- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Visualization of RNA 3’ ends in Escherichia coli Using 3’ RACE Combined with Primer Extension
Published: Vol 8, Iss 5, Mar 5, 2018 DOI: 10.21769/BioProtoc.2752 Views: 8999
Reviewed by: Gal HaimovichYi ZhangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
In this assay, 3’ RACE (Rapid Amplification of cDNA 3’ Ends) followed by PE (primer extension), abbreviated as 3’ RACE-PE is used to identify the mRNA 3’ ends. The following protocol describes the amplification of the mRNA 3’ ends at the galactose operon in E. coli and the corresponding visualization of the PCR products through PE. In PE, the definite primer is 5’ end-labeled using [γ-(32) P] ATP and T4 polynucleotide kinase, which anneals to the specific DNA molecules within the PCR product of the 3’ RACE. The conventional PE can only be used to locate the 5’ end of an mRNA transcript since reverse transcriptase (RTase) polymerizes only in the 5’ → 3’ direction. Thus, Taq polymerase is used instead of RTase, PCR is performed. Therefore, we are able to locate the 3’ end of the mRNA using this assay. The relative amount of the 3’ end can be directly visualized and quantified by way of separating DNA products in a denaturing 8% urea-PAGE (Polyacrylamide Gel Electrophoresis) gel. The exact position of the 3’ ends can be sequenced by comparison of these final DNA products with the corresponding DNA sequencing ladder.
Keywords: 3’ RACEBackground
The synthesis of the mRNA 3’ end is an important step in E. coli that produces a stable messenger RNA (mRNA). In eukaryotic cells, the mRNA 3’ end formation is through a cleavage from an internal phosphodiester bond, followed by the addition of a poly (A) tail; whereas in prokaryotic cells, the 3’ ends of mRNAs are generated by termination of transcription or by processing of the primary transcript (Altman and Robertson, 1973; Nudler and Gottesman, 2002; Zhao et al., 1999). Therefore, it is important to analyze the exact position and relative quantity of mRNA 3’ end to understand the mechanism of mRNA generation.
3’ RACE assay is a particular procedure to obtain the 3’ end sequence information of a defined RNA transcript (Sambrook and Russell, 2006). Generally, the experiment procedure starts with ligating the 3’ end of RNA to a synthetic RNA oligo, followed by the synthesis of cDNA using RTase and a complementary primer (3RP) to the RNA oligo. Subsequently, specific cDNA is amplified by PCR using the gene-specific primer and the primer, 3RP. Usually, RACE products are directly sequenced, however, based on our modified procedure, PCR products undergo another concluding step of primer extension (PE), which uses Taq polymerase instead of RTase. The labeled primer integrated into the PCR products are extended in a denaturing PAGE gel which makes us visualize and quantify each product. The scheme of 3’ RACE-PE is presented in Figure 1. The polycistronic gal operon encodes amphibolic enzymes for the amphibolism of the sugar D-galactose (Holden et al., 2003). Using this method, we have identified and quantitated the 3’ ends of the gal operon mRNAs in wild type and mutant strains (Lee et al., 2008; Wang et al., 2014 and 2015).
Figure 1. An illustration of the procedure 3’ RACE-PE in E. coli
Materials and Reagents
- Pipettes tips (DNase/RNase-free, Sorenson Bioscience)
- 1.5 ml centrifuge tube (SARSTEDT, catalog number: 72.690.001 )
- Cell culture flasks (Corning, catalog number: 3056 )
- 20 x 150 mm Test tube (Karter Scientific Labware Manufacturing, catalog number: 212W5 )
- Sephadex G-50 column (GE Healthcare, catalog number: 27-5330-01 )
- Whatman 3MM paper (GE Healthcare, catalog number: 3017-915 )
- Kodak CL-XPosure Film (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 34090 )
- Gloves (Ultra TEX Glove, Taeshin Bio Science, catalog number: UT-11 )
- E. coli strains (CGSC, the coli genetic stock center) (an example)
- Lysozyme (Roche Diagnostics, catalog number: 10837059001 )
- TRIZOL (Molecular Research Center, catalog number: TR 118 )
- Chloroform (Merck, Sigma-Aldrich, catalog number: C2432 )
- Isopropanol (Merck, Sigma-Aldrich, catalog number: I9516 )
- Ethanol (Merck, Sigma-Aldrich, catalog number: E7023 )
- RNA storage solution (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM7001 )
- Alkaline phosphatase (Takara Bio, catalog number: 2250A )
- DNase I (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM2222 )
- RNasin® Ribonuclease Inhibitors (Promega, catalog number: N2111 )
- PCI (Phenol:Chloroform:Isoamyl Alcohol) Solution 25:24:1 (Merck, Sigma-Aldrich, catalog number: 77617 )
- Sodium acetate (Merck, Sigma-Aldrich, catalog number: S2889 )
- RNase and DNase-free water (Bioneer, catalog number: C-9011 )
- T4 RNA ligase (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM2141 )
- The synthesized 3’ RACE RNA oligo: 3’-inverted deoxythymidine (3’-idT) RNA (Dharmacon)
- 3RP primer (5’AGCATGCGGCCGCTAAGAAC3’)
- dNTP mix (10 mM each) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0191 )
- HotStarTaq Plus DNA Polymerase (QIAGEN, catalog number: 203603 )
- Gal-specific primers (Table 1)
Table 1. Sequence of each galactose operon gene specific primers are listed
- T4 Polynucleotide Kinase (New England Biolabs, catalog number: M0201L )
- ATP, [γ-32P]- 6,000 Ci/mmol (PerkinElmer, catalog number: NEG002Z250UC )
- 5’ end labeled PE primer (5’ GAGAGTAGGGAACTGCCA 3’)
- 5x Developer (Vivid X-RAY DEVELOPER, Duksan (DS) Lab, catalog number: 0514_00004 )
- 5x Fixer (Vivid X-RAY RAPID FIXER, Duksan (DS) Lab, catalog number: 0514_00003 )
- Tryptone (BD, DifcoTM, catalog number: 211705 )
- Yeast extract (BD, DifcoTM, catalog number: 212750 )
- Sodium chloride (NaCl) (Merck, Sigma-Aldrich, catalog number: 746398 )
- Galactose (Merck, Sigma-Aldrich, catalog number: G5388 )
- Acrylamide (Merck, Sigma-Aldrich, catalog number: V900845 )
- Bis-acrylamide (Merck, Sigma-Aldrich, catalog number: V900301 )
- 5x TBE (Bioneer, catalog number: C-9002 )
- Urea (Merck, Sigma-Aldrich, catalog number: U5128 )
- Ammonium persulfate (Merck, Sigma-Aldrich, catalog number: 248614 )
- TEMED (Merck, Sigma-Aldrich, catalog number: T9281 )
- Formamide (Merck, Sigma-Aldrich, catalog number: F9037 )
- 0.5 M EDTA, pH 8.0 (Bioneer, catalog number: C-9007 )
- Xylene cyanol (Sigma-Aldrich, catalog number: X4126 )
- Bromophenol blue (Sigma-Aldrich, catalog number: B0126 )
- 1 M Tris-HCl, pH 8.0 (Bioneer, catalog number: C-9006 )
- Sucrose (Merck, Sigma-Aldrich, catalog number: S7903 )
- Omniscript RT Kit (QIAGEN, catalog number: 205111 )
- Sigmacote (Sigma-Aldrich, catalog number: SL2 )
- LB + 0.5% galactose (see Recipes)
- 8% sequencing solution (see Recipes)
- 8% Urea-PAGE gel (see Recipes)
- 2x gel loading mix (see Recipes)
- Protoplasting buffer (see Recipes)
Equipment
- Automatic pipette aid (Thermo Fisher Scientific, Thermo ScientificTM, model: S1 Pipette Filler )
- Pipettes (Gilson Company, USA)
- 37 °C shaking incubator (Hanbaek Scientific Co., Korea (S))
- 37 °C and 42 °C heat block (FINEPCR, Korea (S))
- Spectrophotometer (BECKMAN COULTER, USA)
- Vortex-Genie 2 (Scientific Industries, model: Vortex-Genie 2 )
- 4 °C micro-centrifuge (Beckman Coulter, USA)
- NanoDrop 1000 (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 1000 )
- PCR machine (Bio-Rad Laboratories, USA)
- Shallow fixer tray (Bio-Rad Laboratories, USA)
- Sequencing gel electrophoresis apparatus (LABREPCO, USA)
- X-ray cassette (Duksan (DS) Lab, Korea (S))
- Power supply (Bio-Rad Laboratories, USA)
- Autoclave (Dong Won Scientific Corp, Korea (S))
Software
- ImageJ
Procedure
- RNA extraction
- Streak out the E. coli strain to be used for RNA extraction on an LB plate and incubate overnight at 37 °C.
- Select a single colony from the streaked LB agar plate. Inoculate E. coli strain in 3 ml fresh LB medium supplemented with 0.5% galactose, and then grow for 16 h. The next day, assess the bacterial growth by measuring the OD600, usually it is around 4.
- Dilute the culture in 50 ml pre-warmed (37 °C) LB medium supplemented with 0.5% galactose to give an OD600 reading of about 0.05.
- Grow the bacterial culture at 37 °C in a shaking incubator. Assess the OD600 till it reaches 0.6, usually it takes 1 h 50 min for the wild type E. coli cell. At this time, the number of cells in 1 ml culture is about 2 x 108.
- Collect 1 ml cells in a microcentrifuge tube, pellet the cells by centrifugation (12,000 x g, 1 min, 20 °C).
- Discard the supernatant by pipetting, resuspend the cell pellet in 50 µl protoplasting buffer, and add 5 µl lysozyme (50 mg/ml), mix thoroughly, incubate for 5 min at room temperature.
- Add 500 µl Trizol, mix thoroughly by vortexing for 15 sec. incubate e the lysed sample for 5 min at room temperature.
- Add 100 µl chloroform to the lysate, shake vigorously for 15 sec. incubate the resulting mixture at room temperature for 5 min, and centrifuge at 12,000 x g for 15 min at 4 °C.
- Following the centrifugation process, the mixture separates into 3 layers: a clear upper aqueous layer (containing RNA), an interphase and a red lower organic layer, carefully transfer 250 µl of the upper aqueous phase to a fresh tube.
- Precipitate RNA from the aqueous phase by mixing 250 µl (equal volume) of isopropanol.
- Store samples at room temperature for 10 min and centrifuge at 12,000 x g for 15 min at 4 °C.
- Decant the supernatant and wash the RNA pellet by vortexing in 1 ml 75% ethanol.
- Centrifuge the RNA suspension at 12,000 x g for 5 min at 4 °C.
- Remove the remaining ethanol by repeating the centrifugation process and briefly air-dry the RNA pellet for 5 min (air-dry can be carried on at room temperature).
- Dissolve RNA in 30 µl RNA storage solution.
- Measure the concentration of RNA by NanoDrop. Typically, 800-1,000 ng/µl RNA can be isolated from 1 ml of E. coli culture.
- Streak out the E. coli strain to be used for RNA extraction on an LB plate and incubate overnight at 37 °C.
- DNase I and Alkaline Phosphatase treatment
- Resuspend 25 µg RNA in 1x AP Reaction Buffer to a final volume of 50 µl.
- Add 3 U of DNase I, and 45 U of Alkaline Phosphatase, 20 U of RNasin® Ribonuclease Inhibitors, mix thoroughly and incubate at 37 °C for 30 min.
- Add 50 µl PCI, mix thoroughly by vortexing for 10 sec.
- Centrifuge at 12,000 x g for 5 min.
- Collect 30 µl of supernatant, and transfer to a Sephadex G-50 column.
- Centrifuge at 2,000 x g for 2 min, collect the pass through.
- Measure the RNA concentration by NanoDrop. Typically, 300-400 ng/µl RNA can be collected.
- Resuspend 25 µg RNA in 1x AP Reaction Buffer to a final volume of 50 µl.
- Ligation
- Deprotection of 3’-idT RNA oligo
- Centrifuge the tube briefly. Add 400 µl of deprotection buffer to the tube, vortex for 10 sec and centrifuge for 10 sec.
- Incubate at 60 °C for 30 min.
- Perform phenol extraction and ethanol precipitation as follows: add 400 µl of PCI solution to the tube, vortex for 5 sec.
- Centrifuge at 12,000 x g for 5 min at room temperature.
- Carefully remove the 300 µl upper aqueous phase to a new tube.
- Add 2.5 volume of ice cold 100% ethanol, 0.1 volume of 3 M sodium acetate, and vortex to mix thoroughly.
- Precipitate at -80 °C for 20 min.
- Centrifuge at 12,000 x g for 20 min at 4 °C.
- Decant the supernatant and carefully pipet off the remaining supernatant, wash pellet twice by adding 500 µl ice cold 70% ethanol.
- Air dry the pellet at room temperature and resuspend in 100 µl of RNase and DNase-free water.
- Centrifuge the tube briefly. Add 400 µl of deprotection buffer to the tube, vortex for 10 sec and centrifuge for 10 sec.
- Set up a 20 μl reaction as follows:
2 µl 10x RNA ligase buffer
0.25 µl RNasin (40 U/µl)
1 μl T4 RNA ligase (5 U/µl)
2.5 µg RNA
0.5 µl 3’ RACE RNA oligo (100 nM)
RNase-free water - Incubate at 37 °C for 3 h.
The sequence of 3’ RACE RNA oligo is 5’ P. UUCACUGUUCUUAGCGGCCGCAUGCUC.idT - Apply the ligation mix to a G-50 column for buffer exchange, centrifuge at 2,000 x g for 2 min and collect the pass through in a new micro centrifuge tube.
- Deprotection of 3’-idT RNA oligo
- Reverse transcription
- Assemble 3RP primer, dissolve the primer in RNase- and DNase-free water to make a 100 µM stock solution (= concentration). A working solution of 25 µM was made by diluting in RNase and DNase-free water from this stock.
Sequence of 3RP primer (5’AGCATGCGGCCGCTAAGAAC3’) - Set up a 20 μl reaction as follows:
2 µl 10x RTase buffer
0.25 µl RNasin (40 U/µl)
1 μl RTase (4 U/µl)
Ligation product 13.75 µl
1 µl 3RP primer (25 µM)
2 µl dNTP mix (5 mM) - Incubate at 37 °C for 2 h.
- Assemble 3RP primer, dissolve the primer in RNase- and DNase-free water to make a 100 µM stock solution (= concentration). A working solution of 25 µM was made by diluting in RNase and DNase-free water from this stock.
- PCR
- Assemble primers, as in Table 1.
- Set up a 50 μl reaction as follows:
5 µl 10x PCR buffer
0.5 µl HotStarTaq plus DNA polymerase (5 U/µl)
1 µl dNTP mix (10 mM)
1 µl 3RP primer (25 µM)
2.5 µl gal-specific primer (10 µM)
2 µl cDNA
38 µl RNase-free water - Transfer PCR tubes to a PCR machine with the lid pre-heated to 105 °C and run the thermocycling process. Thermocycling conditions for PCR are shown in Table 2.
Table 2. Thermocycling conditions for PCR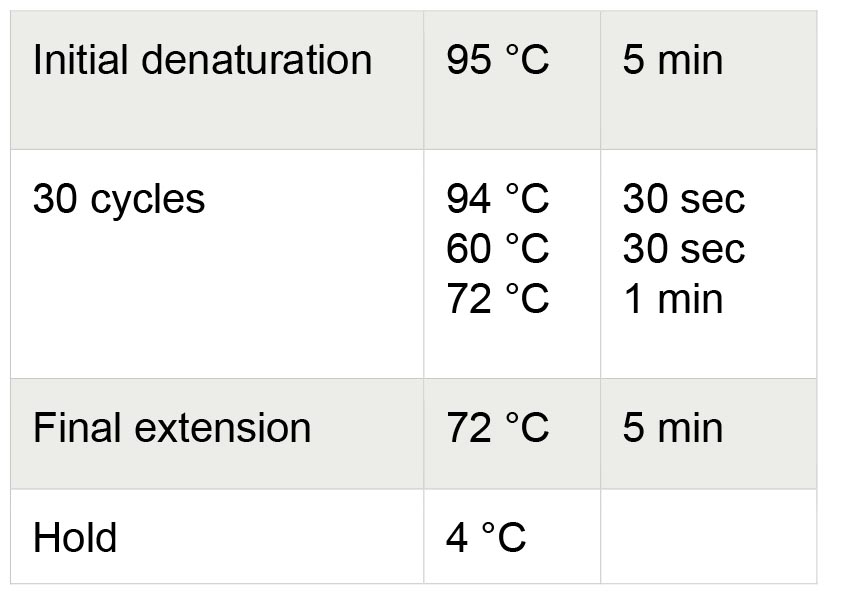
- Assemble primers, as in Table 1.
- Primer end labeling
- Set-up the following reaction to a final volume of 20 µl:
2 µl 10x kinase buffer
1 µl kinase (10 U/µl)
2 µl primer (10 µM)
2 µl [γ-32P] ATP
13 µl RNase-free water - Incubate at 37 °C for 30 min.
- Inactivate the enzyme at 65 °C for 30 min.
- Add 30 µl RNase and DNase-free water to the reaction mix, pass through the G-50 column by centrifuge at 2,000 x g for 2 min.
- Store the labeled primer at 4 °C.
- Set-up the following reaction to a final volume of 20 µl:
- Primer extension
- Set-up the following reaction to a final volume of 20 µl:
2 µl 10x PCR buffer
0.2 µl HotStarTaq plus DNA polymerase (5 U/µl)
0.3 µl dNTP mix (10 mM)
0.3 µl 5’ end labeled PE primer
2 µl DNA template
15.2 µl RNase-free water - Transfer PCR tubes to a PCR machine with the lid preheated to 105 °C and run the thermocycling process. Thermocycling conditions for PCR are shown in Table 3.
Table 3. Thermocycling conditions for PE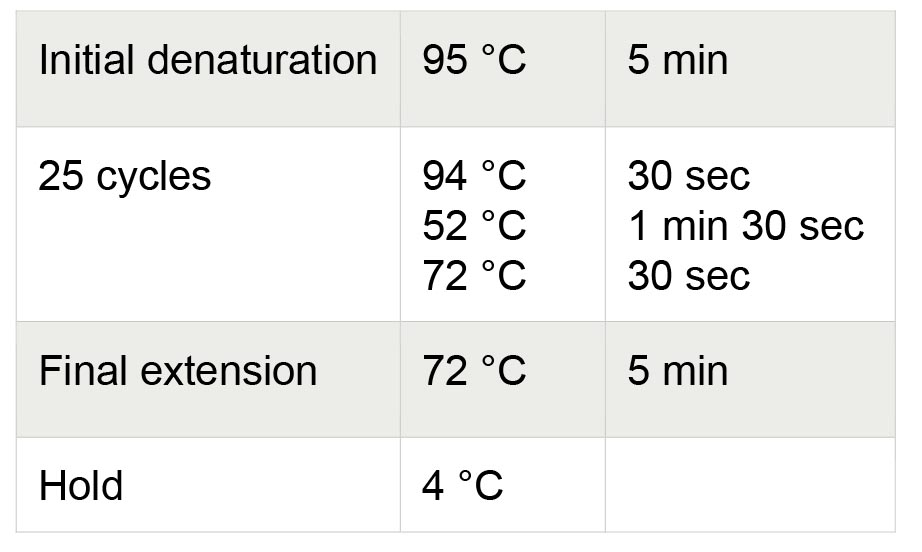
- Set-up the following reaction to a final volume of 20 µl:
- Gel electrophoresis
- Gel casting
- Assemble the gel according to manufacturer’s description and fix the gel in the gel-casting chamber. Use 0.4 mm thick spacers, glass plates (Inner plate x outer plate (W x H): 31 x 38.5 cm), 4 x 0.4 mm (14 cm) gel comb).
- Prepare 50 ml of the 8% urea-PAGE solution.
- Pour the gel immediately using an automatic pipette aid between the two glass plates. Avoid introducing air bubbles. Insert the comb and let the gel polymerize for 60 min. The assembled gel is indicated as in Figure 2.

Figure 2. A picture of assembled gel in the gel-casting chamber with the automatic pipette aid
- Assemble the gel according to manufacturer’s description and fix the gel in the gel-casting chamber. Use 0.4 mm thick spacers, glass plates (Inner plate x outer plate (W x H): 31 x 38.5 cm), 4 x 0.4 mm (14 cm) gel comb).
- Set up the electrophoresis apparatus and pre-run the gel
- Dismount the gel from the casting chamber and assemble it to the gel apparatus according to the manufacturer’s instructions.
- Fill the lower buffer chamber with 1x TBE running buffer, so that the glass plates will be submerged 2-3 cm with the buffer. Fill the upper buffer chamber up to the top of the gel with 1x TBE running buffer.
- Carefully remove the comb and rinse the wells with running buffer by using a pipette and gel loading tips.
- Attach the lid of the gel system and plug in the cables to a high voltage power supply. Before loading the samples you have to prerun the gel for at least 30 min to heat up the gel and to remove the remaining urea from the gel. The optimal temperature should be between 45-55 °C. Avoid temperatures higher than 60 °C as bands could smear or the glass plates could crack. Choose a constant watt for the prerun (30 W). The assembled electrophoresis apparatus is indicated as in Figure 3.

Figure 3. A picture of the assembled electrophoresis apparatus
- Dismount the gel from the casting chamber and assemble it to the gel apparatus according to the manufacturer’s instructions.
- Sample preparation
- Add 15 µl 2x gel loading mix to 15 µl sample.
- Denature the sample by heating at 95 °C for 3 min.
- Add 15 µl 2x gel loading mix to 15 µl sample.
- Load and run the gel
- Remove the lid and rinse the wells thoroughly as described earlier as urea will leach into the wells.
- Load 3 µl of samples carefully from the bottom of the wells. Avoid introducing air bubbles.
- Assemble the lid and run the gel at 30 W to maintain a gel temperature of 55 °C similar to the prerun. Observe the migration of the marker dyes until the dye front reached the lower end of the gel. A run can last 2-4 h.
- Remove the lid and rinse the wells thoroughly as described earlier as urea will leach into the wells.
- Process the gel
- Remove the gel cassette from the chamber by loosening the clamps. Pull away the spacers and carefully disassemble the glass plates. If necessary, cut away the upper well containing the gel part.
- Cut a Whatman 3MM paper to the same size of the gel, attach it to the gel, and carefully remove the gel from the glass plate.
- Carefully attach a saran wrap on top of the gel, seal it with scotch tape. A picture of the Whatman 3MM paper cut and saran wrapped is indicated as in Figure 4.
- Move the sealed gel to the X-ray cassette, put in a single piece of X-ray film, expose at -80 °C for 16 h.

Figure 4. A picture showing the Whatman 3MM paper cut the same size as the outer plate or gel, saran wrapped sealed with scotch tape placed in the X-ray cassette - After the exposure time, take out the cassette from the refrigerator, wait about 1 h until it warms up to the room temperature.
- Develop the X-ray film in a dark room. Firstly, dilute one-part concentrate of the developer to 4 parts of water. Mix and store it in a bottle. Prepare the fixer in the same way. Then, pour the developer into a container, take the exposed X-ray film out of the cassette, immerse the film into the developer. After several minutes, when the dark bands appear, rinse the film with distilled water and then fix the film in the fixer.
- Remove the gel cassette from the chamber by loosening the clamps. Pull away the spacers and carefully disassemble the glass plates. If necessary, cut away the upper well containing the gel part.
- Gel casting
Data analysis
The data can be scanned and quantified using the software ‘ImageJ’ (https://imagej.nih.gov/ij/). ImageJ is a Java based software by Wayne Rasband and others from National Institute of Health (USA).
- Scanning the film and setting the measurement criteria
- Create high resolution scans from the film in .tiff file format. Keep this as your source image, and use it for the quantification analysis.
- Start the ImageJ analysis software and load the previously saved .tiff file from the destination folder using the ‘File’ menu. Change the picture format to .jpeg and the picture mode to ‘grayscale’.
- Under the ‘Analyze’ menu, choose ‘Set Measurements’. From the checkboxes have the ‘Area’ checked (Figure 5).
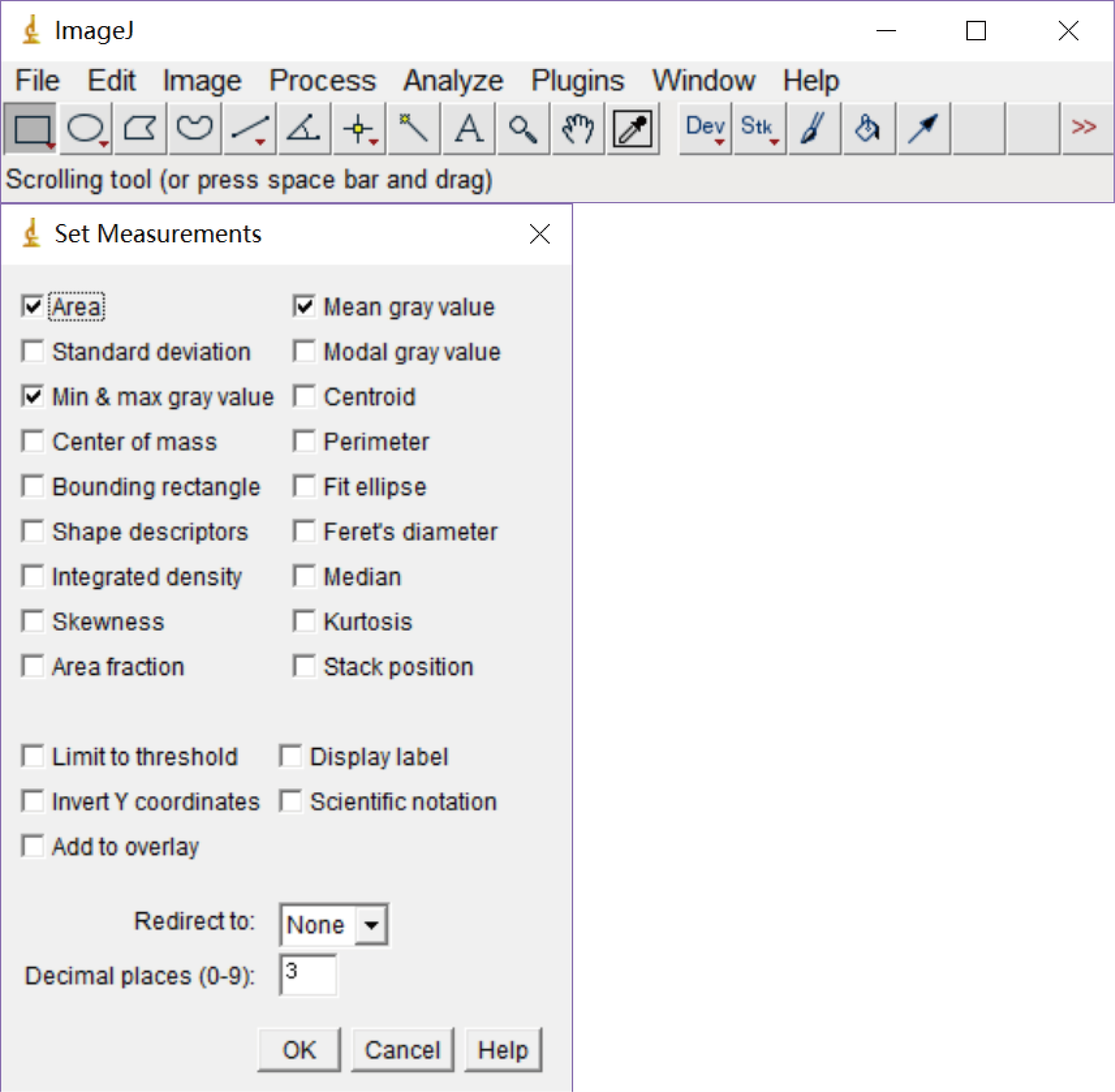
Figure 5. ImageJ software interface showing the Menu and tool features. The bottom picture explaining the different types of ‘Set Measurements’ options, checked ‘Area’ for our specific quantification of the 3’ end bands.
- Create high resolution scans from the film in .tiff file format. Keep this as your source image, and use it for the quantification analysis.
- Selection for measurements
- ImageJ selection of measurements defines a rectangle area of interest across the lanes of the final PE products of the 3’ RACE. Choose the ‘rectangle’ tool from ImageJ and draw a frame around the bands of the samples.
Notes:- You can trail around and resize the frame.
- Adjust it so that it covers the area to contain the whole of the largest and the smallest band of the sample row.
- You can trail around and resize the frame.
- For each of the analysis to take measurements, for instance in Figure 6, the first lane will be the control and all the other bands across the lanes will be the samples to quantify and compare. Centre the band inside the frame and utilize the ‘Ctrl’ + ‘M’ to record a measurement or choose ‘Measure’ from the ‘Analyze’ menu.
- This will spread out the measurement window and display the area peaks of the samples in order. Make a closed peak of the samples by using the ‘Straight’ tool to draw freehand lines to find the area of the individual sample peaks. Applying the ‘Wand’ tool obtains the area of the individual sample peaks as shown in Figure 6.
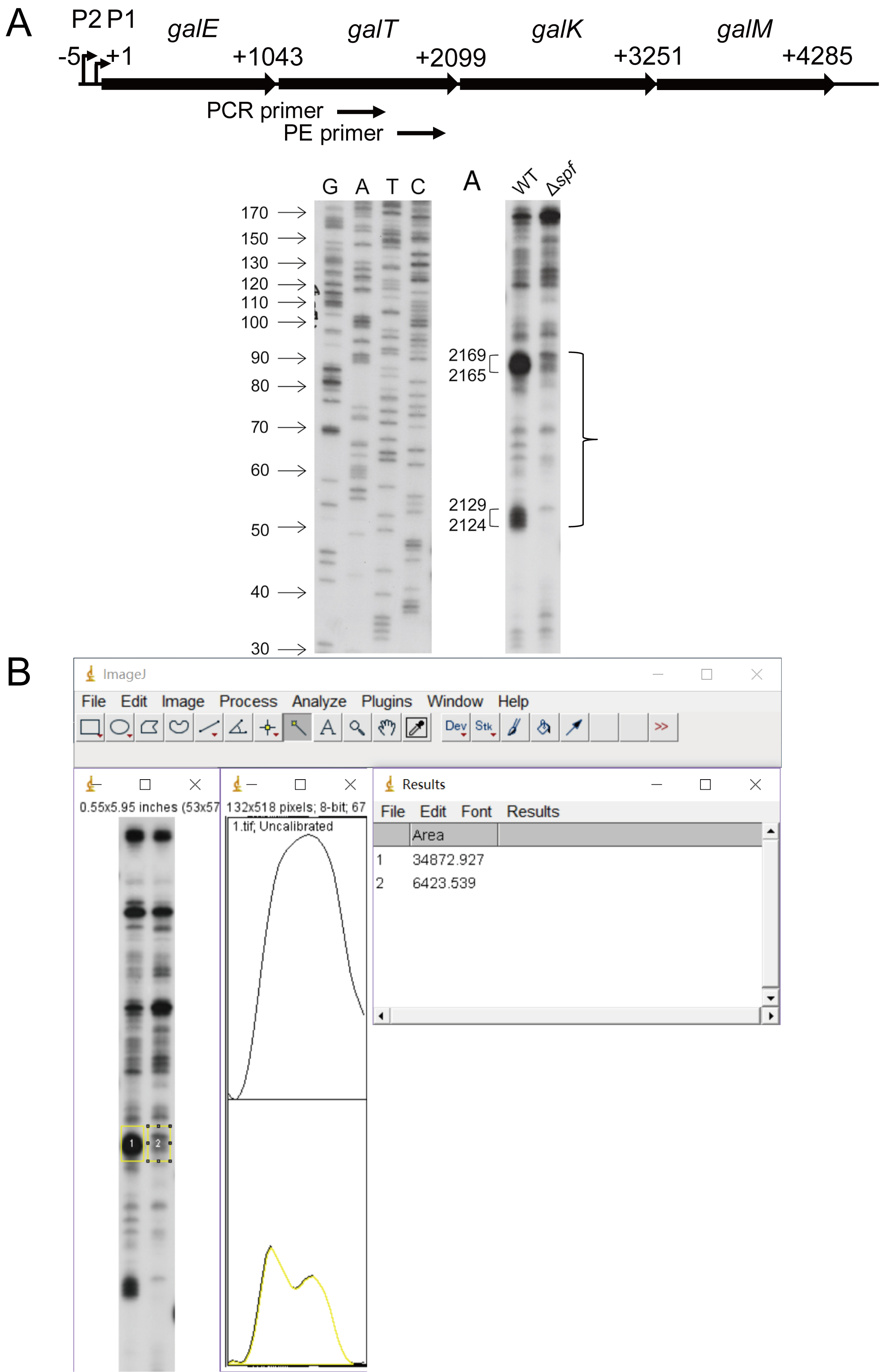
Figure 6. An example of quantifying 3’ RACE-PE results using ImageJ. A. A schematic of the galactose operon. The transcription initiation site from the P1 promoter is marked as + 1. The positions of primers used to amplify the 3’ ends of mT1 are indicated by small arrows. The gel image showing the 3’ ends of galT mRNA in WT and Δspf strains by 3’ RACE-PE. The numbers on the sides of each gel picture indicate relative positions to the P1 transcription start site (+1) (Lee et al., 2008). B. The peaks and area table of the mT1 bands. Bands framed by the ‘Rectangle’ tool in yellow is used to obtain the peaks and to find the area of these 3’ end bands.
- ImageJ selection of measurements defines a rectangle area of interest across the lanes of the final PE products of the 3’ RACE. Choose the ‘rectangle’ tool from ImageJ and draw a frame around the bands of the samples.
- Spreadsheets and calculations
- When the area of the peaks of the bands along with the loading control band is obtained, the data are transferred to an Excel sheet.
- When the area of sample bands with respect to the control band is recorded, a ratio of samples band value over the control band of that lane is calculated (Figure 6. Lane 2, 3, 4 over Lane 1 (Control)).
- The final relative quantitative values are the proportion of the sample bands to the control band.
- A bar (or other) graph is constructed to compare the samples with respect to the control.
- When the area of the peaks of the bands along with the loading control band is obtained, the data are transferred to an Excel sheet.
Notes
- The amount of starting sample should not exceed the capacity of Trizol-reagent. Overloading will reduce the quality of RNA.
- Use RNase-free reagents and filter tips during the whole process of the experiment. Wear gloves and keep the tubes closed throughout the procedure.
- PCR cycles can be modified depending on the amount of product.
- The time of exposure on the X-ray films from the gel at -80 °C can be modified according to the intensity of band.
- A specific area of the laboratory should be designated for P-32 handling. When handling radioactive materials, always wear protective gloves and clothing, use a Plexiglas shield to prevent radiation. Try to keep your exposure time as short as possible. The radioactive materials should be kept in lead-lined containers, and experiment wastes should be discarded in designated containers. Use the Geiger counter to check the radiation level in front of the shield and your gloves frequently for contamination.
Recipes
- LB + 0.5% galactose (1 L)
10 g tryptone
5 g yeast extract
10 g NaCl
Add distilled water up to 1 L
Autoclave
Add 25 ml filtered 20% galactose
Store at room temperature - 8% sequencing solution (1 L)
76 g acrylamide
4 g Bis-acrylamide
200 ml 5x TBE
500 g urea
Add distilled water up to 1 L
Dissolve by heating and stirring - 8% Urea-PAGE gel (50 ml)
49.67 ml 8% sequencing solution
300 µl 10% APS solution
30 µl TEMED - 2x gel loading mix
9 ml formamide
200 µl 0.5 M EDTA
100 µl 1% xylene cyanol
100 µl 1% bromophenol blue
Add RNase and DNase-free water up to 10 ml - Protoplasting buffer
750 µl 1 M Tris-HCl (pH8.0)
7.7 g sucrose
800 µl 0.5 M EDTA (pH8.0)
Add RNase and DNase-free water up to 50 ml
Acknowledgments
This work was supported by the National Science Foundation for Young Scientists of China (grant 31600061) and the China Postdoctoral Science Foundation (grants 2015M582234, 2017T100562). This research was also supported by a grant from Chungnam National University (2015-1420-01). This protocol was adapted from our publication in J Mol Biol (Lee et al., 2008), J Bacteriol (Wang et al., 2014) and PNAS (Wang et al., 2015). The authors declare no conflict of interest.
References
- Altman, S. and Robertson, H. D. (1973). RNA precursor molecules and ribonucleases in E. coli. Mol Cell Biochem 1(1): 83-93.
- Holden, H. M., Rayment, I. and Thoden, J. B. (2003). Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 278(45): 43885-43888.
- Lee, H. J., Jeon, H. J., Ji, S. C., Yun, S. H. and Lim, H. M. (2008). Establishment of an mRNA gradient depends on the promoter: an investigation of polarity in gene expression. J Mol Biol 378(2): 318-327.
- Nudler, E. and Gottesman, M. E. (2002). Transcription termination and anti-termination in E. coli. Genes Cells 7(8): 755-768.
- Sambrook, J. and Russell, D. W. (2006). Rapid amplification of 3' cDNA ends (3'-RACE). CSH Protoc 2006(1).
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
- Wang, X., Ji, S. C., Jeon, H. J., Lee, Y., Lim, H. M. (2015). Two-level inhibition of galK expression by Spot 42: Degradation of mRNA mK2 and enhanced transcription termination before the galK gene. Proc Natl Acad Sci U S A 112: 7581-7586.
- Wang, X., Ji, S. C., Yun, S. H., Jeon, H. J., Kim, S. W., and Lim, H. M. (2014). Expression of each cistron in the gal operon can be regulated by transcription termination and generation of a galk-specific mRNA, mK2. J Bacteriol 196: 2598-2606.
- Zhao, J., Hyman, L. and Moore, C. (1999). Formation of mRNA 3' ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 63(2): 405-445.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wang, X., Jeon, H. J., Abishek N, M. P., He, J. and Lim, H. M. (2018). Visualization of RNA 3’ ends in Escherichia coli Using 3’ RACE Combined with Primer Extension. Bio-protocol 8(5): e2752. DOI: 10.21769/BioProtoc.2752.
Category
Microbiology > Microbial biochemistry > RNA
Molecular Biology > RNA > 3' end analysis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.