- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolate and Sub-fractionate Cell Membranes from Caulobacter crescentus

Published: Vol 2, Iss 20, Oct 20, 2012 DOI: 10.21769/BioProtoc.276 Views: 14849
Original research article
The authors used this protocol in:

Jun 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Cell membranes from Caulobacter can be isolated and separated into inner and outer membranes according their characteristic buoyant densities on a sucrose gradient. Fractionation can be used to determine the localisation of uncharacterised proteins and to enrich protein complexes present in either of these membranes for biochemical analysis such blue-native PAGE and immunoprecipitation.
Keywords: BacteriaMaterials and Reagents
- 0.2% Bacto Peptone
- 0.1% yeast extract
- MgSO4
- CaCl2
- Tris (Sigma-Aldrich)
- Sucrose (Sigma-Aldrich)
- Lysozyme (Sigma-Aldrich)
- EDTA-free protease inhibitors (F. Hoffmann-La Roche, catalog number: 11873580001 )
- Dnase I (Sigma-Aldrich, catalog number: DN25 )
- MgCl2 (Sigma-Aldrich)
- EDTA (Sigma-Aldrich)
- Na2HPO4
- KH2PO4
- NH4Cl
- FeSO4 (EDTA chelate) (Sigma-Aldrich, catalog number: F0518 )
- Glucose
- Spheroplasting buffer
- Growth media (Rich PYE or Minimal M2G) (see Recipes)
- Spheroplasting buffer (see Recipes)
- Lysis buffer (see Recipes)
- Growth media (Rich PYE or Minimal M2G) (see Recipes)
- Sucrose solutions (see Recipes)
- TEM buffer (see Recipes)
Equipment
- Centrifuges
- Peristaltic pump (Bio-Rad Laboratories, catalog number: 731-8140EDU )
- High-pressure homogenizer (Avestin Emulsiflex, EmulsiFlex-C5)
- 7 and 15 ml Dounce homogenizers (WHEATON)
- BECKMAN centrifuge, fixed angle rotor (Ti45 or Ti60), swing-out rotor (SW40) and appropriate tubes
- Density gradient fractionation system (Brandel, model: BR188 )
- Beckman Ultra-Clear tubes
- SW40 rotor tubes
Procedure
- Preparation of membranes
- Grow Caulobacter in PYE media at 30 °C till OD660 is 0.6-0.8.
- Spin 2 L of Caulobacter culture at 10,000 x g for 12 min at 4 °C. For all following steps, keep everything on ice.
- Resuspend cell pellet with spheroplasting buffer. The volume of spheroplasting buffer required (in ml) = [(OD660 x ml culture volume)/100] x 7. For example, a cell pellet from a 2 L culture with an OD660 of 0.8 will be resuspended by 112 ml of spheroblasting buffer.
- Add 100 mg/ml lysozyme (dissolved in spheroplasting buffer). Leave on ice for 2 min. For cultures grown in M2G minimal media, use less lysozyme (50 mg/ml) as cell grown under these conditions have weaker cell walls. M2G minimal media can be used to examine differential expression of outer membrane proteins as a result of carbon starvation.
- Add 1 tablet of protease inhibitor cocktail, 25 μg/ml Dnase I (final concentration) and 10 mM MgCl2 (final concentration).
- Add 2 volumes of cold lysis buffer to the cells. This must be done slowly to avoid localised cell lysis. Add the lysis buffer at a rate of 4-6 ml/min, either drop-wise with a pipette or using a peristaltic pump. The cell solution must be constantly swirled during the addition of lysis buffer to avoid localised lysis.
- Completely lyse the cells with the Avestin Emulsiflex using homogenizing pressure set to ~12,000-15,000 psi.
- Pellet insoluble debris at 10,000 x g, 20 min at 4 °C. Collect the supernatant (contains membranes).
- Spin the supernatant in the Beckman ultracentrifuge using the Ti45 rotor (36,000 rpm, 1.5 h, 4 °C). After the spin, keep the membrane pellet and discard the supernatant.
- To wash membranes, resuspend the membrane pellets in 50-100 ml of spheroblasting buffer and repeat step 9. Addition of 1x protease inhibitor cocktail to the spheroblasting buffer for the wash step is optional.
- Resuspend the membrane pellet using ~ 6-7 ml of 25% sucrose (w/v). Addition of 1x protease inhibitor cocktail to the sucrose solution is optional but unnecessary.
- Grow Caulobacter in PYE media at 30 °C till OD660 is 0.6-0.8.
- Fractionation of cell membranes
- Prepare the sucrose gradients using the Beckman Ultra-Clear tubes. Use Beckman tubes (14 x 95 mm) for the SW40 rotor. The sucrose gradient consists of 1.7 mL each of 60, 55, 50, 45, 40, 35, 30% sucrose. To pour the sucrose gradient, pipette the required volume for each sucrose concentration into an eppendorf tube. Using a plastic pasteur pipette, transfer 60% sucrose solution from the eppendorf tube to the bottom of the Ultra-clear tube. Hold the Ultra-clear tube at 45° angle before transferring the 55% sucrose solution by slowly adding it drop-by-drop near the 60% sucrose solution. Then add 50, 45, 40, 35 and 30% sucrose. I found this particular method minimised disruption to the gradient.
- Add ~ 1.2 ml of membrane sample (from step 10) on top of the sucrose gradient. Make sure to balance the sucrose gradient tubes with the SW40 rotor tubes.
- Spin sucrose gradients for 40 h in a SW40 rotor at 34,000 rpm, 4 °C to fractionate the membranes by equilibrium sedimentation. Use slow break so the sucrose gradient is maintained.
- The gradient should have a yellow-brown region (inner membranes) and a cloudy white region (outer membranes). Isolate 0.5 ml fractions using the Brandel/Foxy Jr fractionator and 2 M sucrose (in 5 mM EDTA, pH 7.5) as the displacing fluid. Alternatively, collect 0.5 ml fractions from the top of the gradient. Fractions 9-12 generally contain inner membranes and fractions 16-18 contain outer membranes from a total of approximately 23 fractions. To confirm, perform western blot analysis using an antibody against an inner and an outer membrane protein (see figure below showing 20 μl of each fraction for SDS-PAGE and analysis by Coomassie; and 5 μl of each fraction analysed by western blotting using our in-house antibodies).
- Freeze fractions in liquid N2 and store at -80 °C. For further analysis, pool relevant fractions and wash in TEM buffer before resuspending membranes in the buffer of choice. To wash, mix fractions with at least 5 volumes of TEM buffer and spin in the Beckman ultracentrifuge (36,000 rpm, 1.5 h, 4°C).
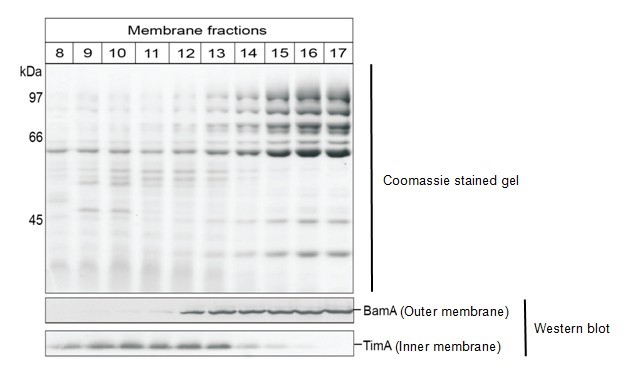
Figure 1. Membranes from wild-type C. crescentus were fractionated on sucrose gradient and analysed by SDS-PAGE. Coomassie Brilliant Blue staining (upper panel) reveals separation of the membrane protein profiles and immunoblotting (lower panel) for the inner membrane protein TimA and the outer membrane protein BamA.
- Prepare the sucrose gradients using the Beckman Ultra-Clear tubes. Use Beckman tubes (14 x 95 mm) for the SW40 rotor. The sucrose gradient consists of 1.7 mL each of 60, 55, 50, 45, 40, 35, 30% sucrose. To pour the sucrose gradient, pipette the required volume for each sucrose concentration into an eppendorf tube. Using a plastic pasteur pipette, transfer 60% sucrose solution from the eppendorf tube to the bottom of the Ultra-clear tube. Hold the Ultra-clear tube at 45° angle before transferring the 55% sucrose solution by slowly adding it drop-by-drop near the 60% sucrose solution. Then add 50, 45, 40, 35 and 30% sucrose. I found this particular method minimised disruption to the gradient.
Recipes
- Growth media (Rich PYE or minimal M2G)
PYE (peptone yeast extract) broth
0.2% Bacto peptone
0.1% yeast extract
1 mM MgSO4
0.5 mM CaCl2
M2G (M2 minimal salt medium with glucose as the sole carbon source)
6.1 mM Na2HPO4
3.9 mM KH2PO4
9.3 mM NH4Cl
0.5 mM MgSO4
10 uM FeSO4 (EDTA chelate)
0.5 mM CaCl2
0.2% glucose - Spheroplasting buffer
10 mM Tris (pH 7.5)
0.75 M sucrose - Lysis buffer
1.5 mM EDTA (pH 7.5) - Sucrose solutions
25% sucrose (w/v) in 5 mM EDTA (pH 7.5)
60, 55, 50, 45, 40, 35, 30% (w/v) sucrose in 5 mM EDTA (pH 7.5)
2 M sucrose displacing fluid for fractionation - TEM buffer
10 mM Tris (pH 7.5)
1 mM EDTA
10 mM MgCl2
10% glycerol
Acknowledgments
The author acknowledges the help and support of Professor Trevor Lithgow and Dr. Xenia Gatsos.
References
- Anwari, K., Poggio, S., Perry, A., Gatsos, X., Ramarathinam, S. H., Williamson, N. A., Noinaj, N., Buchanan, S., Gabriel, K., Purcell, A. W., Jacobs-Wagner, C. and Lithgow, T. (2010). A modular BAM complex in the outer membrane of the alpha-proteobacterium Caulobacter crescentus. PLoS One 5(1): e8619.
- Anwari, K., Webb, C. T., Poggio, S., Perry, A. J., Belousoff, M., Celik, N., Ramm, G., Lovering, A., Sockett, R. E., Smit, J., Jacobs-Wagner, C. and Lithgow, T. (2012). The evolution of new lipoprotein subunits of the bacterial outer membrane BAM complex. Mol Microbiol 84(5): 832-844.
Article Information
Copyright
© 2012 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Anwari, K. (2012). Isolate and Sub-fractionate Cell Membranes from Caulobacter crescentus. Bio-protocol 2(20): e276. DOI: 10.21769/BioProtoc.276.
Category
Biochemistry > Protein > Isolation and purification
Cell Biology > Organelle isolation > Membrane
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.