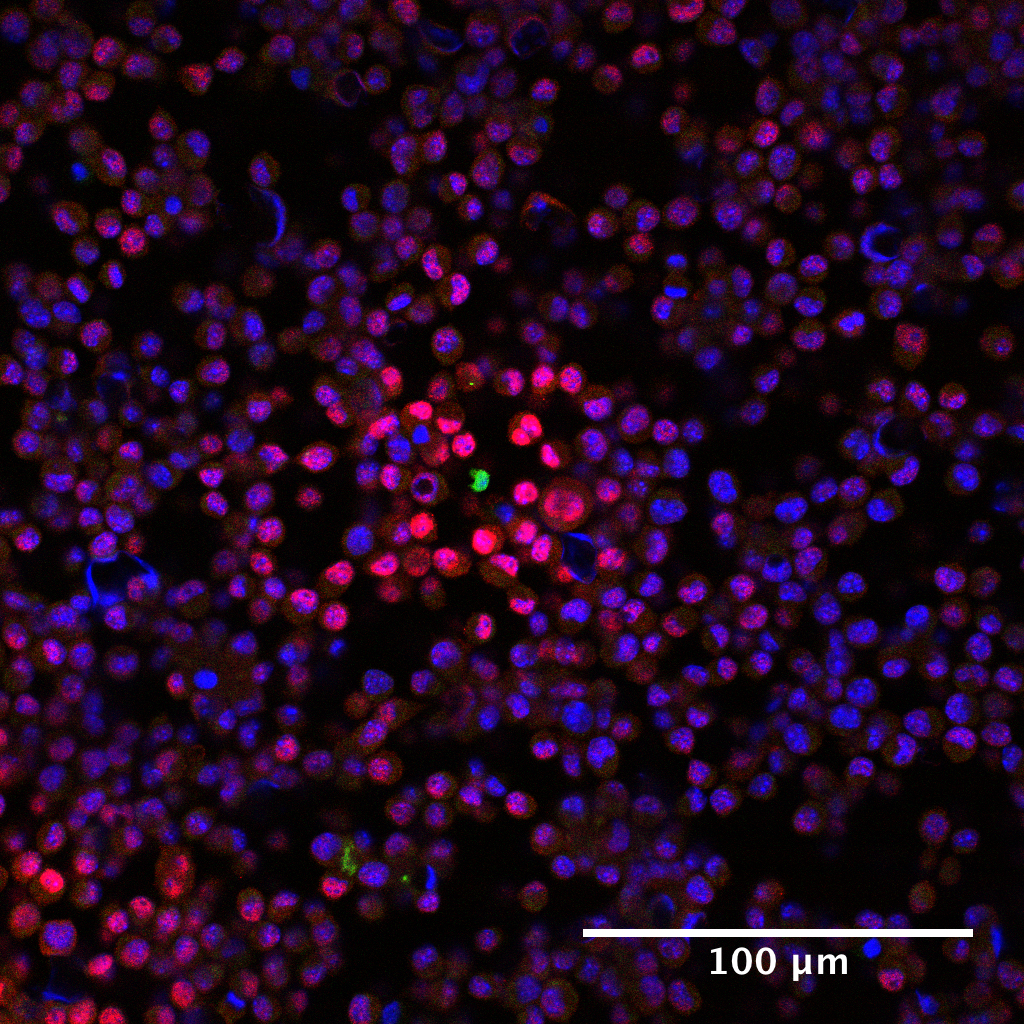
Figure 4. pSTAT1 distribution around IFNγ producer. Immunofluorescence staining of cell preparation containing SIINFEKL pulsed B16-10A cells spiked with a small amount of OT-I cells was prepared as described in Procedure D. Blue: DAPI, Red: pSTAT1, Green: DDAO-SE. - To control for background transcription factor phosphorylation and cytokine specificity, devices are loaded with consuming cell cultures containing no producers. The cells are then covered with either fresh media or media containing 10 nM of cytokine to serve as negative and positive controls, respectively. These samples showed effectively no signal for the negative control and bright, uniform, signal for the positive control (Oyler-Yaniv et al., 2017).
- The system is incubated at 37 °C for 1 h. After that, media is carefully aspirated off the devices and the cells are fixed for 20 min at 37 °C in 4% PFA. PFA is then removed and the cells are permeabilized by using 200 μl ice cold 90% MeOH for 10 min to allow for intracellular immunostaining. Special attention should be given during this stage to minimally disrupt the cell pellets.
- After fixation and permeabilization, the PDMS well and the membrane are removed using forceps. At this stage, the cells would be tightly bound to the glass slide and standard immunostaining protocols can be used. Nonspecific antibody binding is blocked by a 1 h incubation in 5% FBS and 0.3% Triton X-100 in PBS at room temperature. Primary antibodies (Rabbit anti-pSTAT5 1:200) are applied in a moist chamber for 1 h at room temperature. Fluorophore-conjugated antibodies (Goat anti-Rabbit Alexa 488, Rat anti-IL-2Rα R-PE, 1:300) are applied for 1 h at room temperature. Cells are then briefly stained with DAPI and a coverslip is mounted using Fluoromount (Figure 1B) for fluorescent imaging.
- For measuring IL-2 concentration fields (Figure 3), 2 x 105 IL-2 consuming T cells, or a combination of 10% consuming T cells (2 x 104) and 90% inert cells (1.8 x 105) are mixed with 0.1% IL-2 producing T cells (200 cells), each in a total volume of 20 μl, and deposited in a monolayer. Then, 10 more layers (2 x 106) of cells containing no producers and either a 1:0 or a 1:9 ratio of consumers to inert cells, respectfully, each in a volume of 180 μl, are added on top, forming a three dimensional strata with the producing cells dispersed on the bottom (Figure 2). A semipermeable membrane is placed on the cells to preserve their positions during further processing.
Data analysis
Images are processed by segmenting individual cells (Gonzalez and Woods, 2007), then determining whether each cell is a consumer based on IL-2Rα expression. On each consumer, the total level of pSTAT5 expression and the center-of-mass is calculated and logged. An example of the procedure is shown in Figure 5. Profiles of pSTAT5 expression on individual cells as a function of distance from the nearest producer are then generated (Figure 6).
Figure 5. Image processing procedure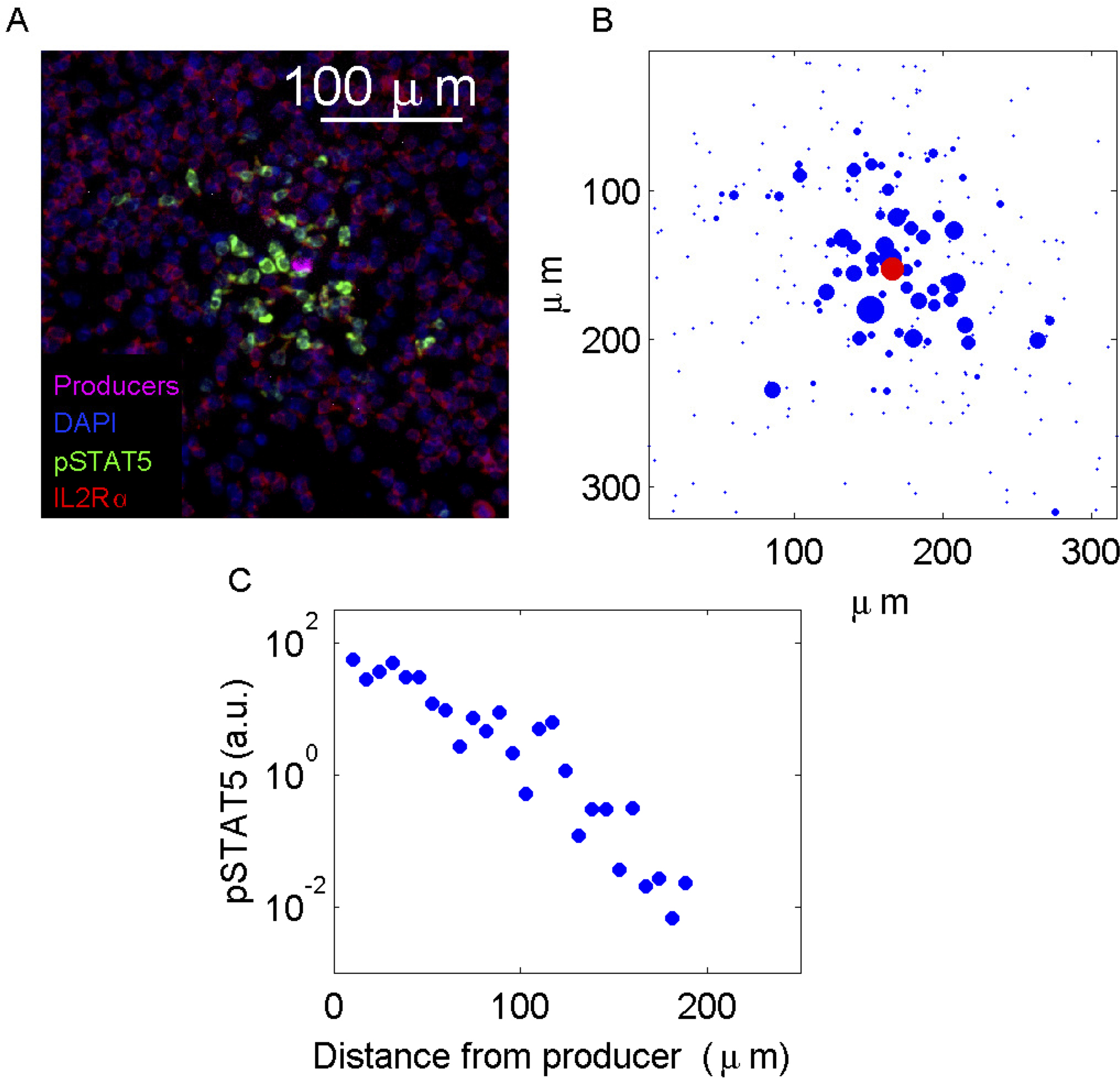
Figure 6. Analysis of pSTAT5 distributions. A. A sample containing 100% IL-2 consuming T cells spiked with a small amount of IL-2 producing cells was prepared as described in Procedure D. B. Image reconstruction based on total pSTAT5 and center of mass per cell. The radius of each circle is proportional to the pSTAT5 level. C. pSTAT5 profile as a function of the distance from the cytokine producer.
Recipes
- Complete RPMI
RPMI 1640 media supplemented with:
Heat-inactivated 10% fetal bovine serum
2 mM L-glutamine
10 mM HEPES
0.1 mM non-essential amino acids
1 mM sodium pyruvate
100 µg/ml of penicillin
100 µg/ml of streptomycin
50 µM β-mercaptoethanol
Acknowledgments
This work has been done in collaboration with Grégoire Altan-Bonnet (NIH) whom we would like to thank for the overall support and, in particular, for the critical reading of the manuscript. We are also grateful to U.S.-Israel Binational Science Foundation (grant #2012327 to G. Altan-Bonnet and O.K.) for funding. The protocol has been adapted from Oyler-Yaniv et al., 2017.
The Authors declare no conflicts of interest or competing interests.
References
- Alves, N. L., Richard-Le Goff, O., Huntington, N. D., Sousa, A. P., Ribeiro, V. S., Bordack, A., Vives, F. L., Peduto, L., Chidgey, A., Cumano, A., Boyd, R., Eberl, G. and Di Santo, J. P. (2009). Characterization of the thymic IL-7 niche in vivo. Proc Natl Acad Sci U S A 106(5): 1512-1517.
- Böyum, A. (1968). Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 97: 77-89.
- Busse, D., de la Rosa, M., Hobiger, K., Thurley, K., Flossdorf, M., Scheffold, A. and Hofer, T. (2010). Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci U S A 107(7): 3058-3063.
- Feinerman, O., Jentsch, G., Tkach, K. E., Coward, J. W., Hathorn, M. M., Sneddon, M. W., Emonet, T., Smith, K. A. and Altan-Bonnet, G. (2010). Single-cell quantification of IL-2 response by effector and regulatory T cells reveals critical plasticity in immune response. Mol Syst Biol 6: 437.
- Gonzalez, R. C. and Woods, R. E. (2007). Digital Image Processing (3rd Edition).
- Höfer, T. O. Krichevsky, O, and Altan-Bonnet, G. (2012). Competition for IL-2 between regulatory and effector T cells to chisel immune responses. Front Immunol 3: 268.
- Liu, Z., Gerner, M. Y., Van Panhuys, N., Levine, A. G., Rudensky, A. Y. and Germain, R. N. (2015). Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528(7581): 225-230.
- Müller, A. J., Filipe-Santos, O., Eberl, G., Aebischer, T., Spath, G. F. and Bousso, P. (2012). CD4+ T cells rely on a cytokine gradient to control intracellular pathogens beyond sites of antigen presentation. Immunity 37(1): 147-157.
- Oyler-Yaniv, A., Oyler-Yaniv, J., Whitlock, B. M., Liu, Z., Germain, R. N., Huse, M., Altan-Bonnet, G. and Krichevsky, O. (2017). A tunable diffusion-consumption mechanism of cytokine propagation enables plasticity in cell-to-cell communication in the immune system. Immunity 46(4): 609-620.
- Thurley, K., Gerecht, D., Friedmann, E. and Hofer, T. (2015). Three-dimensional gradients of cytokine signaling between T cells. PLoS Comput Biol 11(4): e1004206.
- Tokoyoda, K., Egawa, T., Sugiyama, T., Choi, B. I. and Nagasawa, T. (2004). Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20(6): 707-718.
- Weist, B. M., Kurd, N., Boussier, J., Chan, S. W. and Robey, E. A. (2015). Thymic regulatory T cell niche size is dictated by limiting IL-2 from antigen-bearing dendritic cells and feedback competition. Nat Immunol 16(6): 635-641.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Oyler-Yaniv, A. and Krichevsky, O. (2018). Imaging Cytokine Concentration Fields Using PlaneView Imaging Devices. Bio-protocol 8(7): e2788. DOI: 10.21769/BioProtoc.2788.
Category
Immunology > Immune cell function > Cytokine
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.