- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Hydrogen Deuterium Exchange Mass Spectrometry of Oxygen Sensitive Proteins
Published: Vol 8, Iss 6, Mar 20, 2018 DOI: 10.21769/BioProtoc.2769 Views: 10250
Reviewed by: Vamseedhar RayaproluPaul FinchAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The protocol detailed here describes a way to perform hydrogen deuterium exchange coupled to mass spectrometry (HDX-MS) on oxygen sensitive proteins. HDX-MS is a powerful tool for studying the protein structure-function relationship. Applying this technique to anaerobic proteins provides insight into the mechanism of proteins that perform oxygen sensitive chemistry. A problem when using HDX-MS to study anaerobic proteins is that there are many parts that require constant movement into and out of an anaerobic chamber. This can affect the seal, increasing the likelihood of oxygen exposure. Exposure to oxygen causes the cofactors bound to these proteins, a common example being FeS clusters, to no longer interact with the amino acid residues responsible for coordinating the FeS clusters, causing loss of the clusters and irreversible inactivation of the protein. To counteract this, a double vial system was developed that allows the preparation of solutions and reaction mixtures anaerobically, but also allows these solutions to be moved to an aerobic environment while shielding the solutions from oxygen. Additionally, movement isn’t limited like it is in an anaerobic chamber, ensuring more consistent data, and fewer errors during the course of the reaction.
Keywords: HDX-MSBackground
Many oxygen sensitive proteins are required for organisms to thrive in an anoxic environment. Some of these proteins provide an alternative supply of energy to anaerobic microbes through a process known as Flavin-based electron bifurcation (FBEB) (Lubner et al., 2017). FBEB generates reduced ferredoxin, which can be oxidized to produce energy. Proteins that are capable of reducing ferredoxin are of great interest and have been the focus of recent studies using HDX-MS (Demmer et al., 2016; Lubner et al., 2017; Berry et al., 2018). HDX-MS is a powerful technique for investigating protein stability, dynamics, and ligand binding providing information about the relationship between structure and function. HDX-MS uses the intrinsic property of amide hydrogens to exchange with hydrogens in solution to track changes in the structure and dynamics of a protein/protein complex. By preparing buffers with heavy water (D2O) instead of monoisotopic water (H2O), amide hydrogens on a protein will exchange with the deuterium in solution. The rate of exchange for a given amino acid is influenced by the stability of hydrogen bonds in the secondary structure, as well as the tertiary and quaternary interactions within a single protein or protein complex. Using mass spectrometry, deuterium incorporation is determined by measuring the shift in isotope distribution between deuterated and non-deuterated samples. HDX-MS has been applied to a large number of proteins and protein complexes across a wide range of conditions. To successfully study these proteins with HDX-MS, it was imperative to establish a means of performing this reaction on the benchtop to avoid heavy traffic into and out of an anaerobic chamber which is time consuming and burdensome. The problem was then how to allow manipulation of the sample while keeping the protein sample anaerobic for an extended period of time in an aerobic environment. To solve this problem, the reaction mixture and protein stock solutions were placed into a double vial system that allowed addition and removal of sample while maintaining strict anaerobic conditions. The logic behind the setup was to create an airlock. Vials are placed under positive pressure with nitrogen gas with a screw cap vial, inside a larger crimp vial that contains reductant. With this double barrier system, small volumes of air can be trapped in the outer vial and do not contact the sample.
Materials and Reagents
- VerexTM vial kit, 9 mm, screw top, polypropylene, 300 μl + PTFE/silicone cap, blue, 1,000/pk (Phenomenex, catalog number: AR0-9991-13 )
- Clear glass serum vial with 20 mm crimp top finish, 10 ml, 100/case (DWK Life Sciences, WHEATON, catalog number: 225278 )
- Curwood Parafilm MTM laboratory wrapping film (Bemis, catalog number: PM996 )
- Costar Microcentrifuge Tubes 0.65 ml, 500/bag (Corning, catalog number: 3208 )
- Onyx Monolithic C18 column, 100 x 2 mm (Phenomenex, catalog number: CH0-8467 )
Alternative column (see Notes for a detailed explanation): Onyx Monolithic C18 column, 100 x 3 mm (Phenomenex, catalog number: CH0-8158 ) - Model 1701 and 1702 small RN syringes, 10 μl (26s gauge) and 25 μl (22s gauge), 2” needle point style 2 (Hamilton, catalog numbers: 80030 and 80230 )
- Unlined aluminum open-top seals, 20 mm, 1,000/case (DWK Life Sciences, WHEATON, catalog number: 224178-05 )
- 20 mm stopper, straight plug, ultra-pure (DWK Life Sciences, WHEATON, catalog number: W224100-405 )
- 200 μl Pipet Tips (VWR, catalog number: 53508-810 )
- Liquid nitrogen
- Purified ferredoxin (Fd) in 50 mM Ammonium Acetate buffer at pH 6.8 in H2O (stock concentration 150 μM) from Pf
Note: pH adjusted with 1 N HCl. - Purified NADH-dependent ferredoxin-NADP+ oxidoreductase (Nfn) in 20 mM Tris, 150 mM NaCl buffer at pH 8 in H2O (stock concentration 16.5 mg/ml) from the organism Pyrococcus furiosus (Pf)
Note: pH adjusted with 1 N HCl. - Sodium dithionite (Merck, catalog number: 1065051000 )
- Nicotinamide adenine dinucleotide (NAD+, Cayman Chemical, catalog number: 16077 , 500 mg)
- Nicotinamide adenine dinucleotide (NADH, Cayman Chemical, catalog number: 16078 , 500 mg)
- Nicotinamide adenine dinucleotide phosphate (NADP+, Cayman Chemical, catalog number: 10004675 , 50 mg)
- Nicotinamide adenine dinucleotide phosphate (NADPH, Cayman Chemical, catalog number: 9000743 , 25 mg)
- 99.5% formic acid (Fisher Scientific, catalog number: A117-50 )
- Pepsin from porcine gastric mucosa (Sigma-Aldrich, catalog number: P6887-1G )
- Sodium acetate trihydrate (Fisher Scientific, catalog number: S607-500 )
- Ammonium acetate, ≥ 99% (Sigma-Aldrich, catalog number: 09689-250G )
- 37% hydrochloric acid (= 12.1 M) (Merck, catalog number: HX0603-3 )
- Sodium hydroxide (Fisher Scientific, catalog number: BP359-500 )
- Deuterium oxide (Sigma-Aldrich, catalog number: 151882-100G )
- Tris base (Merck, catalog number: 648311-5KG )
- Sodium chloride (Fisher Scientific, catalog number: BP358-212 )
- HPLC grade water (Fisher Scientific, catalog number: W5-4 )
- HPLC grade acetonitrile (Fisher Scientific, catalog number: A998-4 )
- Nanopure water (purified in-house using a Millipore Q-Gard 2)
- Tris base, NaCl buffer (pH 7.0) in deuterium oxide (see Recipes)
- Tris base, NaCl buffer (pH 7.0) in H2O (see Recipes)
- Tris base, NaCl, sodium dithionite buffer (pH 7.0) in H2O (see Recipes)
Equipment
- HPLC stack for separation of peptides generated via pepsin digestion (e.g., 1290 Infinity series HPLC stack manufactured by Agilent Technologies) (Agilent Technologies, model: 1290 Infinity Series )
- LC/MS Q-TOF system for sample analysis/data acquisition (e.g., 6538 UHD Accurate-Mass Q-TOF LC/MS manufactured by Agilent Technologies) (Agilent Technologies, model: 6538 UHD Accurate-Mass Q-TOF LC/MS )
- Glove box capable of maintaining anaerobic conditions under positive inert gas pressure (e.g., MBraun, model: UNIlab Plus Glove Box Workstation )
- Nitrogen tank
- 20 mm Kebby standard crimper for aluminum seals (Medical Laboratory Supply, catalog number: 2001-00-C01A )
- Fisher Scientific isotemp 110 water bath (Fisher Scientific, model: FisherbrandTM IsotempTM, catalog number: S63077Q )
Note: This product has been discontinued. - Milli-Q purification system (Merck, catalog number: QGARD00D2 )
- Pipettes (10 μl and 100 μl) (Eppendorf, catalog numbers: 022478886 and 022478924 )
Software
- Microsoft Excel 2016 on Windows 7
- UCSF Chimera v. 1.11.2
- MassHunter Workstation Software Qualitative Analysis v. B.06.00 (Agilent Technologies)
- HDExaminer v. 1.3.0 beta 6 (Sierra Analytics, Inc.)
- MassHunter Workstation Software LC/MS Data Acquisition for 6200 series TOF/6500 series Q-TOF v. B.05.01 (Agilent Technologies)
- Peptide Analysis Worksheet Freeware Edition (PAWs, ProteoMetrics–freeware edition)
- SearchGUI v. 3.2.18 (Compomics)
- Peptide Shaker v. 1.16.9 (Compomics)
Procedure
- Reaction preparation
- Before preparation of stock solutions and reaction mixtures, determine the number of time points. The number of time points dictates the volume of the reaction mixtures. For every time point, 10 μl must be available to add to the quench solution, plus an additional 10 μl for the 24 h/fully deuterated time point. For instance, in the HDX experiment of Nfn, five time points were measured (1 min, 3 min, 15 min, 60 min, 3 h, and 24 h) (Berry et al., 2018). An additional 10 μl of reaction solution is included to prevent withdrawing the full reaction solution, thus bringing the total volume to 70 μl.
Note: Time points are chosen to provide data on fast, medium, and slow exchanging residues. Data spanning multiple time scales facilitates a deeper interpretation of the results. While short time points can be very informative on protein dynamics, care should be taken to allow time to withdraw sample from the reaction chamber and quench in a reproducible manner. - Prepare stock solutions and reaction mixtures under anaerobic conditions, using an MBraun glove box fed by a nitrogen tank set to 40 PSI. The anaerobic atmosphere inside the glove box is maintained by keeping a positive nitrogen pressure. The exact gas flow will depend on the specific set up, however, the gas sensor monitoring conditions inside the glove box should register 0 ppm O2 and H2O and a positive pressure of 2 mbar.
- Prepare two anaerobic aliquots of purified Nfn (stock concentration 16.5 mg/ml) in an MBraun glove box by adding 30 μl of Nfn into one Verex vial, and 10 μl of Nfn into another Verex vial. Purified Fd is then added to the 10 μl aliquot of Nfn in a 1:1 molar ratio. Using a double vial system, cap the Verex vials, and place inside a clear glass serum vial that contains 1 ml of degassed Tris-HCl buffer with sodium dithionite, and seal using a crimper. Remove sealed vials from the glove box, wrap in Parafilm, and store at 4 °C prior to reaction initiation.
Note: Only 1 ml of reductant containing buffer is added to the glass serum vial. Any more volume causes the Verex vial to slightly float, making it difficult to withdraw sample at the designated time points. - Make deuterated buffers with 1 mM of each pyridine nucleotide in the following combinations: NAD+, NADH, NADP+, NADPH, NADH + NADP+, and NADPH + NAD+. Also prepare an additional stock of the same buffer without pyridine nucleotides to test the Nfn:Fd complex.
Note: A 5 ml stock of deuterated buffer was made and subsequently aliquoted out with each nucleotide combination to make a 500 µl stock of each condition to use to set up reaction mixtures. For a condition run in triplicate, 500 µl should be sufficient volume to set up reaction mixtures. These volumes can/should change depending on the number of conditions and time points being tested. - Prepare reaction vials by first degassing the deuterated buffers using four-10 min cycles on a Schlenk line. The Schlenk line functions by alternating between a vacuum to remove the oxygen, and nitrogen to place the solutions under positive pressure. After degassing, transfer reaction solutions to the glove box.
- Place 63 μl of each reaction buffer into three Verex vials, cap, place into clear glass serum vials, and seal using a crimper. This volume of buffer accommodates the volume for all time points while also leaving room for the addition of the protein. Seal the reaction vials, remove from the glove box, and store at room temperature prior to reaction initiation.
- Prepare a quench solution for each time point, and keep the solutions on ice before and during the reaction. Prepare solutions in Costar microcentrifuge tubes by mixing 1% formic acid (45 μl) and porcine pepsin (15 μl of 1 mg/ml in 50 mM sodium acetate pH 4.5 (H2O; pH set with 0.1 N HCl); final concentration: 0.2 mg/ml). This final concentration of pepsin gives sufficient coverage of the target proteins (~100%) in the limited digestion time. Prepare a separate quench solution with 10 μl of reaction buffer to test the pH of the solution. The target pH for HDX quenching is pH 2.5.
Notes:- Quench solutions do not need to be prepared anaerobically because the reaction is over at this point.
- Pepsin digestion of the protein localizes the location of exchange onto certain regions of the protein while under low pH quench conditions.
- Quench solutions do not need to be prepared anaerobically because the reaction is over at this point.
- Before preparation of stock solutions and reaction mixtures, determine the number of time points. The number of time points dictates the volume of the reaction mixtures. For every time point, 10 μl must be available to add to the quench solution, plus an additional 10 μl for the 24 h/fully deuterated time point. For instance, in the HDX experiment of Nfn, five time points were measured (1 min, 3 min, 15 min, 60 min, 3 h, and 24 h) (Berry et al., 2018). An additional 10 μl of reaction solution is included to prevent withdrawing the full reaction solution, thus bringing the total volume to 70 μl.
- Hydrogen Deuterium exchange reaction
- Begin the HDX reaction by adding protein via a 1:10 (v/v) dilution into the deuterated buffer using gastight Hamilton syringes (this results in a final Nfn concentration of 1.6 mg/ml). This ensures that D2O is the dominant solvent species. A workflow depicting the HDX reaction steps is shown in Figure 1.
Note: The reaction for Nfn is performed at 60 °C. Regulation of this temperature was through a Fisher Scientific isotemp 110 water bath. This temperature was chosen to simulate the high temperature conditions where the organism, Pyrococcus furiosus lives. - At specific time points (for this reaction: 1 min, 3 min, 15 min, 60 min, 3 h, and 24 h), remove 10 μl of the reaction mixture using a gas tight, Hamilton syringe, and add to the quench solution (pH 2.5, 0 °C; Concentration of Nfn in quench solution: ~0.23 mg/ml).
Notes:- This halts the exchange reaction, so any damage to the protein caused by low pH, temperature, or oxygen exposure will not impact data interpretation.
- Pepsin digestion is allowed to proceed for 2 min before flash freezing the solution in liquid nitrogen and storing at -80 °C.
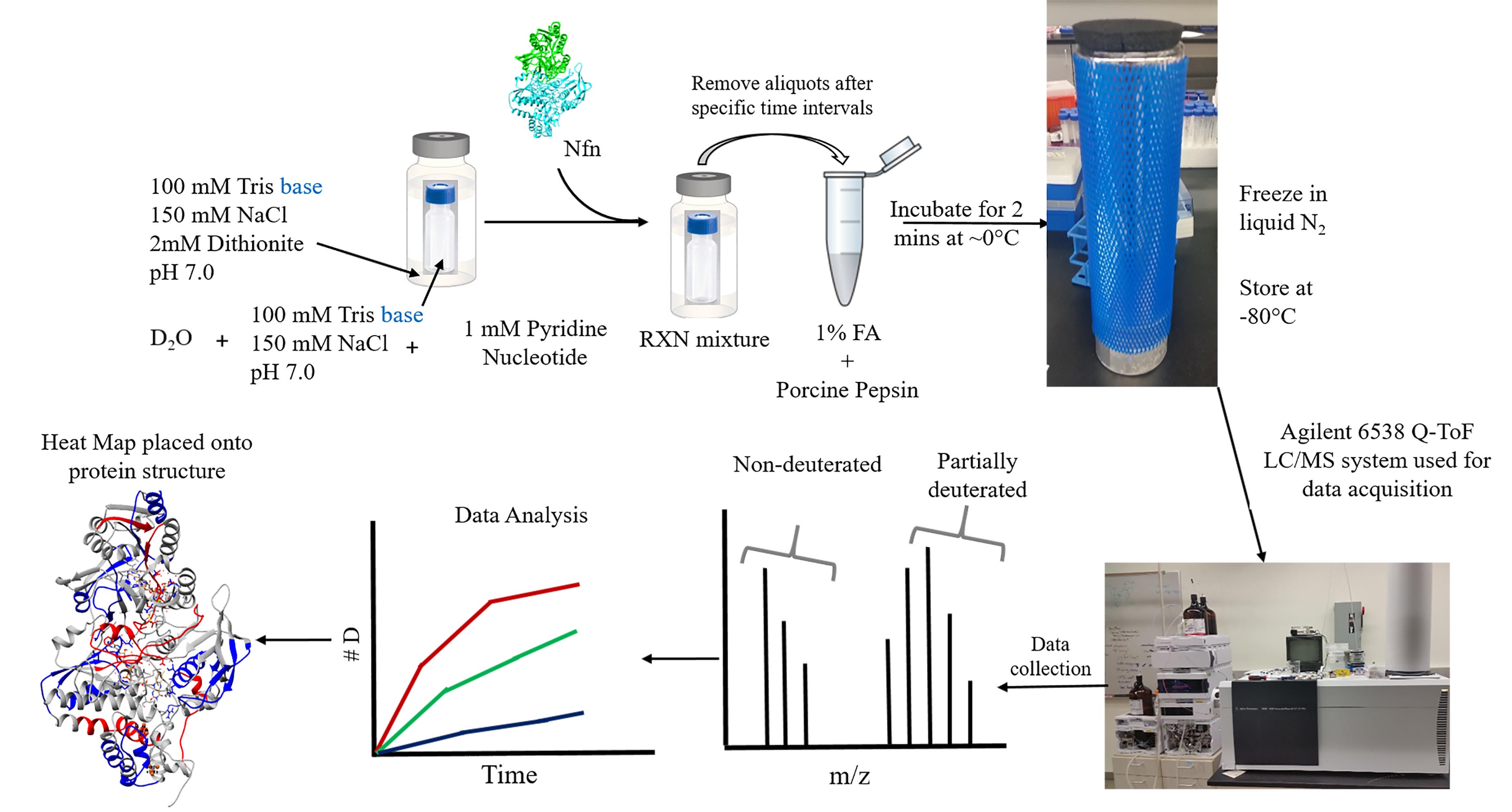
Figure 1. HDX-MS sample prep and work flow. Samples are kept anaerobic using a double vial system in which all reaction components are added to a small screw cap vial, which are sealed anaerobically in a slightly larger clear glass crimp vial. Protein is injected into the reaction mixture using gas tight Hamilton syringes to initiate the reaction. At designated time points, 10 μl of sample is extracted from the reaction mixture and added to an aerobic quench solution, containing formic acid and porcine pepsin. The acid not only locks in any deuterium that has exchanged onto the protein, but also serves to partially denature the protein complex, making it more accessible to pepsin digestion. Additional unfolding occurs upon exposure to oxygen. After 2 min of digestion, samples are flash frozen in liquid N2 and stored at -80 °C until data acquisition on Agilent 6538 Q-TOF. After acquisition, data is processed to display deuterium uptake over time in the form of uptake curves. Deuterium incorporation can be localized onto the structure of a protein by mapping the deuterium incorporation onto a 3D structure of the protein (if one is available).
- This halts the exchange reaction, so any damage to the protein caused by low pH, temperature, or oxygen exposure will not impact data interpretation.
- Begin the HDX reaction by adding protein via a 1:10 (v/v) dilution into the deuterated buffer using gastight Hamilton syringes (this results in a final Nfn concentration of 1.6 mg/ml). This ensures that D2O is the dominant solvent species. A workflow depicting the HDX reaction steps is shown in Figure 1.
- Data acquisition and processing
- Conduct liquid chromatography using an Onyx monolithic C18 column with the following gradient of A (H2O, 0.1% formic acid) and B (ACN, 0.1% formic acid) solvents over 10 min: 0.0-1.0 min, 5% B; 1.0-9.0 min, 5-45% B; 9.1-9.8 min, 95% B; 9.8-9.9 min, 5% B. Set the flow rate to 500 μl/min. Inject 10 μl, resulting in the injection of approximately 2.3 μg of protein onto the column. Keep the solvents cool (~0 °C) by storing the LC bottles on ice before and during data acquisition. Additionally, set the column compartment temperature to 1 °C and the auto-sampler temperature to 4 °C to help maintain a low temperature in the samples prior to injection on the mass spectrometer.
Notes:- Low temperatures in the HPLC stack helps minimize the amount of back exchange during chromatographic separation of peptides.
- The HPLC stack used in this protocol does not allow lower temperatures in the auto-sampler and lower temperatures in the column compartment are not stable, which can result in variations in the data.
- Low temperatures in the HPLC stack helps minimize the amount of back exchange during chromatographic separation of peptides.
- For data acquisition on an Agilent 6538 Q-TOF operating with MassHunter Workstation Software, scans are collected at 2 Hz over a scan range 50-1,700 m/z in positive mode. Set electrospray settings to the following: nebulizer gas at 3.7 bar, drying gas at 8.0 L/min, drying temperature at 350 °C, capillary voltage at 3.5 kV, and the acquisition mode was set to MS (seg).
Note: Agilent Technologies supplies detailed information on its software for new users. For more information about different software options including Masshunter, please refer to Agilent Technologies’ website (Agilent Software: Software & Informatics). - Identify compounds in the mass spectra by molecular feature in Agilent MassHunter Qualitative Analysis using the following settings: Extraction algorithm target data type, Small molecules; Peak filters, ≥ 500 counts; Ion species, +H and +Na; Isotope grouping, peak spacing tolerance 0.0025 m/z + 7.0 ppm, isotope model: peptides; Charge state assignment limited to a maximum of 5.
Note: This is also a good chance to review the data, and look in the mass spectra for signs that deuterium exchange took place (this can also be done during data acquisition). Find an isotope envelope of a peptide in the non-deuterated sample. Then look at the same retention time in the deuterated samples to see if the isotope distribution has shifted. If it has, the experiment worked, if not, it is crucial to determine why the exchange either didn’t work, or why back exchange occurred causing a loss of deuterium ions. An example isotope comparison can be seen in Figure 2A.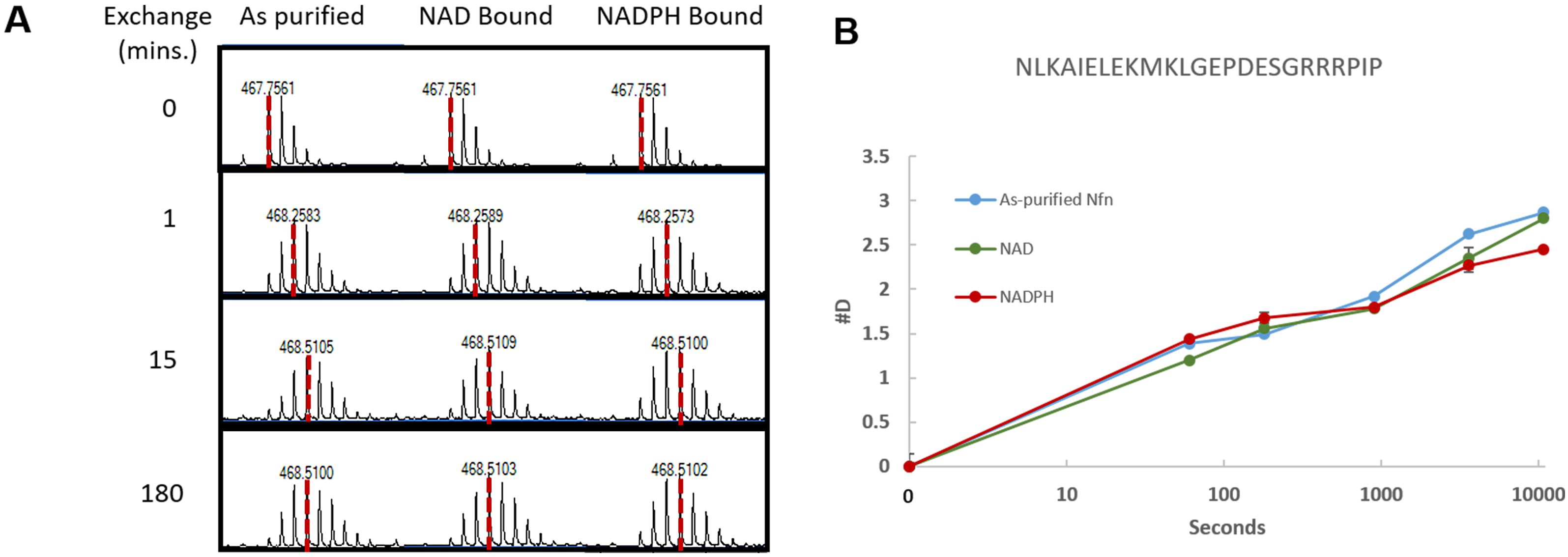
Figure 2. Isotope distribution and uptake curve of HDX-MS data. A. Example of the shift in isotope distribution for a peptide from Nfn-L. As the time for exchange increases (0-180 min) the centroid shifts to the right due to the addition of deuterium, as indicated by the dashed red line. B. Uptake curves showing the deuterium incorporation for four tested conditions. This data is for residues 124-144 of Nfn-L, the amino acid sequence located above the uptake curve. - Create compound lists for non-deuterated digests of the protein sample, and export as a compound summary CSV (.csv) and import into Microsoft Excel 2016.
- Use the compound summary lists in a two-step process to identify peptides produced by pepsin digestion.
Notes:- The first step identifies unique peptides detected with mass spectrometry. These are masses that only have one match with the sequence of Nfn, while also falling in the mass error tolerance.
- The second step determines the identity of masses containing multiple peptide matches within the error tolerance by collecting fragmentation data to sequence the peptide.
- The first step identifies unique peptides detected with mass spectrometry. These are masses that only have one match with the sequence of Nfn, while also falling in the mass error tolerance.
- Place the protein sequence into PAWs to create a theoretical cleavage map using pepsin cleavage sites and an error of 100 ppm to account for the non-specific nature of pepsin cleavage.
Note: Pepsin cleavage sites are after the following residues: L, W, I, A, F, Y, N, T, C, V, S, Q, G, E, D, R, M, K, H, and P. Cleavage sites were chosen based on outputs provided by the program PeptideShaker. - PAWs identifies unique peptides for each of the subunits present in the protein complex of interest. Unique peptides are those who only have one match with the sequence of Nfn within the set error tolerance (100 ppm). Add each unique peptide to an Excel spreadsheet along with the corresponding sequence, charge, and retention time.
Notes:- Exclude any masses with multiple matches within the error tolerance.
- Video 1 shows how to set up PAWs for identification of unique peptides.
- Exclude any masses with multiple matches within the error tolerance.
- Save the peptide list as a comma delimited (.csv) file.
 Video 1. PAWs set-up. Walkthrough of how to set up protein analysis worksheet (PAWs) for the identification of unique peptides. Walkthrough includes how to load a sequence, develop theoretical cleavage map, and input mass lists for peptide identification.
Video 1. PAWs set-up. Walkthrough of how to set up protein analysis worksheet (PAWs) for the identification of unique peptides. Walkthrough includes how to load a sequence, develop theoretical cleavage map, and input mass lists for peptide identification.
- Conduct liquid chromatography using an Onyx monolithic C18 column with the following gradient of A (H2O, 0.1% formic acid) and B (ACN, 0.1% formic acid) solvents over 10 min: 0.0-1.0 min, 5% B; 1.0-9.0 min, 5-45% B; 9.1-9.8 min, 95% B; 9.8-9.9 min, 5% B. Set the flow rate to 500 μl/min. Inject 10 μl, resulting in the injection of approximately 2.3 μg of protein onto the column. Keep the solvents cool (~0 °C) by storing the LC bottles on ice before and during data acquisition. Additionally, set the column compartment temperature to 1 °C and the auto-sampler temperature to 4 °C to help maintain a low temperature in the samples prior to injection on the mass spectrometer.
- Peptide fragmentation data acquisition and processing
- Several peptides can have the same m/z, therefore in order to successfully identify these peptides fragmentation data was collected on the Agilent 6538 Q-TOF instrument. Collision energies for fragmentation were set as a gradient over the data acquisition scan range (50-1,700 m/z) ranging from 4 V to 68 V.
Note: The HPLC and MS settings that were used are the same as discussed previously in Procedure C, Steps 1 and 2 with the exception of the data acquisition mode, which is set to auto MS/MS (seg). - Open the data files in the Masshunter Qualitative Analysis software to identify fragmentation features using the command ‘Find by Auto MS/MS’. Then export the data files as a Mascot Generic File (.mgf).
- Use SearchGUI to identify peptides based on the fragmentation data.
- To set up SearchGUI, start by defining the protein sequence. For this experiment, the sequence for the Nfn small and large subunits from Pyrococcus furiosus is used. SearchGUI will ask to make decoy sequences, which will help identify false positives.
- Next, set the search parameters for SearchGUI. For this experiment use the following settings: Digestion: Enzyme; Enzyme: Pepsin A; Specificity: Semi-Specific; Max Missed Cleavages: 5; Fragment Ion Types: b and y; Precursor m/z Tolerance: 20 ppm; Fragment m/z Tolerance: 20 ppm; Precursor Charge: 1-5; Isotopes: 0-1.
- Choose both the output folder, as well as the search engines. For this experiment, X!Tandem, MyriMatch and MS Amanda are chosen for the peptide search.
- Select PeptideShaker for post processing. PeptideShaker provides an easy to follow output of the fragment ion identifications, and provides the sequence, m/z, and retention time of the identified peptides.
- Combine the peptides identified from the fragmentation data with the unique peptides identified in PAWs. The combined list of identified peptides is saved as a .csv file, and used as the input file for deuterium incorporation calculations.
Note: Video 2 shows how to set up SearchGUI and Peptide Shaker for the identification of peptides from fragmentation data. Video 2. SearchGUI/PeptideShaker set-up. Walkthrough of how to set up SearchGUI to identify peptides based on fragmentation data collected using MS/MS. The search results from SearchGUI will be opened in PeptideShaker. PeptideShaker provides a user friendly interface for the examination of identified peptides, which shows the protein coverage and number of peptides identified.
- To set up SearchGUI, start by defining the protein sequence. For this experiment, the sequence for the Nfn small and large subunits from Pyrococcus furiosus is used. SearchGUI will ask to make decoy sequences, which will help identify false positives.
- Several peptides can have the same m/z, therefore in order to successfully identify these peptides fragmentation data was collected on the Agilent 6538 Q-TOF instrument. Collision energies for fragmentation were set as a gradient over the data acquisition scan range (50-1,700 m/z) ranging from 4 V to 68 V.
- Calculation of deuterium uptake using HDExaminer
- Deuterium incorporation is calculated by measuring the shift in the centroid of the isotope distribution for a given peptide. While this can be done manually, it is extremely time consuming. Several programs are available that will calculate the deuterium incorporation for HDX data. HDExaminer is a commercially available program by Sierra Analytics Inc. that accomplishes this.
Note: Sierra Analytics has several tutorial videos to show the features of HDExaminer and how to set up the program (HDExaminer: The Basics). - To set up HDExaminer, first add the sequence for the protein of interest and add a list of tested conditions on the ‘proteins’ page. This allows the program to calculate the deuterium incorporation in each condition.
- Next, add the list of peptides under the ‘peptides’ page, and select the column which has the charge, sequence, and retention time of each peptide. Then add the peptides to the peptide pool.
- Finally, on the ‘analysis’ page add the data files for each condition. All conditions will have the non-deuterated files, which are added first. Next add the fully deuterated data files (24 h time point), followed by the partially deuterated data files (1 min, 3 min, 15 min, 1 h, 3 h). To begin calculations automatically select the ‘auto-calculate’ box in the bottom right-hand corner of the screen, otherwise calculations will have to be initiated manually. A progress bar in the bottom right-hand corner indicates when calculations are underway.
Note: Alternatively, the maximum number of deuterium can be calculated from the number of exchangeable amide hydrogens within the peptide sequence. - Once the calculations are complete, manually verify the calculations.
- Under the peptide tab of HDExaminer, each peptide has a dropdown menu showing the different conditions. Each condition has an additional dropdown menu showing the different charge states of the peptide based on the peptide pool.
- If HDExaminer successfully identifies the location of the peptide within the data files, clicking on a charge state shows the isotope distribution of the peptide in each data file on the left-hand side. The right-hand side shows the chromatogram for each data file and two red arrows indicate the retention time window containing the peptide isotope pattern.
- To verify the data, examine the retention time window for the peptides, they should be the same, or within a few seconds of one another for each data file. The search window can be adjusted by moving either of the red arrows. If the partially deuterated or fully deuterated data files are changed, the deuterium incorporation for only those files will be re-calculated. However, if the non-deuterated files search retention time is changed, the deuterium incorporation for all data files for that peptide will be re-calculated.
- Manually verifying the data ensures the same retention time search window is used for all data files for a peptide. This gives the most accurate measurement of the deuterium incorporation and helps minimize the error between replicates.
- Under the peptide tab of HDExaminer, each peptide has a dropdown menu showing the different conditions. Each condition has an additional dropdown menu showing the different charge states of the peptide based on the peptide pool.
- After manual verification, export the peptide pool results for each condition, this provides the peptide identification, search retention time and charge. This also provides the start and end retention time, amount of deuterium incorporated (#D), percent deuterium incorporated (%D), score and confidence for each time point and replicate.
- The data provided in the peptide pool results can be used in a variety of ways.
- The easiest means of visualizing the data are uptake curves which show the #D incorporated into a peptide over time. Generate a curve for each peptide in order to compare the deuterium incorporation over time for each condition tested. Figure 2B displays an example of an uptake curve that shows a comparison of four conditions.
- An additional way to analyze the data is to calculate the %D incorporated and measure the percent difference to determine the relative change in accessibility to exchange between conditions. Calculate the %D by dividing the #D for a time point by the #D from the fully deuterated sample and multiply by 100. Then calculate the percent difference by subtracting the %D for two conditions. This allows comparisons in the deuterium incorporation between peptides of different length.
- Map the percent difference onto the structure of the protein (if available) to show the areas that are impacted by the different conditions. Mapping the exchange data onto the structure can also reveal the presence of allostery, as well as which regions of the protein are communicating with one another to enact the allosteric mechanism.
Note: For a detailed explanation on mapping exchange data onto a protein structure, see Data analysis.
- The easiest means of visualizing the data are uptake curves which show the #D incorporated into a peptide over time. Generate a curve for each peptide in order to compare the deuterium incorporation over time for each condition tested. Figure 2B displays an example of an uptake curve that shows a comparison of four conditions.
- Deuterium incorporation is calculated by measuring the shift in the centroid of the isotope distribution for a given peptide. While this can be done manually, it is extremely time consuming. Several programs are available that will calculate the deuterium incorporation for HDX data. HDExaminer is a commercially available program by Sierra Analytics Inc. that accomplishes this.
Data analysis
- Use MassHunter Qualitative Analysis to verify the data during data acquisition by examining the deuterated samples for evidence of exchange in the isotope distribution. This is done by comparing the deuterated samples to the non-deuterated samples. Figure 2A shows an example of this by displaying the isotope distribution for the non-deuterated, 1 min, 15 min, and 3 h time points. This allows for a comparison in the shift in the isotope distribution as more deuterium is incorporated between the tested conditions.
- After calculation of the deuterium incorporation, use Microsoft Excel or a similar spreadsheet program to generate uptake curves to compare deuterium incorporation over time in the tested conditions within a peptide. Figure 2B shows an example uptake curve.
- To compare deuterium incorporation between multiple peptides and/or conditions, convert the #D to %D and calculate the difference between two peptides. Additionally, the percent difference can be mapped onto a 3D structure of a protein using UCSF Chimera as shown in Figure 3 (Pettersen et al., 2004).
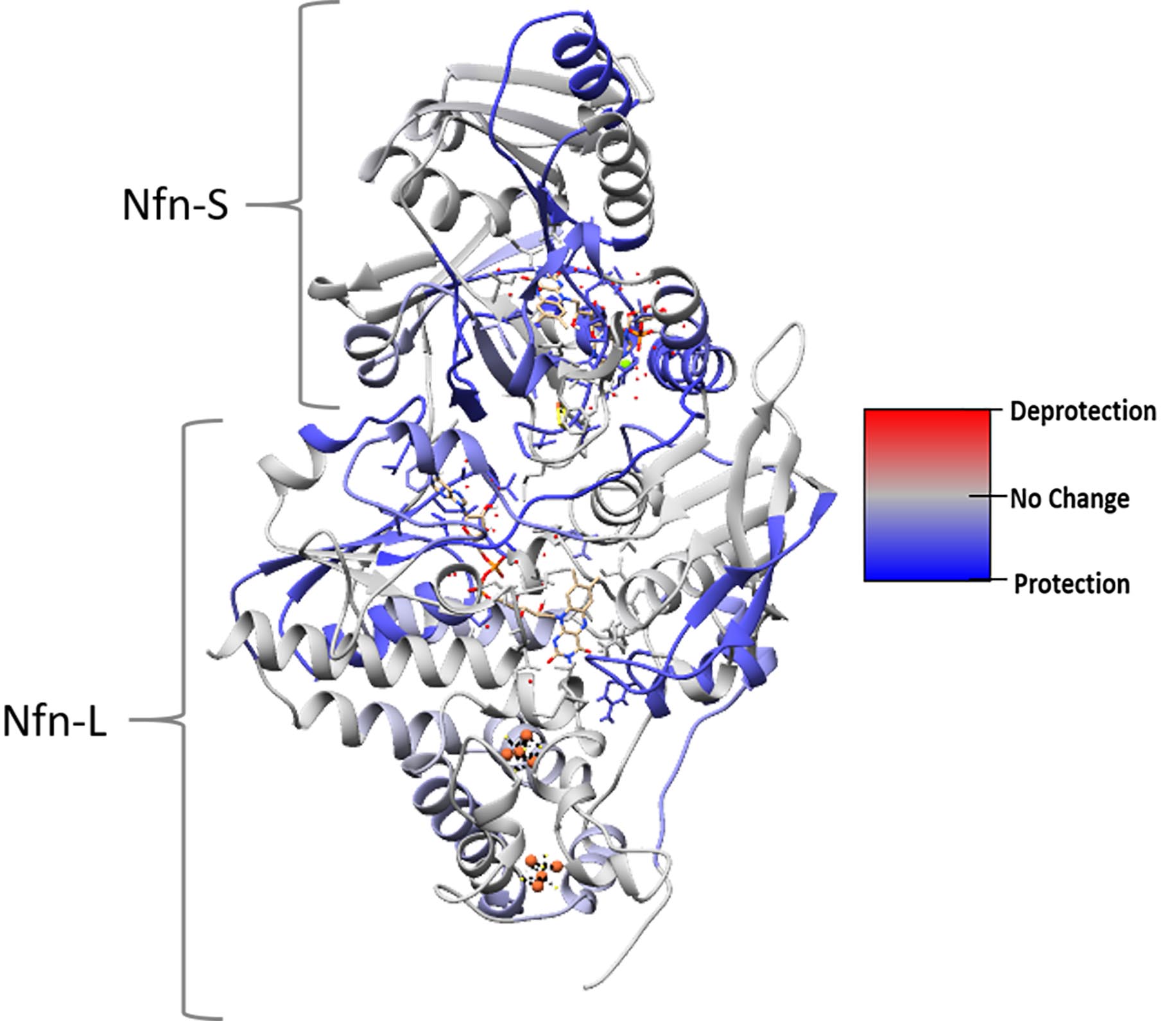
Figure 3. HDX-MS data mapped onto a protein structure. This structural heat map shows the influence of NADPH and NAD+ binding on exchange of Nfn. H/D exchange data collected under two different conditions was mapped onto the protein structure using UCSF Chimera (see Notes). The heat map was calculated from the difference between exchange in as-purified and NADPH + NAD+ bound reactions. Color code was set to red (100% difference, increased exchange), blue (-100% difference, protection), and gray (0% difference, no change). In this example, the NADPH + NAD+ bound condition incorporates less deuterium than the as-purified protein. - Generate a structure-based heat map of the HDX data in UCSF Chimera (Pettersen et al., 2004).
- Create a txt file that lists the residue number and the %D that correlates to that residue in the following format:
Attribute: nad_nadph-as_purified_nfn_3hrs_percent_deuterium
match mode: 1-to-1
recipient: residues
:1.A 20
:2.A 20
:3.A 45
:4.A 45
:5.A 45
The header names the attribute that will be displayed onto the model and defines that the residues are what will be colored according to the attribute (rather than atoms). The ‘:1.A’ defines the residue number and chain name. A tab separates the value from the residue identifier. These columns can easily be generated in Excel or a similar spreadsheet program and either saved as a txt file or copy/pasted into a text file. If there is no data for a residue, leave empty space next to the residue identifier or completely remove the line. - Open the pdb protein structure file in Chimera. Go to Tools > Structure Analysis > Define Attribute. Select the .txt file generated in step a.
Note: An error will open in a pop up window if data is listed for residues that are not resolved in the structure file. This error can be ignored. - Once the attribute is defined, the Render/Select by Attribute window opens. Use this window to determine which colors will represent which %D values.
- Residues that are listed in the txt file but do not have data associated with them will be colored according to the highest %D color unless an additional color is added above this value.
- To add additional color bars use Ctrl + left click in the histogram plot. Ctrl + left click on existing color bars to remove them.
- Move color bars either by the mouse or by entering a value in the ‘Value’ box.
- Residues that are listed in the txt file but do not have data associated with them will be colored according to the highest %D color unless an additional color is added above this value.
- Click ‘Apply’ to color the protein structure according to the data file.
- Figure 3 shows an example of a colored structure.
Note: Video 3 shows how to map HDX-MS data onto a 3D structure of a protein. Video 3. UCSF Chimera protein mapping. Walkthrough showing how to format data to map onto a 3D protein structure in UCSF Chimera. Walkthrough also shows the correct menus to select to map data onto the structure, as well as how to set up a color gradient and specify what colors to use. - Create a txt file that lists the residue number and the %D that correlates to that residue in the following format:
Notes
- The recommended alternative column for HPLC separation of peptides is the same as the one used in the protocol described here, however the internal diameter is 1 mm larger. This will influence the flow rate of solvent through the column, which can affect the elution of peptides from the column. It is highly recommended to test different flow rates and gradients to optimize separation of peptides prior to performing the exchange reaction.
- The measured pH of the deuterated buffers must be corrected to pD by adding the correction factor (0.4 pH units) to the measured pH. For this experiment the tris base buffer prepared in D2O had a measured pH of 7.0, therefore the pD of the buffer is 7.4 (Krezel and Bal, 2004).
- Before beginning the exchange reaction, check that the quench solution with formic acid, pepsin, and reaction buffer are at pH 2.5 so back exchange does not occur during protein digestion.
- Place HPLC solvents on ice, set the column compartment to 0 °C, and the auto-sampler to 4 °C to help reduce back exchange of the deuterated amides.
- HDX-MS can be performed under a wide range of temperatures. While exchange rate does have a dependence on temperature, pH can affect it much more (Walters et al., 2012). However, it should be noted that a comparative analysis between HDX-MS reactions run at different temperatures is difficult because of the change in exchange rate as the temperature is varied. HDX-MS is most commonly run at 25 °C (room temperature), however, some cases are reported to run as low as 0 °C and as high as 60 °C.
- When planning an HDX-MS experiment, it is highly recommended that the starting concentration of the protein of interest be approximately 10 mg/ml, this ensures that approximately 1.5 μg of protein is injected onto the instrument.
- Lower amounts of protein injected onto the instrument will cause peptides to be detected at low intensity, which in turn can prevent accurate deuterium incorporation calculations.
- A 1:10 dilution of protein into D2O buffer is encouraged, it is possible to do less if a particular protein is not easy to obtain aliquots with a high concentration. It is then recommended to do some pilot experiments to ensure that deuterium incorporation is satisfactory, and the extra water present in the reaction is not causing back exchange of the deuterium ions.
- Lower amounts of protein injected onto the instrument will cause peptides to be detected at low intensity, which in turn can prevent accurate deuterium incorporation calculations.
Recipes
- Tris base buffer in deuterium oxide (shelf life ~one week)
100 mM Tris-HCl
150 mM NaCl
Adjust pH to 7.0 with 1 N HCl - Tris base buffer in H2O
100 mM Tris-HCl
150 mM NaCl
Adjust pH to 7.0 with 1 N HCl - Tris base buffer w/sodium dithionite
100 mM Tris-HCl
150 mM NaCl
2 mM sodium dithionite
Adjust pH to 7.0 with 1 N HCl
Acknowledgments
This work was supported as part of the Biological and Electron Transfer and Catalysis (BETCy) EFRC, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science (DE-SC0012518). The Mass Spectrometry Facility at MSU was supported in part by the Murdock Charitable Trust and an NIH IDEA program grant P20GM103474. The work detailed in this protocol was adapted from work described in recent publications (Lubner et al., 2017; Berry et al., 2018). The authors declare that they do not have any conflicts of interest.
References
- Berry, L., Poudel, S., Tokmina-Lukaszewska, M., Colman, D. R., Nguyen, D. M. N., Schut, G. J., Adams, M. W. W., Peters, J. W., Boyd, E. S. and Bothner, B. (2018). H/D exchange mass spectrometry and statistical coupling analysis reveal a role for allostery in a ferredoxin-dependent bifurcating transhydrogenase catalytic cycle. Biochim Biophys Acta 1862(1): 9-17.
- Demmer, J. K., Rupprecht, F. A., Eisinger, M. L., Ermler, U. and Langer, J. D. (2016). Ligand binding and conformational dynamics in a flavin-based electron-bifurcating enzyme complex revealed by Hydrogen-Deuterium Exchange Mass Spectrometry. FEBS Lett 590(24): 4472-4479.
- Krezel, A. and Bal, W. (2004). A formula for correlating pKa values determined in D2O and H2O. J Inorg Biochem 98: 161-166.
- Lubner, C. E., Jennings, D. P., Mulder, D. W., Schut, G. J., Zadvornyy, O. A., Hoben, J. P., Tokmina-Lukaszewska, M., Berry, L., Nguyen, D. M., Lipscomb, G. L., Bothner, B., Jones, A. K., Miller, A. F., King, P. W., Adams, M. W. W. and Peters, J. W. (2017). Mechanistic insights into energy conservation by flavin-based electron bifurcation. Nat Chem Biol 13(6): 655-659.
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. and Ferrin, T. E. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25(13): 1605-1612.
- Walters, B. T., Ricciuti, A., Mayne, L. and Englander, S. W. (2012). Minimizing back exchange in the hydrogen exchange-mass spectrometry experiment. J Am Soc Mass Spectrom 23(12): 2132-2139.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Berry, L., Patterson, A., Pence, N., Peters, J. W. and Bothner, B. (2018). Hydrogen Deuterium Exchange Mass Spectrometry of Oxygen Sensitive Proteins. Bio-protocol 8(6): e2769. DOI: 10.21769/BioProtoc.2769.
Category
Biochemistry > Protein > Interaction > Protein-ligand interaction
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.