- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Dual-sided Voltage-sensitive Dye Imaging of Leech Ganglia

Published: Vol 8, Iss 5, Mar 5, 2018 DOI: 10.21769/BioProtoc.2751 Views: 8235
Reviewed by: Oneil G. BhalalaMenghon CheahCarey Y. L. Huh
Original research article
The authors used this protocol in:

Sep 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
In this protocol, we introduce an effective method for voltage-sensitive dye (VSD) loading and imaging of leech ganglia as used in Tomina and Wagenaar (2017). Dissection and dye loading procedures are the most critical steps toward successful whole-ganglion VSD imaging. The former entails the removal of the sheath that covers neurons in the segmental ganglion of the leech, which is required for successful dye loading. The latter entails gently flowing a new generation VSD, VF2.1(OMe).H, onto both sides of the ganglion simultaneously using a pair of peristaltic pumps. We expect the described techniques to translate broadly to wide-field VSD imaging in other thin and relatively transparent nervous systems.
Keywords: Voltage-sensitive dye imagingBackground
A double-sided microscope is a wide-field fluorescence imaging system consisting of a pair of microscopes precisely aligned for viewing a neuronal preparation from opposite sides and with distinct focal planes at once (Tomina and Wagenaar, 2017). By combining this optical system with a new-generation voltage-sensitive dye (VSD), VoltageFluor (Miller et al., 2012; Woodford et al., 2015), fluorescence signals that encode membrane voltages with high fidelity can be simultaneously captured from neurons at different depths. We applied this pan-neuronal recording system to the nervous system of the medicinal leech, in which we elicited fictive behaviors and quantitatively manipulated membrane potential of identifiable neurons using electrophysiological methods (Tomina and Wagenaar, 2017). Fictive behaviors were induced in an isolated nervous system: local bending was elicited by intracellular stimulation of a pressure-sensitive neuron, while swimming and crawling were elicited by extracellular stimulation of a lateral nerve of a segmented ganglion and nerves of tail brain, respectively. We were able to analyze the dynamics of almost all individual identifiable neurons within a functional unit of the leech nervous system, allowing us to construct functional maps of the roles played by these neurons in various behaviors. The imaging technique potentially is applied to other nervous systems that have multiple layers of somata such as the pedal ganglia of Aplysia.
For successful VSD imaging with a double-sided microscope, three procedures are critical: (1) dissection of the target nervous system, (2) dye loading, and (3) VSD imaging itself. These procedures were not explained in detail in our previous paper (Tomina and Wagenaar, 2017), nor in other studies using the same type of dyes (Miller et al., 2012, Moshtagh-Khorasani et al., 2013, Woodford et al., 2015, Frady et al., 2016).
As part of the dissection process, removing the sheath from the surface of the target ganglion is necessary to ensure that the dye can reach the neurons. For keeping a preparation in a healthy state, it is important to handle the preparation in an adequate way and to load the dye into cells for an appropriate length of time. During VSD imaging, the nervous system must be strictly immobilized in order to suppress motion artifact. All those steps are critical for successful whole-ganglion VSD imaging using a double-sided microscope. This protocol provides the detailed procedures to realize wide-field VSD imaging in a whole ganglion of the leech.
Materials and Reagents
- Dissection
- Microdissection container, comprising:
- The lid of a 35-mm plastic Petri dish (e.g., Nunclon Delta, Thermo Fisher Scientific, catalog number: 153066 )
- 1.5-mm thick layer of transparent PDMS (Sylgard 184 elastomer, 0.5 mg kit, Dow Corning, catalog number: 4019862 )
- Disk of transparent PDMS (13 mm diameter, 0.65 mm thick) with a rectangular window (1.6 x 2.6 mm) cut out of it (Sylgard 184, as above)
- The lid of a 35-mm plastic Petri dish (e.g., Nunclon Delta, Thermo Fisher Scientific, catalog number: 153066 )
- Nylon head insect pins #6 (Emil Artl Elephant brand)
- Small pins (Fine Science Tools, catalog number: 26002-10 )
- Tungsten wires, 50 μm diameter (California Fine Wire, catalog number: MS138 )
- Artificial pond water (0.1 % ocean strength salt solution) (e.g., Sea salt, Instant Ocean, catalog number: SS15-10 )
- Nervous system of a leech
Note: We use adult medicinal leeches Hirudo verbana (2.5-3 years old), a standard species in neuroethology field, purchased from Niagara Leeches (Niagara Falls, NY, www.leeches.bi). H. medicinalis would be equally acceptable. Leeches were maintained in an aquarium tank (250 x 500 x 300 mm) half filled with artificial pond water with aeration at 15 °C and subjected to 12:12 light:dark cycle. - Coarse dissection container, comprising:
- Enclosure (AN-1321, Bud Industries, Digi-Key Electronics, catalog number: 377-1740-ND )
- Small aquarium pebbles (e.g., Imagitarium Mini White Aquarium Gravel, PetCo)
- Dark PDMS (Sylgard 170 silicone, 0.9 kg kit, Dow Corning, catalog number: 1696157 )
- Enclosure (AN-1321, Bud Industries, Digi-Key Electronics, catalog number: 377-1740-ND )
- Microdissection container, comprising:
- Dye loading
- Dye-loading dish connected with an outflow capillary, comprising:
- The lid of a 35-mm plastic Petri dish (e.g., Nunclon Delta, Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 153066 )
- Capillary glass, standard 1.2 x 0.68 mm, 4” (A-M Systems, catalog number: 627000 )
- Epoxy glue (e.g., Slow-cure 30 min Epoxy, Bob Smith Industries Inc.)
- Transparent PDMS (Sylgard 184 elastomer, 0.5 kg kit, Dow Corning, catalog number: 4019862 )
- The lid of a 35-mm plastic Petri dish (e.g., Nunclon Delta, Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 153066 )
- Tube for perfusion (Masterflex C-flex tubing, Cole-Parmer Instrument, catalog number: 06424-13 )
- Volume-restricting well:
A 3-mm thick slab of PDMS, 25 mm in diameter, with a 16-mm diameter hole cut out of it - 1,000 μl pipette tips (e.g., VWR, catalog number: 83007-380 )
- 200 μl pipette tips (e.g., VWR, catalog number: 53508-783 )
- 10 μl pipette tips (e.g., Thermo Fisher Scientific, Thermo Scientific, catalog number: 490014-502 )
- 70% EtOH
- Dye-loading dish connected with an outflow capillary, comprising:
- VoltageFluor solution
- VF2.1(OMe).H (provided by Evan Miller, University of California, Berkeley. Reported in Woodford et al., 2015)
Note: An alternative product can be commercially purchased: FluoVolt Membrane Potential Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: F10488 ). - Dimethyl Sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D4540 )
- Pluronic acid (PowerLoadTM Concentrate 100x, Thermo Fisher Scientific, InvitrogenTM, catalog number: P10020 )
- Hirudo verbana physiological saline (see Recipes)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P3911 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: 223506 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M9272 )
- Glucose (Sigma-Aldrich, catalog number: DX0145 )
- HEPES (Sigma-Aldrich, catalog number: H3375 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888 )
- VF2.1(OMe).H solution (see Recipes)
- VF2.1(OMe).H (provided by Evan Miller, University of California, Berkeley. Reported in Woodford et al., 2015)
- VSD imaging
Note: Refer to Tomina and Wagenaar (2017) for details on building a double-sided microscope. Here we describe the use of the microscope.- Recording dish, comprising:
- Medical dressing (Tegaderm transparent film dressing, 3M, catalog number: 9505W , e.g., Amazon)
- Modeling clay (Crayola, any color is fine)
- Lens cleaning paper (e.g., Tiffen, catalog number: EK1546027T , e.g., Adorama)
- Glass capillaries for intracellular recording (standard-wall 1.00 x 0.75 mm with filament, 4”, A-M Systems, catalog number: 615000 )
- Suction electrodes for extracellular recording, comprising:
- Glass capillary (standard-wall 1.20 x 0.68 mm, 4”, A-M Systems, catalog number: 627000 )
- Stainless steel blunt needle with Luer (Component Supply, catalog number: NE-252PL )
- 2.5-mm mono jack (e.g., CUI, Digi-Key Electronics, catalog number: MJ-2506 )
- 5-ml Luer-Lok tip Syringe (BD, catalog number: 309646 )
- Silver wire (200 μm diameter, California Fine Wire, catalog number: 100183 )
- Cast acrylic base, 2” x 0.5” x 0.25”
- Glass capillary (standard-wall 1.20 x 0.68 mm, 4”, A-M Systems, catalog number: 627000 )
- Petroleum jelly (e.g., Vaseline)
Equipment
- Dissection
- Microsurgical knife, 5.0 mm blade (SharpPoint, Surgical specialties, catalog number: 72-1551 )
- Coarse dissection scissors (e.g., Heritage, model: Electricians Scissors 103C ) for cutting body wall tissue
- Medium dissection scissors (e.g., Natsume Seisakusho, catalog number: MB-54-1 ) for dissecting connective and nerve tissues
- Microdissection scissors: 2-mm cutting edge vannas spring scissors (Fine Science Tools, catalog number: 15000-03 ) for desheathing of a leech ganglion
- Fine forceps (e.g., Dumont #5 Dumostar, Fine Science Tools, catalog number: 11295-10 )
- Coarse forceps (e.g., Peer-Vigor Tweezer Swiss #5, Peer, catalog number: 57.0805 )
- Stereomicroscope
Note: We use a ZEISS Stemi 2000-CS with W10x/21 eye pieces at 6.5-50x magnification for most of the dissection and a ZEISS SteREO Discovery V8 with 10x/23 eye pieces and a custom focus drive at 80x magnification for desheathing (ZEISS, models: Stemi 2000-CS and SteREO Discovery.V8 ). - LED illuminator with dual gooseneck light guide (Dolan-Jenner Fiber-Lite, model: Mi-LED-US-DG )
- Cooling plate base for dissection:
- DC Power supply (30 V x 10 A DC, e.g., Yescom, model: 15SDS019-DCP3010D-09 )
- Peltier modules (TE Technology, model: HP-127-1.4-2.5-72P )
- 0.25”-thick aluminum
- 18-gauge stainless steel
- Aquarium glue
- DC Power supply (30 V x 10 A DC, e.g., Yescom, model: 15SDS019-DCP3010D-09 )
- Microsurgical knife, 5.0 mm blade (SharpPoint, Surgical specialties, catalog number: 72-1551 )
- Dye loading
- Perfusion pump head (Easy-Load II Head, Masterflex L/S, Cole-Parmer Instrument, catalog number: EW-77201-60 )
- Perfusion pump driver (Economy drive, Masterflex L/S, Cole-Parmer Instrument, catalog number: 07554-80 )
- Miniature dovetail stage (Siskiyou, catalog number: DT3-100 )
- Glass cutting ceramic tile (Sutter Instrument, catalog number: CTS )
- Pipettor 100-1,000 μl (e.g., VWR, catalog number: 89079-974 )
- Pipettor 20-200 μl (e.g., VWR, catalog number: 89079-970 )
- Pipettor 0.5-10 μl (e.g., VWR, catalog number: 89079-960 )
- Vortexer (e.g., VWR, model: VM-3000 , catalog number: 58816-121)
Note: This product has been discontinued. - Centrifuge (e.g., Spectrafuge, Labnet International, model: C1301-R )
- LED illuminator with dual gooseneck light guide (Dolan-Jenner Fiber-Lite, model: Mi-LED-US-DG )
- Red filter for illuminator (660-nm long-pass, 0.5” diameter, Edmund Optics, catalog number: 66-045 )
- Instrument for drilling a hole in dye-loading/recording dishes (e.g., a lathe such as: Jet Tools, model: BDB-1340A , catalog number: 321102AK)
- Perfusion pump head (Easy-Load II Head, Masterflex L/S, Cole-Parmer Instrument, catalog number: EW-77201-60 )
- Optical imaging
- Upright fluorescent microscope (Olympus, model: BX51 )
- Fluorescence train of an inverted fluorescent microscope (Olympus, model: IX51 )
- Water-immersion 20x objective lens for top microscope (Olympus, model: XLUMPlanFLN20XW )
- 5x objective lens top microscope (Olympus, model: MPlan FLN )
- 20x objective lens for bottom microscope (Olympus, model: UCPlanFLN )
- Vibration isolation table (e.g., Newport, models: LW3048B-OPT or VIS3048-SG2-325A )
- Blue LED (LedEngin, model: LZ1-10B200 )
- LED controller (according to Wagenaar, 2012)
- CCD cameras (Photometrics, model: QuantEM 512SC )
- Intracellular amplifiers (A-M Systems, model: Model 1600 )
- Four-channel differential amplifier (A-M Systems, model: Model 1700 )
- Flaming/Brown micropipette puller (Sutter Instrument, model: P-97 )
- Data acquisition board (National Instruments, model: NI USB-6221 )
- Upright fluorescent microscope (Olympus, model: BX51 )
Procedure
- Making tools
- Coarse dissection container:
- Fill the base of the container with aquarium pebbles to about half its height.
- Pour black Sylgard (PDMS) into the container up to 10 mm from the top.
Note: It is critical that the pebbles are generously covered. - Leave the PDMS to set overnight at room temperature or in a lab oven set to 65 °C.
- Fill the base of the container with aquarium pebbles to about half its height.
- Microdissection container:
- Pour 0.65-mm thick layer of transparent Sylgard (PDMS) into one plastic Petri dish, and a 2-mm thick layer into another.
- Allow the PDMS to set (as above).
- Use a microsurgical knife to cut a disk (13 mm in diameter) with an open window (1.6 x 2.6 mm) out of the 0.65-mm thick PDMS layer.
- Pour 0.65-mm thick layer of transparent Sylgard (PDMS) into one plastic Petri dish, and a 2-mm thick layer into another.
- Dye-loading dish (Figure 1):
- Into the center of a plastic Petri dish, drill a hole of just over 1.2 mm diameter for a capillary to fit through using a belt drive bench lathe.
- Heat capillary glass over a gas flame and shape it into an ‘L’ with a short leg of 7 mm. Cut the long leg to 10 mm.
- Insert the short leg of the L into the hole from the outside.
- Glue the capillary to the dish with epoxy glue.
- Connect the end of the capillary with a short tube connected to a short (18 mm) straight capillary. This short capillary is connected to the tubing of the perfusion pump.
- Pour a 2-mm-thick base layer of PDMS into the Petri dish and let it set overnight.
- Cut a 2-mm-diameter hole out of the PDMS directly over the capillary and verify that liquid can pass through the capillary.

Figure 1. Double-sided dye-loading system. A. Top view of a dye-loading dish. In the center of the dish there is an outflow hole for loading dye onto the bottom surface of the ganglion. The hole is connected to capillary and tube for circulation pumping. A well formed with PDMS substrate around the hole helps reduce the required volume of VSD solution. The dish is placed on two aluminum blocks so that the dish, the capillary, and the tube are stably positioned. B. Side view of a dye-loading dish. Glass capillary inserted into the bottom hole of the dish is stabilized with epoxy glue (yellow matter). (For cleaning, the dish and tube can be separated by pulling out the non-glued part of capillary and tube.) C. Spatial arrangement of outflows and intakes of the dye-loading system. PDMS disk with a target ganglion on its center window is put in the center of the well so that the outflow from the bottom side reaches the bottom surface of the ganglion. Intake capillaries are positioned close to the edge of the well so as not to bump the PDMS disk. Top outflow is placed just above the top surface of the ganglion.
- Into the center of a plastic Petri dish, drill a hole of just over 1.2 mm diameter for a capillary to fit through using a belt drive bench lathe.
- Recording dish
- Make a hole (14 mm in diameter) in the center of the plastic dish.
Note: This can be easily done using a laser cutter, a lathe, or a mill. We have found it difficult to achieve clean holes with a handheld drill. - Place an 18 mm diameter round cover glass on the bottom of the center hole, apply PDMS around the rim of the cover glass to adhere it to the Petri dish, and leave it to set overnight.
- Pour transparent Sylgard (PDMS) into the dish to a thickness of 1.5 mm, again leaving it to set overnight.
- Cut a circle out of the PDMS slightly larger than the hole in the bottom of the dish.
- Completely remove any pieces of PDMS substrate from the cover glass.
- Make a hole (14 mm in diameter) in the center of the plastic dish.
- Coarse dissection container:
- Dissection (Video 1: Coarse dissection, Video 2: Microdissection)
 Video 1. Isolation of the leech nervous system (a short chain of ganglia). 1. Anesthetize a leech in ice-cold saline. 2. Pin down the leech’s head and tail, using insect pins on a coarse dissection container. 3. Cut the dorsal skin along with the midline with a pair of dissection scissors. 4. Cut open the skin and pin it down. 5. Find a target ganglion for imaging. 6. Use a microsurgical knife to expose a lateral nerve root to be extracellularly recorded, if needed. 7. Dissect away the blood sinus surrounding the target ganglion. 8. Dissect away the blood sinus with the lateral nerves of the ganglion. 9. Isolate a short chain of ganglia. 10. Aspirate the isolated preparation into a pipette. Video 2. Double-sided desheathing of a leech ganglion. 1. Position a disk of transparent PDMS in the center of the container. 2. Transfer the ganglion into a microdissection container filled with cold saline. 3. Place the ganglion over the disk of transparent PDMS. 4. Use short pieces of tungsten wire to pin down sinus (the ganglion’s ventral side is up). 5. Stretch the connectives to apply tension to the ganglion’s sheath. 6. Zoom in on the target ganglion. 7. Remove the ventral sheath from the glial packet in the following order: the posterior, the central, and the lateral glial packets. 8. Check that the cells are not displaced or damaged. 9. Unpin the ganglion, flip the dorsal side up, then pin it down. 10. Zoom in on the ganglion. 11. Remove the dorsal sheath from the lateral glial packets (posterior to anterior). 12. Check that the cells are not displaced or damaged. 13. Unpin the ganglion, flip the ventral side up, then pin it down. 14. Check that the cells are not displaced or damaged.
Video 1. Isolation of the leech nervous system (a short chain of ganglia). 1. Anesthetize a leech in ice-cold saline. 2. Pin down the leech’s head and tail, using insect pins on a coarse dissection container. 3. Cut the dorsal skin along with the midline with a pair of dissection scissors. 4. Cut open the skin and pin it down. 5. Find a target ganglion for imaging. 6. Use a microsurgical knife to expose a lateral nerve root to be extracellularly recorded, if needed. 7. Dissect away the blood sinus surrounding the target ganglion. 8. Dissect away the blood sinus with the lateral nerves of the ganglion. 9. Isolate a short chain of ganglia. 10. Aspirate the isolated preparation into a pipette. Video 2. Double-sided desheathing of a leech ganglion. 1. Position a disk of transparent PDMS in the center of the container. 2. Transfer the ganglion into a microdissection container filled with cold saline. 3. Place the ganglion over the disk of transparent PDMS. 4. Use short pieces of tungsten wire to pin down sinus (the ganglion’s ventral side is up). 5. Stretch the connectives to apply tension to the ganglion’s sheath. 6. Zoom in on the target ganglion. 7. Remove the ventral sheath from the glial packet in the following order: the posterior, the central, and the lateral glial packets. 8. Check that the cells are not displaced or damaged. 9. Unpin the ganglion, flip the dorsal side up, then pin it down. 10. Zoom in on the ganglion. 11. Remove the dorsal sheath from the lateral glial packets (posterior to anterior). 12. Check that the cells are not displaced or damaged. 13. Unpin the ganglion, flip the ventral side up, then pin it down. 14. Check that the cells are not displaced or damaged.- Place a leech in a saline-filled plastic container with an ice cube made of frozen saline and allow the leech to cool down for 10-20 min before dissection.
Note: When anesthetized sufficiently, leeches do not show the fast reflexive twisting-like response that unanesthetized leeches display when pinned down at their head or tail. - Transfer the leech to a ‘coarse dissection container’ (see ‘Materials and Reagents’) that has been kept in a fridge or freezer.
- Use insect pins to immobilize the leech for dissection.
Note: Throughout the dissection, keep the container set on a cooling plate (see ‘Equipment’) and use a stereomicroscope for all procedures. - Cut away tissue as needed to isolate the central nervous system of the leech (either the whole nerve cord or a short chain of ganglia).
Note: The blood sinus surrounding the nervous system needs to be dissected away only around the ganglion targeted for imaging, not around any other ganglia. - Aspirate the isolated preparation into a pipette and transfer it into a microdissection container (see ‘Materials and Reagents’) filled with saline.
- Place this container on a cooling plate and use a high-magnification stereomicroscope for the remaining steps.
- Position the disk of transparent PDMS (see ‘Materials and Reagents’) in the center of the container.
- Place the target ganglion over disk, ventral side up, centered over the window so that the PDMS substrate does not touch the dorsal side of the ganglion.
- Use short pieces of tungsten wire to pin down blood sinus tissue that surrounds the lateral nerve roots to the disk.
- Using micro-scissors or a microsurgical knife, carefully and cleanly remove the sheath from the ventral and dorsal surfaces of the ganglion with the following steps:
- Remove the ventral sheath (Figure 2A).
- Carefully unpin the ganglion as necessary to flip it upside down over the window in the PDMS disk, taking care not to damage any neurons in the process.
- Remove the dorsal sheath (Figure 2B).
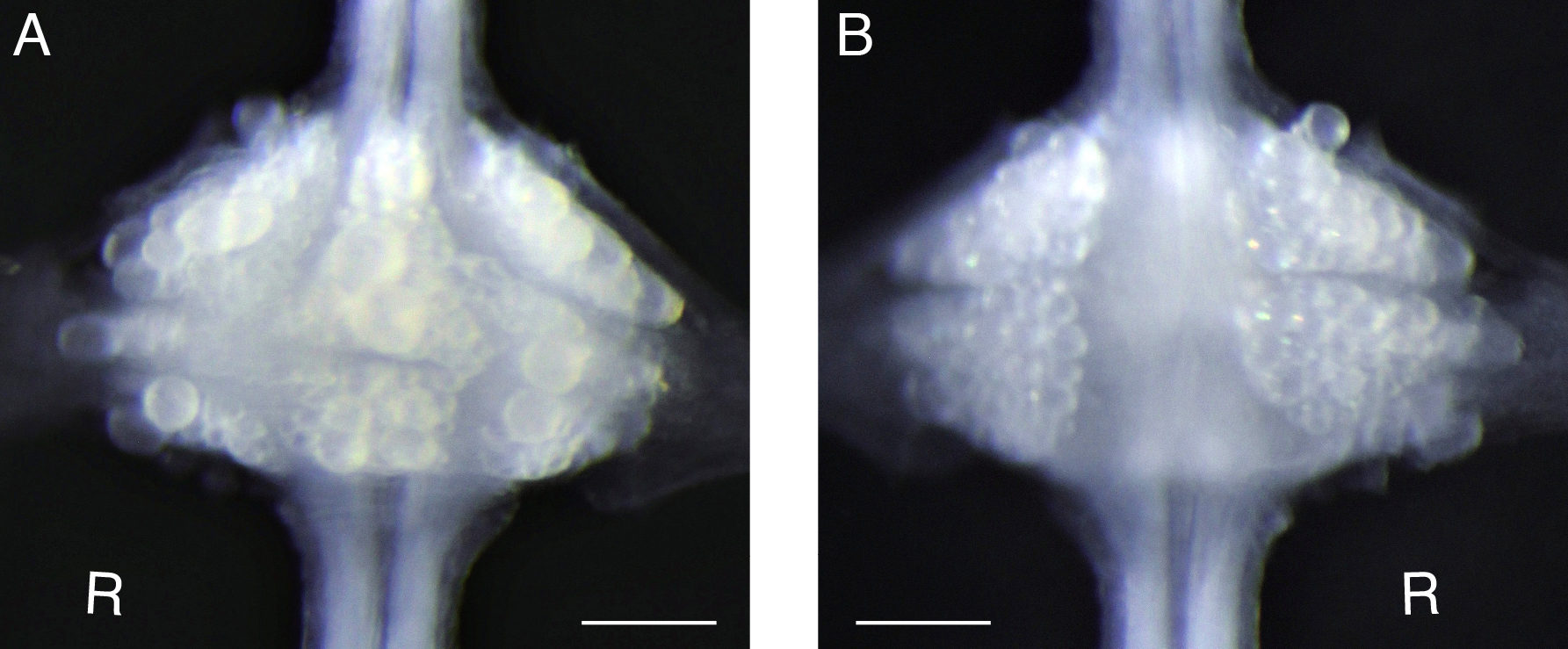
Figure 2. Desheathed ganglion. For voltage-sensitive dye loading, sheath tissue covering the target ganglion (e.g., the 10th segmental ganglion) is removed from both ventral (A) and dorsal (B) sides. ‘R’ indicates the right side of the ganglion, i.e., the animal’s right side when oriented dorsal side up. Scale bars = 100 μm. - Remove the ventral sheath (Figure 2A).
- Place a leech in a saline-filled plastic container with an ice cube made of frozen saline and allow the leech to cool down for 10-20 min before dissection.
- Dye loading
- Prepare 2 ml of VSD solution (800 nM VF2.1(OMe).H) in leech saline containing 1% pluronic acid, see Recipes) and leave it at room temperature for at least an hour in the dark.
Note: Using freshly made cold VSD solution results in significantly compromised staining and hence a dramatic drop in signal quality. Make sure that dye loading is carried out under dim red light to avoid bleaching the dyes. - Place the ‘volume-restricting well’ into the ‘dye-loading dish’ (see ‘Materials and Reagents’), centered over the bottom outflow capillary.
- Position the top outflow capillary loosely over the center of the well.
- Fill the well with VSD solution, and dip the two intake capillaries into the solution near the edge of the well.
- Set both peristaltic pumps to a flow rate of about 1.1 ml/min (speed knob level 2), and verify that perfusion works well.
Note: Make sure that no bubbles accumulate inside the outflow capillaries. If bubbles are stuck inside the capillaries, temporarily increase the pumping speed to eject the bubbles. Removing bubbles must be done before initiation of VSD staining. - Transfer the PDMS disk with the ganglion still attached to it into the well in the dye-loading dish and use one or two insect pins to secure it with the ganglion hanging over the outflow capillary (Figure 1A).
Note: The ganglion is ‘hanging’ below the disk at this time with the dorsal side facing down. - Bring the top outflow capillary close to the ganglion (about 1-mm distance; Figure 1C) and use both peristaltic pumps to circulate the VSD solution (1.1 ml/min flow rate) for 8 min.
- Flip the PDMS disk with the ganglion attached to it upside down within the well (so the ganglion is on top of the disk and the dorsal side is facing up), taking care to keep it centered over the bottom outflow capillary.
- Use both peristaltic pumps to circulate the VSD solution for a further 12 min.
Note: For well-balanced brightness on the both sides of the imaged preparation, what will be the bottom of the preparation must be stained more strongly than the top because the excitation light onto the top focus plane is brighter than that onto the bottom focus plane. The top outflow more efficiently stains a ganglion than the bottom outflow, so the choice of 8 min circulation in Step C7 and 12 min in Step C9 causes the dorsal side to be more strongly stained than the ventral side. This is by design: it compensates for the reduced excitation efficiently at what will be the bottom side of the ganglion during imaging. - Gently wash the preparation with cold saline after dye loading and completely replace VSD-containing saline with normal cold saline.
- Clean the outflows tubes and capillaries by circulating with 70% EtOH and distilled water.
Note: This can be done after imaging.
- Prepare 2 ml of VSD solution (800 nM VF2.1(OMe).H) in leech saline containing 1% pluronic acid, see Recipes) and leave it at room temperature for at least an hour in the dark.
- Optical imaging
- Stabilize the target ganglion for imaging by tightly pinning down blood sinus tissue that surrounds the nerve roots to the PDMS disk and by sandwiching adjacent connectives between small pieces of medical dressing (Figure 3C), which must also be pinned down, to minimize any motion artifacts.
- Place a small amount of petroleum jelly along the rim of the cover glass of a ‘recording dish’ (see ‘Materials and Reagents’; Figure 3A).
- Place the disk with the ganglion in the center of the recording dish, pushing it down into the petroleum jelly.
- Pull blood sinus tissue around the adjacent connectives from the ganglion so that the tissue can be tightly pinned down onto the periphery PDMS substrate of the recording dish (Figure 3B).
- Clean the bottom surface of the cover glass with lens cleaning paper before imaging.
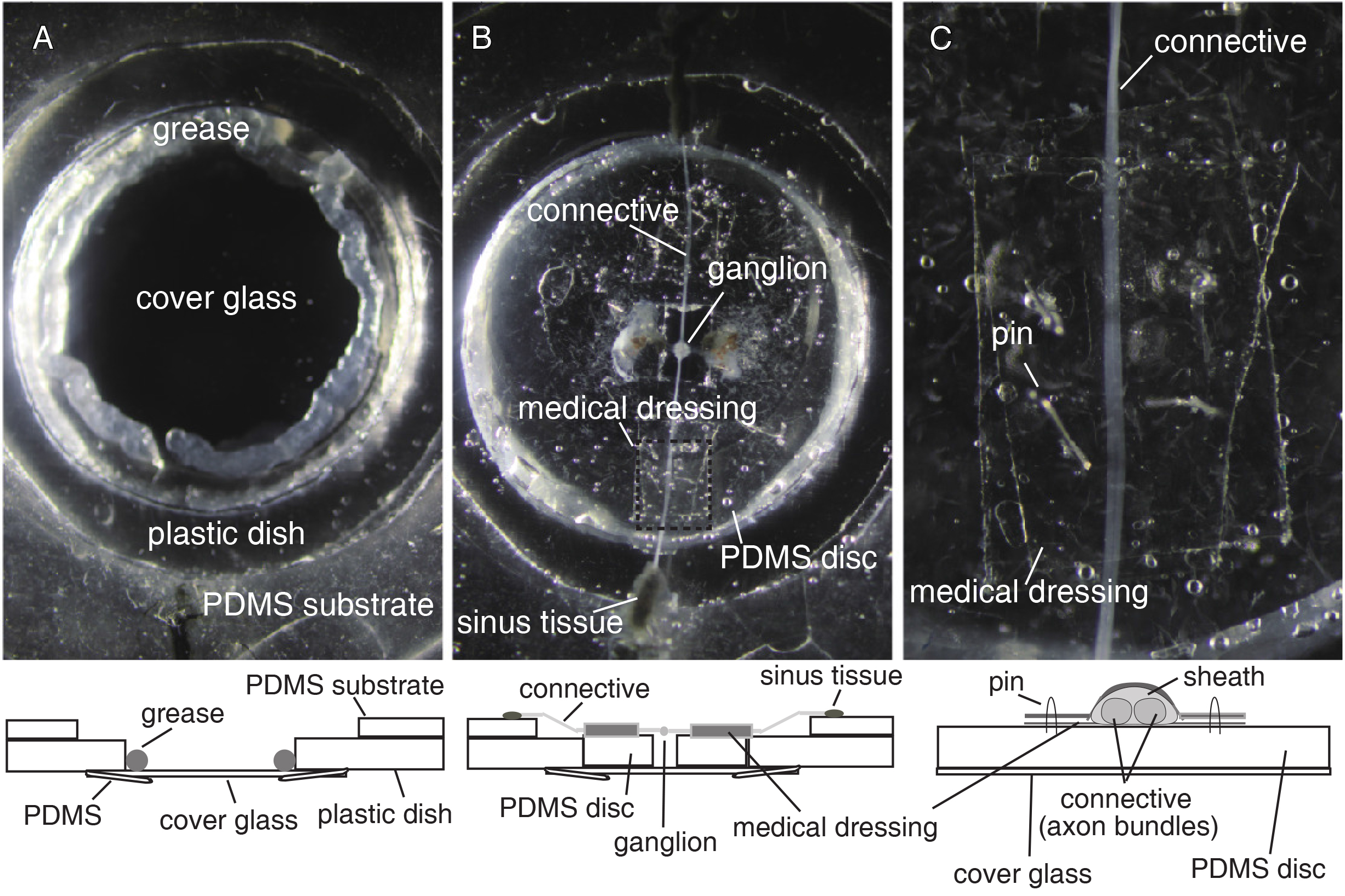
Figure 3. A recording dish for double-sided VSD imaging. A. Top view of the center area of recording dish (top) and a schematic frontal section view (bottom). Before placing a PDMS disk with a preparation into the dish, petroleum jelly is applied to the periphery of the cover glass. B. Top view of PDMS disk with a preparation set on the cover glass (top) and a schematic frontal section view (bottom). To minimize motion artifacts, tension is created by pinning the blood sinus tissues around the adjacent ganglia down onto the PDMS substrate of the outer area of the dish. C. An adjacent connective sandwiched between two pieces of medical dressing (top) and a schematic frontal section view (bottom). The pieces of medical dressing are pinned down onto the PDMS disk. - Place the recording dish on the stage of a double-sided microscope (Figures 4A and 4B).
- Adjust the orientation of preparation using top (upright) microscope under red light exposure.
- Prepare for extracellular and/or intracellular recording by positioning electrodes near target nerves or cell bodies (Figure 4C).
Note: Time interval between the end of dye-loading and the beginning of VSD imaging is typically 20-25 min. Spending much more time here may negatively impact the health of the ganglion.
Figure 4. Double-sided microscope. A. Overview of a double-sided microscope. The fluorescence train of an Olympus BX upright microscope is mounted with a custom focus rack on top of the body of an Olympus IX inverted microscope. Images are acquired with two CCD cameras. B. Top and bottom 20x objectives precisely aligned for dual-sided VSD imaging. A recording dish is put in the cutout (white) of the plastic stage. Sticky clay is used for fixing the position of the dish. C. Simultaneous electrophysiology and double-sided VSD imaging. The top side of the preparation is accessible to both intracellular and extracellular electrodes. - Aspirate target nerves into suction electrodes and/or impale target cells.
- Bring top and bottom surfaces of the ganglion coarsely into focus using top and bottom objectives.
- Adjust the brightness of images to obtain the best brightness but not to saturate the CCD cameras using the intensity control of the LED controller or the aperture stop of the microscope.
- Determine the final focus for top and bottom imaging.
Note (important): Throughout all these preparations, frequently replace saline with cold saline. - Start VSD imaging/electrophysiological trial using VScope (Wagenaar, 2017).
Note: Make sure that optical imaging is carried out in dark surroundings. - After finishing the VSD imaging experiment, acquire snapshots of both top and bottom aspects of the ganglion at different focusing depths for later creation of ‘focus-stacked’ images of the ganglion (Figure 5). These will be used for drawing for Regions of Interest (ROIs) in VScope.
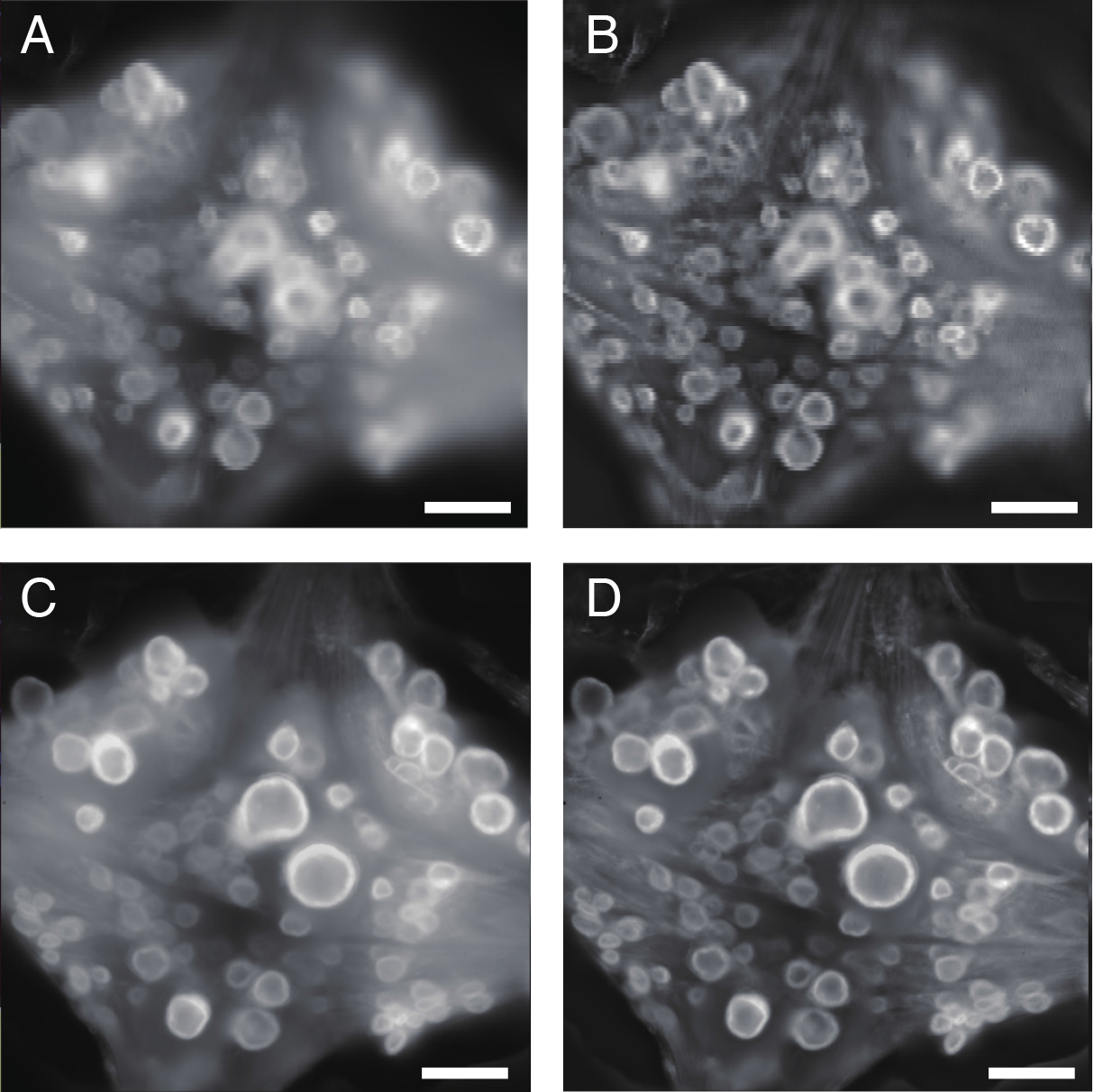
Figure 5. Visualizing the ganglion. A. Single snapshot image of the ventral aspect of the 10th ganglion as captured by top camera. B. The same image after processing with adaptive contrast enhancement (ACE) filter. C. Focus-stacked image constructed from multiple snapshots of the ventral aspect at different depths. D. The same image after processing with ACE filter. Scale bars = 100 μm.
- Stabilize the target ganglion for imaging by tightly pinning down blood sinus tissue that surrounds the nerve roots to the PDMS disk and by sandwiching adjacent connectives between small pieces of medical dressing (Figure 3C), which must also be pinned down, to minimize any motion artifacts.
Data analysis
- Even during an ongoing experiment, VScope (Wagenaar, 2017) can be used to check if the acquired optical signals show neuronal activity (spontaneous activity, stimulus-induced response, or fictive behavior). The rest of the analysis is usually performed after the end of an experiment.
- Draw ROIs in focus-stacked images using VScope’s ‘adaptive contrast enhancement’ filter (Figure 5), and then transfer ROI information to individual trials.
- Load the data into Octave using the functions provided with VScope.
- Clean up VSD imaging data using motion correction and debleaching algorithms (Tomina and Wagenaar, 2017).
- Perform further statistical analysis as relevant to your experiment, e.g., using Octave (Figure 6).
- Check that intracellular recording matches VSD signals from selected neurons to confirm the reliability of optical recording. Also check whether stereotypical motor patterns are observed in representative neurons like ventral/dorsal motor neurons (DI1, VI2, DE3, and VE4) especially when experiments involve fictive behaviors (Figure 6).
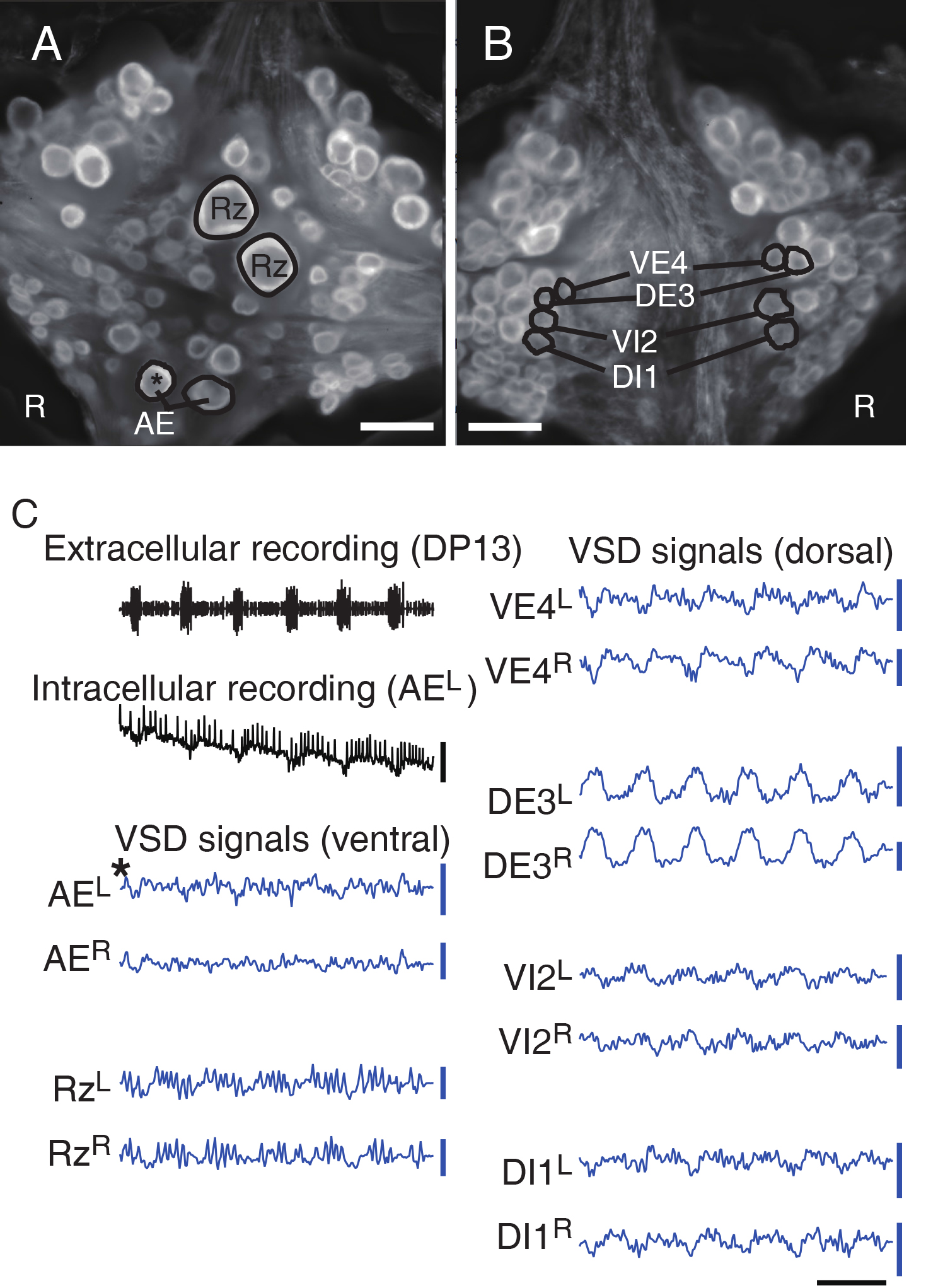
Figure 6. Double-sided VSD imaging. A and B. Dual surface images (ventral in A; dorsal in B) simultaneously captured with two CCD cameras. Scale bars = 100 μm. C. Selected electrophysiological and VSD traces during fictive swimming: Extracellular recording from a nerve root in a posterior segment (DP nerve of 13th ganglion) showing rhythmic dorsal excitatory motor neuron bursts; Intracellular recording and simultaneous optical signal from left AE cell (a motor neuron, marked with *) show matching membrane potential oscillations; VSD signals from the ventral surface: bilateral AE cells and Retzius cells (a neuromodulatory neuron); VSD signals from the dorsal surface: dorsal and ventral inhibitory and excitatory motor neurons DI-1, VI-2, DE-3, and VE-4. Scale bars = 1 sec for time, 10 mV for membrane potential and 0.2% ΔF/F for fluorescence signals.
Note: Sample data, codes and detailed application manual are available in Dryad Digital Repository (https://doi.org/10.5061/dryad.m20kh).
Notes
- Dissection should be done quickly and efficiently (typically 10-30 min for the isolation of the leech nervous system, 15-20 min for double-sided desheathing), and saline should be replaced with cold fresh saline frequently (every 2-3 min) with a pipette, because the health of the leech nervous system is critical for the success of experiments.
- To suppress motion artifact from contractile tissue within sheath around the nerve cords, connectives and lateral nerves should be physically stretched by pinning down the remaining blood sinus onto PDMS substrate after dye loading. If non-negligible motion artifact is observed during imaging, the strength of its stretch should be increased.
- Stocks of VF2.1 (OMe).H dissolved in DMSO should be kept in the refrigerator at 4 °C. To maintain the quality of staining condition, do not keep the dye at room temperature or in the deep freezer.
Note: According to a manual of a commercial product of VoltageFluor (FluoVolt Membrane Potential Kit), this product should be stored at 2-8 °C. - We qualitatively assess how well our specimens were stained using the fluorescent images and signals obtained during the experiment (examples shown in Figures 5 and 6). This is presently the only way to confirm that the staining process went well. Looking for visual changes in a ganglion during dye loading does not provide reliable information regarding the quality of staining.
Note: We recommend that beginners of VSD imaging in the leech first practice to obtain consistent results between intracellular recordings and VSD signals by targeting a Retizus cell or any other cells that are easy to impale.
Recipes
- Hirudo verbana physiological saline
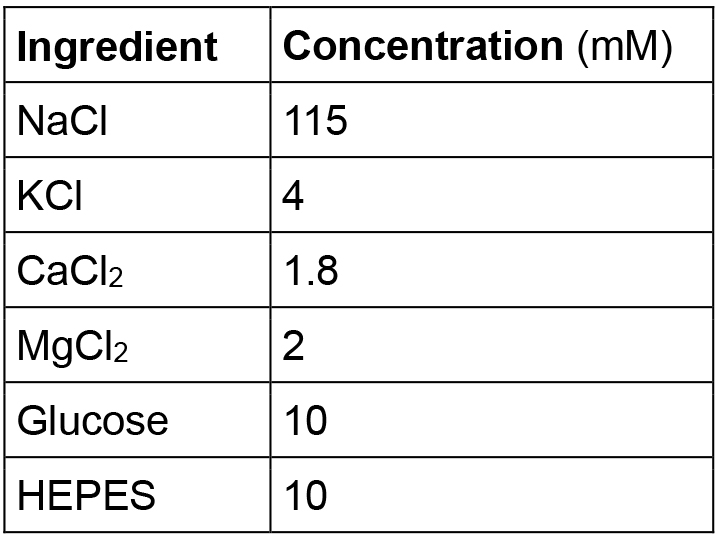
Note: Adjust pH to 7.4 with NaOH or HCl. - 800 nM VF2.1(OMe).H solution (1 ml)
- Aliquot 8 μl of 100 μM VF2.1(OMe).H (stock solution in DMSO, kept in the refrigerator) in a micro-tube and keep them in the refrigerator in advance
- Mix this with 10 μl of pluronic acid
- Add leech saline to make its final volume 1 ml and vortex it
Note: This recipe applies to commercially available VoltageFluor dye. - Aliquot 8 μl of 100 μM VF2.1(OMe).H (stock solution in DMSO, kept in the refrigerator) in a micro-tube and keep them in the refrigerator in advance
Acknowledgments
We thank Evan Miller for supplying the VF2.1(OMe).H dye; Annette Stowasser for her role in developing a prototype of the double-sided microscope and many helpful conversations; Angela Bruno for useful discussions regarding data analysis; Ng Cai Tong for reading and checking the manuscript. This work was supported by the Burroughs Welcome Fund through a Career Award at the Scientific Interface and by the National Institute of Neurological Disorders and Stroke through grant R01 NS094403 (both to DAW). YT was supported by JSPS Overseas Research Fellowships. This protocol was adapted from procedures published in Tomina and Wagenaar (2017). The authors of this work declare no conflicts of interest.
References
- Frady, E. P., Kapoor, A., Horvitz, E. and Kristan, W. B., Jr. (2016). Scalable semisupervised functional neurocartography reveals canonical neurons in behavioral networks. Neural Comput 28(8): 1453-1497.
- Miller, E. W., Lin, J. Y., Frady, E. P., Steinbach, P. A., Kristan, W. B., Jr. and Tsien, R. Y. (2012). Optically monitoring voltage in neurons by photo-induced electron transfer through molecular wires. Proc Natl Acad Sci U S A 109(6): 2114-2119.
- Moshtagh-Khorasani, M., Miller, E. W. and Torre, V. (2013). The spontaneous electrical activity of neurons in leech ganglia. Physiol Rep 1(5): e00089.
- Tomina, Y. and Wagenaar, D. A. (2017). A double-sided microscope to realize whole-ganglion imaging of membrane potential in the medicinal leech. Elife 6.
- Wagenaar, D. A. (2012). An optically stabilized fast-switching light emitting diode as a light source for functional neuroimaging. PLoS One 7(1): e29822.
- Wagenaar, D. A. (2017). VScope – data acquisition and analysis for voltage-sensitive dye imaging using multiple cameras and electrophysiology. Journal of Open Research Software 5: 23.
- Woodford, C. R., Frady, E. P., Smith, R. S., Morey, B., Canzi, G., Palida, S. F., Araneda, R. C., Kristan, W. B., Jr., Kubiak, C. P., Miller, E. W. and Tsien, R. Y. (2015). Improved PeT molecules for optically sensing voltage in neurons. J Am Chem Soc 137(5): 1817-1824.
Article Information
Copyright
![]() Tomina and Wagenaar. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Tomina and Wagenaar. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Tomina, Y. and Wagenaar, D. A. (2018). Dual-sided Voltage-sensitive Dye Imaging of Leech Ganglia. Bio-protocol 8(5): e2751. DOI: 10.21769/BioProtoc.2751.
- Tomina, Y. and Wagenaar, D. A. (2017). A double-sided microscope to realize whole-ganglion imaging of membrane potential in the medicinal leech. Elife 6.
Category
Neuroscience > Neuroanatomy and circuitry > Live-cell imaging
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.