- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Coupling Exonuclease Digestion with Selective Chemical Labeling for Base-resolution Mapping of 5-Hydroxymethylcytosine in Genomic DNA

Published: Vol 8, Iss 5, Mar 5, 2018 DOI: 10.21769/BioProtoc.2747 Views: 7739
Reviewed by: Gal HaimovichOmar AkilAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This protocol is designed to obtain base-resolution information on the level of 5-hydroxymethylcytosine (5hmC) in CpGs without the need for bisulfite modification. It relies on (i) the capture of hydroxymethylated sequences by a procedure known as ‘selective chemical labeling’ (see Szulwach et al., 2012) and (ii) the digestion of the captured DNA by exonucleases. After Illumina sequencing of the digested DNA fragments, an ad hoc bioinformatic pipeline extracts the information for further downstream analysis.
Keywords: 5-HydroxymethylcytosineBackground
The methylation of cytosine in genomic DNA can be read by proteins and is mainly translated into gene silencing. Most CpG dinucleotides in the genome are methylated, including those located in gene regulatory regions such as enhancers. However, when required, these CpGs can be demethylated through oxidation of the methyl group by Ten Eleven Translocation (TET) enzymes and replacement by unmethylated cytosines by the base excision repair system. 5-Hydroxymethylcytosine (5hmC) is the first oxidative derivative of 5-methylcytosine, and mapping this modified base in the genome provides information on the regions undergoing active demethylation. Although selective chemical labeling (SCL) allows very specific detection of 5hmC, the resolution of this technique is limited by the size of the DNA fragments, especially when several CpGs are present in the captured DNA. In order to improve resolution, we have introduced a digestion step using exonucleases which trim the DNA molecule up to close proximity of the hydroxymethylated cytosines (Sérandour et al., 2016). Appropriate bioinformatic treatment of the sequencing reads then assigns hydroxymethylation score to the captured CpGs.
Materials and Reagents
- Pipette tips (TipOne, STARLAB, catalog numbers: S1161-1800 , S1182-1830 , and S1181-3810 )
- 0.65 ml Bioruptor microtubes (Diagenode, catalog number: C30010011 )
- 0.5 ml and 2 ml DNA LoBind tubes (Eppendorf, catalog numbers: 0030108035 and 0030108078 )
- Micro Bio-Spin 6 column (Bio-Rad Laboratories, catalog number: 7326221 )
- 1.5 ml Lobind tubes (Eppendorf, catalog number: 0030108051 )
- 2 ml Lobind tubes (Eppendorf, catalog number: 0030108078 )
- DNeasy Blood & Tissue Kit (QIAGEN, catalog number: 69504 )
- 100-bp DNA marker (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15628019 )
- E-gel EX agarose gel 2% (Thermo Fisher Scientific, InvitrogenTM, catalog number: G401002 )
- β-Glucosyltransferase (β-GT) and associated reaction buffer (New England Biolabs, catalog number: M0357S )
- DBCO-PEG4-Biotin (Sigma-Aldrich, catalog number: 760749 )
- UDP-6-N3-Glc (Active Motif, catalog number: 55020 )
- DMSO (Sigma-Aldrich, catalog number: D8418 )
- QIAquick Nucleotide Removal Kit (QIAGEN, catalog number: 28304 )
- Dynabeads M-280 streptavidin (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11205D )
- NEBuffer 2 (New England Biolabs, catalog number: B7002S )
- 10x NEBuffer 4 (New England Biolabs, catalog number: M0357S )
- ATP (10 mM) (New England Biolabs, catalog number: P0756S )
- dNTP solution mix (New England Biolabs, catalog number: N0447S )
- T4 DNA polymerase (New England Biolabs, catalog number: M0203S )
- DNA Polymerase I, Large (Klenow) Fragment (New England Biolabs, catalog number: M0210S )
- T4 PolyNucleotide Kinase (New England Biolabs, catalog number: M0201S )
- T4 DNA ligase high concentration (New England Biolabs, catalog number: M0202T )
- Nuclease-free water (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9937 )
- SCL-exo P7 adapter: annealing of 2 oligonucleotides (5’ Phos = phosphorylated 5’ end):
P7 exo-adapter reverse: 5’ Phos-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-OH 3’
P7 exo-adapter forward: 5’ OH-GATCGGAAGAGCACACGTCT-OH 3’ - Phi29 polymerase (New England Biolabs, catalog number: M0269S )
- Lambda exonuclease (New England Biolabs, catalog number: M0262S )
- RecJf exonuclease (New England Biolabs, catalog number: M0264S )
- Glycogen (5 mg/ml) (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9510 )
- Sodium chloride (NaCl) (Acros Organics, catalog number: AC207790050 )
- EtOH (100%) (VWR, catalog number: 20821.310 )
- SCL-exo P7 primer:
5’ OH-GACTGGAGTTCAGACGTGTGCT-OH 3’ - Agencourt AMPure XP (Beckman Coulter, catalog number: A63880 )
- Qiagen MinElute PCR Purification Kit (QIAGEN, catalog number: 28004 )
- SCL-exo P5 adapter: annealing of 2 oligonucleotides:
P5 exo-adapter reverse: 5’ OH-AGATCGGAAGAGCG-OH 3’
P5 exo-adapter forward: 5’ OH-TACACTCTTTCCCTACACGACGCTCTTCCGATCT-OH 3’ - NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs, catalog number: M0541S )
- SCL-exo universal P5 PCR primer (* = Phosphorothioates S-linkage):
5’ OH-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACG*A-OH 3’ - SCL-exo index P7 PCR primer (* = Phosphorothioates S-linkage) (index sequences come from TruSeq LT):
Index 2:
5’ OH-CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 4:
5’ OH-CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 5:
5’ OH-CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 6:
5’ OH-CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 7:
5’ OH-CAAGCAGAAGACGGCATACGAGATGATCTGGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 12:
5’ OH-CAAGCAGAAGACGGCATACGAGATTACAAGGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 13:
5’ OH-CAAGCAGAAGACGGCATACGAGATTTGACTGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 14:
5’ OH-CAAGCAGAAGACGGCATACGAGATGGAACTGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 15:
5’ OH-CAAGCAGAAGACGGCATACGAGATTGACATGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 16:
5’ OH-CAAGCAGAAGACGGCATACGAGATGGACGGGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 18:
5’ OH-CAAGCAGAAGACGGCATACGAGATGCGGACGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Index 19:
5’ OH-CAAGCAGAAGACGGCATACGAGATTTTCACGTGACTGGAGTTCAGACGTGTGC*T-OH 3’
Notes (concerning the oligonucleotides):- All oligonucleotides were produced by Sigma-Aldrich, purified by HLPC and resuspended in water at 100 μM final.
- The SCL-exo P7 adapter and the SCL-exo P5 adapter were obtained by mixing pairs of complementary oligonucleotides in 4 volumes of Annealing buffer (see Recipes) and annealed by heating for 5 min at 95 °C then let cool down slowly to room temperature.
- The oligonucleotides designed for SCL-exo were adapted from the P5 and P7 oligonucleotide sequences from Illumina ©2007-2012 Illumina, Inc. All rights reserved. Derivative works created by Illumina customers are authorised for use with Illumina instruments and products only. All other uses are strictly prohibited.
- All oligonucleotides were produced by Sigma-Aldrich, purified by HLPC and resuspended in water at 100 μM final.
- Agilent High Sensitivity DNA Kit (Agilent Technologies, catalog number: 5067-4626 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32854 )
- EDTA (500 mM, pH 8.0) (AppliChem, catalog number: A4892,0500 )
- HEPES (1 M) (GibcoTM, catalog number: 15630056 )
- Na deoxycholate (Sigma-Aldrich, catalog number: D6750 )
- NP-40, IGEPAL® CA-630 (Sigma-Aldrich, catalog number: I8896 )
- Lithium chloride (LiCl) (Sigma-Aldrich, catalog number: 62476 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Merck, catalog number: 442611 )
- Ammonium sulfate ((NH4)2SO4) (Merck, catalog number: 101217 )
- DTT (Sigma-Aldrich, catalog number: D9779 )
- Tris (MP Biomedicals, catalog number: 04819638 )
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: H9892 )
- Formamide for molecular biology (Sigma-Aldrich, catalog number: F9037 )
- 1x PBS (Fisher Scientific, catalog number: BP399 )
- Annealing buffer (see Recipes)
- RIPA buffer (see Recipes)
- Nick Repair buffer low DTT 10x (see Recipes)
- TE buffer (see Recipes)
- Elution buffer (see Recipes)
- Binding & Washing (B&W) buffer (see Recipes)
Equipment
- PIPETMAN ClassicTM Pipets (Gilson, catalog numbers: F123600 , F144801 , F123602 and F123615 )
- Bioruptor Pico with water cooler (Diagenode, catalog numbers: B01060001 and B02010003 )
- E-gel Power Snap Electrophoresis Device (Thermo Fisher Scientific, InvitrogenTM, catalog number: G8100 )
- Qubit 3 Fluorometer (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q33216 )
- Refrigerated centrifuge (Eppendorf, model: 5424 R )
- Thermocycler ProFlex PCR system (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4484073 )
- ThermoMixer C and Eppendorf ThermoTop (Eppendorf, catalog numbers: 5382000015 and 5308000003 )
- DynaMag-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D )
- Speed-Vac Savant (Thermo Fisher Scientific, catalog number: DNA120-115 )
- 2100 Bioanalyzer Instrument (Agilent Technologies, model: 2100, catalog number: G2939BA )
- Mini centrifuge (Bio-Rad Laboratories, catalog number: 1660603 )
Procedure
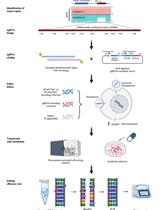
Genomic DNA is extracted using the QIAGEN DNeasy kit and fragmented into 300 bp fragments by sonication. The enzyme β-glucosyltransferase catalyzes the addition of azide-glucose to 5hmCs present in the gDNA fragments. Azide then reacts with a biotin conjugate allowing immobilization of the modified DNA on streptavidin-coated magnetic beads (Figure 1A). After end repair, Illumina P7 adapter ligation and nick repair, the captured DNA is incubated with the 5’ → 3’ exonucleases lambda and RecJf. The lambda exonuclease digests one strand of the double-stranded DNA and stops when it encounters bead-bound biotinylated 5hmC, whereas the RecJf exonuclease digests single-stranded DNA that might result from digestion of unmodified contaminant DNA by the lambda exonuclease. After elution from the beads, the DNA is denatured into single-stranded DNA molecules. This is followed by second strand synthesis, ligation of the Illumina P5 adapter, PCR amplification and Illumina sequencing. Single end sequencing starts from the P5 adapter and identifies the location where the lambda exonuclease stopped digesting and its associated nearest hydroxymethylated CpG (Figure 1B).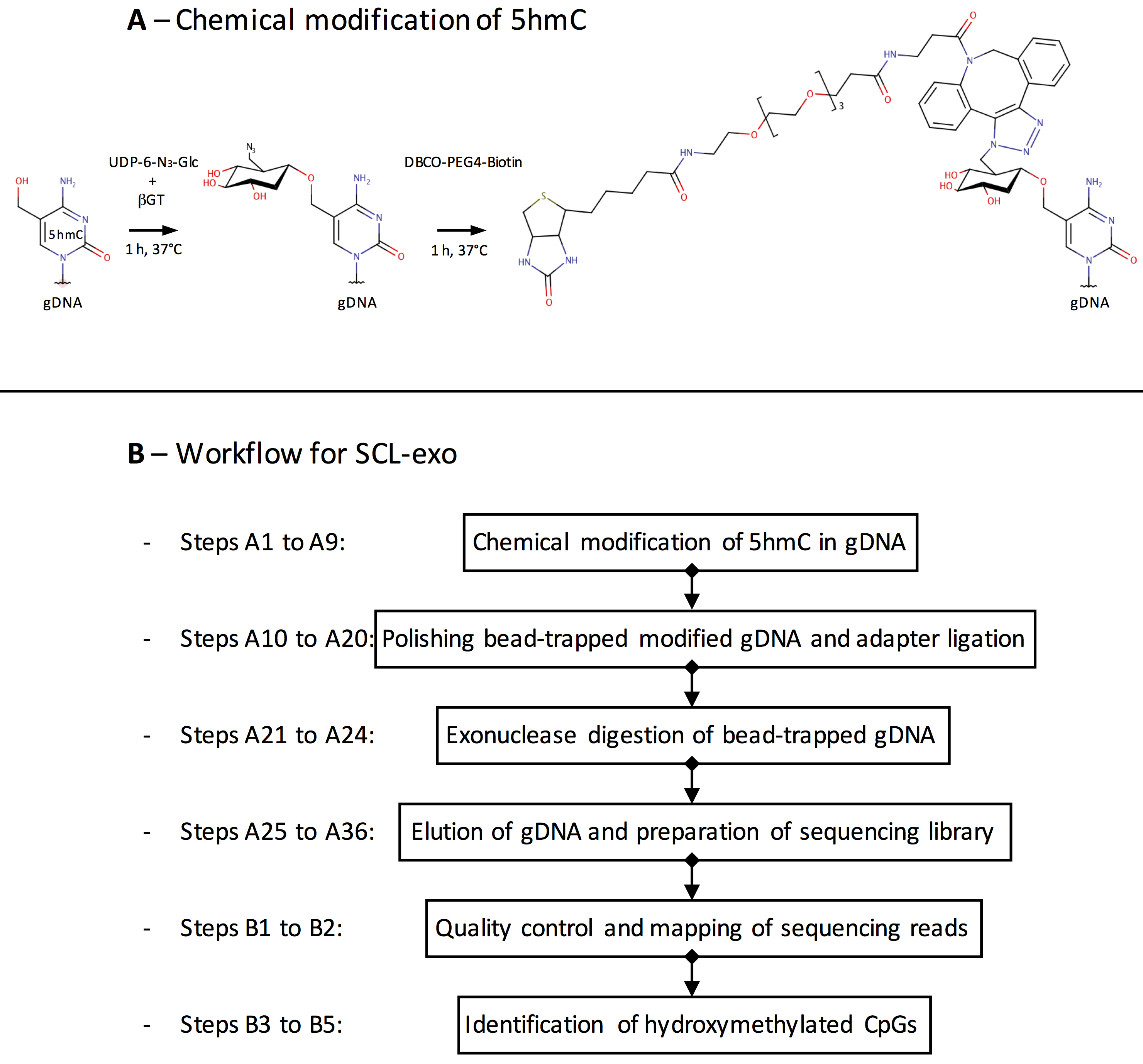
Figure 1. Overview of the SCL-exo procedure. A. As a first step of gDNA chemical modification, β-glucosyltransferase catalyzes the transfer of azide-glucose from UDP-6-N3-Glc to 5hmCs. Click chemistry is then used to add a biotin conjugate (DBCO-PEG4-Biotin) to the N3-Glc-modified 5hmCs. B. Flow chart of the SCL-exo protocol.
- Preparation of samples for Illumina sequencing
We highly recommend using RNA-free genomic DNA (gDNA) for the SCL-exo protocol. We purified RNA-free gDNA of interest by using the QIAGEN DNeasy kit and adding an RNaseA digestion step as described in the manufacturer’s protocol. RNA-free gDNA from any type of tissue or cultured cells can be used for the SCL-exo protocol. However, one should keep in mind that the global amount of 5hmC differs greatly between tissues, therefore the starting amount of gDNA required for SCL-exo might vary according to sample origin. When processing samples from different test conditions, we strongly recommend adding an identical amount of hydroxymethylated DNA standard in each sample after sonication. The number of reads covering this standard can then be used to normalize the SCL-exo signals between samples.- Sonicate 1 μg of gDNA of interest in 10 μl of 10 mM Tris, pH 8 in a 0.65 ml sonication tube using the Bioruptor Pico to obtain DNA fragments of around 300 bp. Sonication cycles should be set at 30 sec off/30 sec on. To ensure a proper and reproducible sonication, we recommend doing 3 cycles of sonication, then a short centrifugation, then again 3 cycles of sonication, then a short centrifugation and finally 4 cycles of sonication.
- The sonication efficiency can be quickly checked by running a 100-bp DNA marker and 0.5 μl of sonicated gDNA (diluted in 19.5 μl water) in an E-gel EX Agarose Gel (2%) for 10 min. You should obtain DNA fragments around 250-300 bp.
- Mix the remaining 9.5 μl of sonicated DNA with: 2 μl of 10x NEB Beta-GT reaction buffer (supplied with the Beta-GT enzyme) + 0.68 μl of UDP-6-N3-Glc (3 mM) + 1 μl NEB Beta-GT enzyme + 6.8 μl water.
- Mix by pipetting and incubate in a thermocycler at 37 °C for 1 h (no heating lid).
- Centrifuge quickly with the mini centrifuge (5 sec at 2,000 x g).
- Prepare a 3 mM working solution of DBCO-PEG4-Biotin conjugate in DMSO by ten-fold dilution of a 30 mM stock solution in DMSO. Store at -20 °C.
- Add 1 μl DBCO-PEG4-Biotin conjugate working solution to the DNA sample from Step A5 to reach a final concentration of 150 μM.
- Mix by pipetting and incubate in a thermocycler at 37 °C for 1 h (no heating lid).
- Centrifuge quickly with the mini centrifuge (5 sec at 2,000 x g) and clean up the reaction with QIAquick Nucleotide Removal Kit. Elute with at least 30 μl water per column.
Note: The biotinylated DNA samples can be conserved at -20 °C for few days. - Wash 25 μl of Dynabeads M-280 Streptavidin three times each with 100 μl of 1x Binding & Washing (B&W) buffer (see Recipes) in a 0.5 ml Lobind tube. Separate the beads from the buffer with a magnetic stand and resuspend the beads in 30 μl of 2x B&W buffer and 140 μl of 1x B&W buffer.
- Add the 30 μl DNA eluate (from Step A9) to the resuspended beads from the previous step. The final concentration of B&W buffer should be 1x.
- Incubate for 30 min at room temperature on rotation.
Note: Prepare the mix of the Step A15 during this step. - Transfer to a 2 ml Lobind tube and wash the beads five times with 1 ml of 1x B&W buffer using the magnetic stand.
- Wash 2 times with 1 ml of 10 mM Tris-HCl pH 8. Do not let the beads dry.
- The beads then undergo 5 successive reactions (in a 2 ml Lobind tube agitated at 900 rpm in a thermomixer) as followed:
End repair: Prepare a mix containing 10 μl of NEB2 buffer (10x), 10 μl of ATP (10 mM), 1 μl of dNTP (10 mM), 5 μl of T4 DNA polymerase (3 U/μl), 1 μl of DNA Polymerase I Large Klenow Fragment (5 U/μl), 5 μl of T4 PolyNucleotide Kinase (T4 PNK) (10 U/μl) and 68 μl of nuclease-free water.
Add the mix to the beads in the 2 ml Lobind. Incubate at 30 °C for 30 min with agitation at 900 rpm in a thermomixer. - Wash 2 times with 1 ml RIPA buffer (see Recipes) and 2 times with 10 mM Tris-HCl, pH 8. After removing the last Tris wash, centrifuge quickly with the mini centrifuge (5 sec at 2,000 x g) and put the tube back in the magnetic stand. Remove the traces of Tris. Make sure you do the same for Steps A18, A20, A22 and A24. Do not let the beads dry.
- Ligation of P7 adapter:
Prepare a mix containing 10 μl of NEB2 Buffer (10x), 10 μl of ATP (10 mM), 15 μl of SCL-exo P7 adapter (10 μM), 1 μl of T4 DNA ligase (2,000 U/μl) and 65 μl of nuclease-free water. Add the mix to the beads in the 2 ml Lobind tube. Incubate at 25 °C for 1 h with agitation at 900 rpm in a thermomixer. - Wash twice with 1 ml of RIPA buffer and twice with 1 ml of 10 mM Tris-HCl, pH 8.
- Nick repair:
Prepare a mix containing 1.5 μl of Phi29 polymerase (10 U/μl), 10 μl of Home-made Nick Repair low DTT buffer (10x) (see Recipes), 1.5 μl of dNTP (10 mM) and 87 μl of nuclease-free water. Add the mix to the beads in the 2 ml Lobind tube. Incubate at 30 °C for 20 min with agitation at 900 rpm in a thermomixer. - Wash twice with 1 ml RIPA buffer and twice with 1 ml of 10 mM Tris-HCl, pH 8.
- Lambda exonuclease digestion:
Prepare a mix containing 2 μl of Lambda exonuclease (5 U/μl), 10 μl of NEB Lambda exonuclease buffer (10x) and 88 μl of nuclease-free water. Add the mix to the beads in the 2 ml Lobind tube. Incubate at 37 °C for 30 min with agitation at 900 rpm in a thermomixer. - Wash twice with 1 ml RIPA buffer and twice with 1 ml of 10 mM Tris-HCl, pH 8.
- RecJf exonuclease digestion:
Prepare a mix containing 1 μl of RecJ exonuclease (30 U/μl), 10 μl NEB2 buffer (10x) and 89 μl nuclease-free water. Add the mix to the beads in the 2 ml Lobind tube. Incubate at 37 °C for 30 min with agitation at 900 rpm in a thermomixer. - Wash twice with 1 ml RIPA buffer and twice with 1 ml of 10 mM Tris-HCl, pH 8.
- Elution:
Incubate the beads in 100 μl of elution buffer (see Recipes) at 90 °C for 5 min, then put directly on ice to cool the sample. - Transfer the 100 μl eluate to a new 1.5 ml Lobind tube and add 300 μl of 10 mM Tris-HCl, pH 8.
- DNA precipitation:
- Add 2 μl of glycogen, 16 μl of NaCl (5 M) and mix well. Add 800 μl of 100% EtOH and mix well.
- Incubate the tube at -80 °C for at least 30 min (overnight if possible).
- Centrifuge at 20,000 x g at 4 °C for 30 min.
- Carefully remove the supernatant without disturbing the pellet.
- Add 500 μl of 70% EtOH.
- Centrifuge at 20,000 x g at 4 °C for 5 min.
- Remove the supernatant carefully.
- Add 500 μl of 100% EtOH.
- Centrifuge at 20,000 x g at 4 °C for 5 min.
- Remove the supernatant carefully.
- Dry pellets 10-20 min in a Speed-Vac at 45 °C and resuspend in 20 μl of 10 mM Tris-HCl, pH 8.
- The purified DNA sample can be conserved for one night at -20 °C. Go to Step A28.
- Add 2 μl of glycogen, 16 μl of NaCl (5 M) and mix well. Add 800 μl of 100% EtOH and mix well.
- DNA denaturation:
- Transfer the 20 μl of DNA solution to a PCR tube and incubate the DNA sample at 95 °C for 5 min in a thermocycler.
- Then put the tube directly on ice to cool the sample.
- Transfer the 20 μl of DNA solution to a PCR tube and incubate the DNA sample at 95 °C for 5 min in a thermocycler.
- Second strand synthesis:
- Add the following reagents to the tube containing the 20 ul of DNA solution: 20 μl of nuclease-free water, 5 μl of the SCL-exo P7 primer (1 μM) and 5 μl of NEB Phi29 Reaction Buffer (10x). Mix gently.
- In a thermocycler, incubate the sample at 65 °C for 5 min and then at 30 °C for 2 min. Pause the PCR program.
- Immediately add 1 μl of Phi29 polymerase (10 U/μl) and 1 μl of dNTP (10 mM), mix gently.
- Restart the PCR program and incubate the sample in a thermocycler at 30 °C for 20 min and then 65 °C for 10 min.
- Add the following reagents to the tube containing the 20 ul of DNA solution: 20 μl of nuclease-free water, 5 μl of the SCL-exo P7 primer (1 μM) and 5 μl of NEB Phi29 Reaction Buffer (10x). Mix gently.
- DNA purification:
- Add 52 μl of room temperature Ampure beads (1 volume) to the 52 μl sample.
- Incubate at room temperature for 15 min.
- Put the tube on the magnetic stand and remove carefully the supernatant. With the tube staying on the magnetic stand, wash the beads twice with freshly made 80% EtOH (wait for at least 30 sec after adding the first ethanol wash).
- Centrifuge with the mini centrifuge (5 sec at 2,000 x g, put the tube back on the magnetic stand and remove the rest of ethanol.
- Leave the tube open on the magnetic stand and let it dry for 10-15 min.
- Add 22 μl of room temperature 10 mM Tris-HCl, pH 8, remove the tube from the magnetic stand and mix well. Make sure that all the beads are resuspended and wet.
- Remove the tube from the magnetic stand and incubate for 3 min at room temperature.
- Put the beads back to the magnetic stand and once they are well packed, pipet carefully 20 μl of the DNA eluate and put it in a new PCR tube.
- Add 52 μl of room temperature Ampure beads (1 volume) to the 52 μl sample.
- Ligation of SCL-exo P5 adapter:
- In a PCR tube, add the following reagents to the 20 μl of DNA sample: 22.5 μl nuclease-free water, 1.5 μl SCL-exo P5 adapter (10 μM), 5 μl of NEB T4 DNA ligase Buffer (10x) and 1 μl of T4 DNA ligase (2,000 U/μl). Mix gently.
- In a thermocycler, incubate at 25 °C for 60 min and then 65 °C for 10 min.
- In a PCR tube, add the following reagents to the 20 μl of DNA sample: 22.5 μl nuclease-free water, 1.5 μl SCL-exo P5 adapter (10 μM), 5 μl of NEB T4 DNA ligase Buffer (10x) and 1 μl of T4 DNA ligase (2,000 U/μl). Mix gently.
- DNA purification:
Add 50 μl of room temperature Ampure beads (1 volume) to the 50 μl sample, and proceed like in Step A30. The resulting 20 μl eluted DNA solution is used for the final PCR. - PCR amplification:
- In a PCR tube, prepare a mix containing 4 μl of nuclease-free water, 25 μl of NEBNext High-Fidelity PCR Master Mix (2x), 0.5 μl of SCL-exo universal P5 PCR primer (25 μM) and 0.5 μl of SCL-exo index P7 PCR primer (25 μM) (choose your index of interest). Add the 20 μl DNA sample and mix gently.
- Put the tube in a thermocycler and run the following program:
98 °C for 30 sec
Then 18 cycles of: 98 °C for 10 sec, 65 °C for 30 sec, 72 °C for 30 sec
72 °C for 5 min
4 °C forever
- In a PCR tube, prepare a mix containing 4 μl of nuclease-free water, 25 μl of NEBNext High-Fidelity PCR Master Mix (2x), 0.5 μl of SCL-exo universal P5 PCR primer (25 μM) and 0.5 μl of SCL-exo index P7 PCR primer (25 μM) (choose your index of interest). Add the 20 μl DNA sample and mix gently.
- DNA purification:
Add 50 μl of room temperature Ampure beads (1 volume) to the 50 μl PCR sample, and proceed like in Step A30. You should get a 20 μl SCL-exo library. - Measure the DNA concentration using Qubit and the dsDNA High Sensitivity kit. Check the library quality on Agilent BioAnalyzer (see Figure 2). In case there is an adapter or a primer contamination, it is advised to redo an Ampure purification (1 volume of beads for 1 volume of DNA). Pool the libraries to multiplex. Get enough index complexity so that the index sequencing is successful. Contact your sequencing facility if you have any doubt.
- Submit for Illumina single-end sequencing MiSeq/GAII/HiSeq to a sequencing facility.
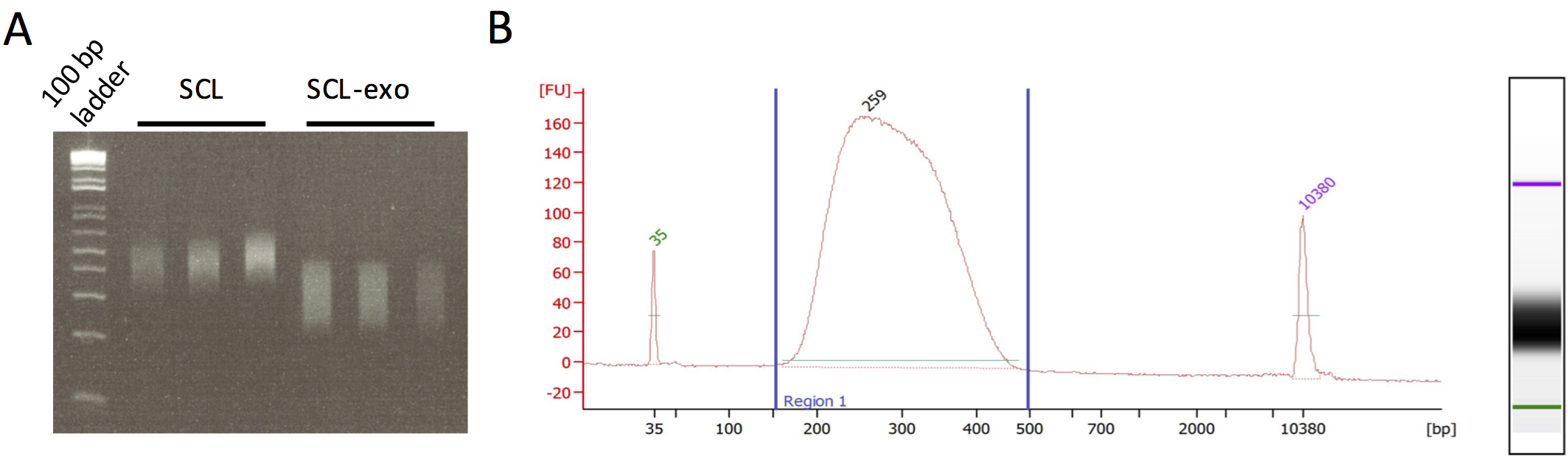
Figure 2. Quality control of SCL-exo libraries. A. Agarose gel electrophoresis of SCL and SCL-exo libraries. SCL libraries were obtained by omitting the exonuclease digestion steps. Note that DNA fragments from the SCL libraries are on average 100 bp longer than in the SCL-exo libraries. B. BioAnalyzer electropherogram profile of a pool of SCL-exo libraries. 1 μl of SCL-exo library was run on an Agilent High Sensitivity DNA chip following the manufacturer’s protocol. The DNA library length should range between 200 and 400 bp. It is important to notice the absence of adapter dimer peak (around 120 bp) and the absence of PCR primers (around 50 bp). If these contaminants are present, we recommend redoing an Ampure purification (1 volume of Ampure beads for 1 volume of DNA library) as in Step A30.
- Sonicate 1 μg of gDNA of interest in 10 μl of 10 mM Tris, pH 8 in a 0.65 ml sonication tube using the Bioruptor Pico to obtain DNA fragments of around 300 bp. Sonication cycles should be set at 30 sec off/30 sec on. To ensure a proper and reproducible sonication, we recommend doing 3 cycles of sonication, then a short centrifugation, then again 3 cycles of sonication, then a short centrifugation and finally 4 cycles of sonication.
- Bioinformatic identification of hydroxymethylated CpGs from SCL-exo fastq files
We conceived and implemented a bioinformatic protocol to identify hydroxymethylated CpGs from SCL-exo fastq files generated in triplicates by a sequencing platform. The protocol involves the following steps:
1) Trimming and filtering the sequence reads with respect to their quality using program SolexaQA (Cox et al., 2010).
2) Mapping high quality reads onto each strand of the genome separately, using the program Bowtie (Langmead et al., 2009), in order to generate sam files for both the forward and reverse strands. Sam files are text files that contain the sequence reads together with their associated genomic localization, if any, and can be parsed to identify reads mapping a unique location on the genome.
3) Creating a hydroxymethylated CpG signal (wig) file for each replicate by directly reading the sequences in the sam files, using our python program generate-SCL-exo signal-from-sams. The program counts the number of reads uniquely overlapping any given CpG, and stores the values into a signal (wig) file at CpG or base-pair resolution. The wig file can be visualized using a genome browser, such as IGB (Nicol et al., 2009).
Note: All our python programs are available at: https://mycore.core-cloud.net/index.php/s/4gyZ9dLTqgo86dt.
4) Identifying putative hydroxymethylated cytosines by retrieving the consensus CpG dinucleotides that are present in at least two of the three replicates, using python program generate-SCL-exo consensus-signal.
5) Determining the set of CpG dinucleotides significantly enriched in 5hmC using a peak-calling algorithm (generate-SCL-exo peaks) with a well-chosen threshold.
Details for each of these steps are given below:- Trimming and filtering the sequenced reads
Only high-quality reads should be retained for sound identification of hydroxymethylated CpGs. Hence we used program SolexaQA (Cox et al., 2010) to trim and filter the reads present in the SCL-exo fastq files. The program takes two parameters: a quality threshold and a minimum length. First, all sequenced nucleotides whose quality is lower than the quality threshold are removed from the reads. Second, reads shorter than the minimum length are deleted. We used value 20 as the minimum nucleotide sequencing quality, corresponding to a p-value of 10-2 (or 1% chance of occurrence of a sequencing error on any given nucleotide) and 17 as the minimum read length. The trimming is achieved by going into the SolexaQA directory and typing under Linux:
perl DynamicTrim.pl fastq -h quality -d.
where fastq is the path and filename of the fastq file and quality is the quality value (e.g., 20). This will generate a trimmed fastq file fastq.trimmed. The filtering is then achieved by typing:
perl LengthSort.pl fastq.trimmed -l minlength
where minlength is the minimum length (e.g., 17) of retained reads. - Mapping filtered reads onto both strands of the genome
Bowtie (Langmead et al., 2009) can be used to map the retained high-quality reads onto the forward and reverse strands of the genome separately, with the following parameters:
-p processors -l length -n nb_mismatches -m 1 --sam --strata --best --norc [or --nofw]
where,
processors: designate the number of computer processors available for the mapping process;
length: the read length taken into account to map the read onto the genome;
nb_mismatches: the allowed number of mismatches;
-m 1 indicates that we only retain reads mapping the genome at a unique location;
--norc (respectively --nofw) that the genome reverse strand (resp. forward strand) is not used for mapping.
Note that Bowtie initially requires indexing the genome fasta files (see Bowtie user guide).
The reads must be mapped onto the forward and reverse strands separately, producing one sam file for each strand. Mapping was launched with Bowtie using the Linux command:
./bowtie -p processors --best -l 28 -n 2 -m 1 --sam --strata --norc genome fastq > fw-sam
to map reads from file fastq onto the forward strand of the indexed genome file (typically a .hs file), so as to generate a forward strand fw-sam file, and:
./bowtie -p processors --best -l 28 -n 2 -m 1 --sam --strata --nofw genome fastq > rv-sam
to map reads from file fastq onto the reverse strand of the indexed genome file, so as to generate a reverse strand rv-sam file. - Parsing single stranded sam files to generate a wig file at CpG or base-pair resolution
- DNA fragments were initially captured according to the presence of 5-hydroxymethylcytosine and then trimmed by exonuclease. As cytosines are mostly hydroxymethylated in a CpG context, 5hmC-positive reads should be enriched in CpGs within a few nucleotides from the start of every sequence (Sérandour et al., 2016). The sam files contain the sequence reads together with their associated genomic localization, if any. Our python program (generate-SCL-exo signal-from-sams) parses both the forward and reverse stranded sam files and considers in turn all reads uniquely mapped on the genome. It checks whether every localized read exhibits a CpG within the first few nucleotides of its sequence. Typically, a 10 base-pair long window, situated at the beginning of the read, is used to attest for the presence of a CpG. Reads not exhibiting any CpG inside the window are discarded. Reads exhibiting two or more CpGs inside the window are kept aside (their CpGs will be stored in a different file), as it is then not possible to determine with certainty which CpG was hydroxymethylated.
- When a single CpG is found within the window, its precise genomic coordinate is determined (from the read localization provided by the sam file and the CpG position within the read) and stored in memory, within a hash table-type structure. The first time a CpG position is encountered, a value of 1 is associated to the genomic position. If a CpG position already contains a value, that value is increased by one, storing effectively the number of reads covering that particular position. Note that the program accounts for the strand associated to the sam file: in the reverse stranded sam file, the read sequences must be read from right to left, while the CpG coordinate must be adjusted (this is automatically accounted for by the program). Once all reads have been parsed, the hash table is stored within a wig file at CpG resolution by default: the genomic positions of every CpG are associated with their number of overlapping reads. By default, our program adds up the number of reads overlapping a given CpG found on both strands. It is possible to use the program so that it produces a different signal value for each stranded cytosine of the CpG, associating each cytosine position with the number of reads overlapping each strand, thus producing a wig file at base-pair resolution.
- Our program is launched using the following Linux command:
python generate-SCL-exo.py signal-from-sams fw-sam rv-sam window SCL-exo-wig [resolution]
where,
fw-sam and rv-sam respectively designate the filenames of the forward and reverse stranded sam files;
window stands for the length (typically 10), expressed in base-pairs, of the window used to identify hydroxymethylated CpG dinucleotides;
resolution is an optional parameter that takes on value 2 or 1, depending on whether the SCL-exo-wig file will be generated respectively at CpG or base-pair resolution (default is 2 if the parameter is not specified).
- DNA fragments were initially captured according to the presence of 5-hydroxymethylcytosine and then trimmed by exonuclease. As cytosines are mostly hydroxymethylated in a CpG context, 5hmC-positive reads should be enriched in CpGs within a few nucleotides from the start of every sequence (Sérandour et al., 2016). The sam files contain the sequence reads together with their associated genomic localization, if any. Our python program (generate-SCL-exo signal-from-sams) parses both the forward and reverse stranded sam files and considers in turn all reads uniquely mapped on the genome. It checks whether every localized read exhibits a CpG within the first few nucleotides of its sequence. Typically, a 10 base-pair long window, situated at the beginning of the read, is used to attest for the presence of a CpG. Reads not exhibiting any CpG inside the window are discarded. Reads exhibiting two or more CpGs inside the window are kept aside (their CpGs will be stored in a different file), as it is then not possible to determine with certainty which CpG was hydroxymethylated.
- Identifying the consensus CpGs found in at least two out of the three replicates
We provide a python program generate-SCL-exo consensus-signal that compares three SCL-exo signal files and returns a wig file containing the hydroxymethylated CpGs identified in at least two of the three replicates, together with their mean signal. Identified CpG positions must exhibit values greater than a minimum threshold min-threshold in at least two of the three files. An example of such consensus signal can be found in Figure 3.
The function can be called thus:
python generate-SCL-exo.py consensus-signal SCL-exo-wig1 SCL-exo-wig2 SCL-exo-wig3 min-threshold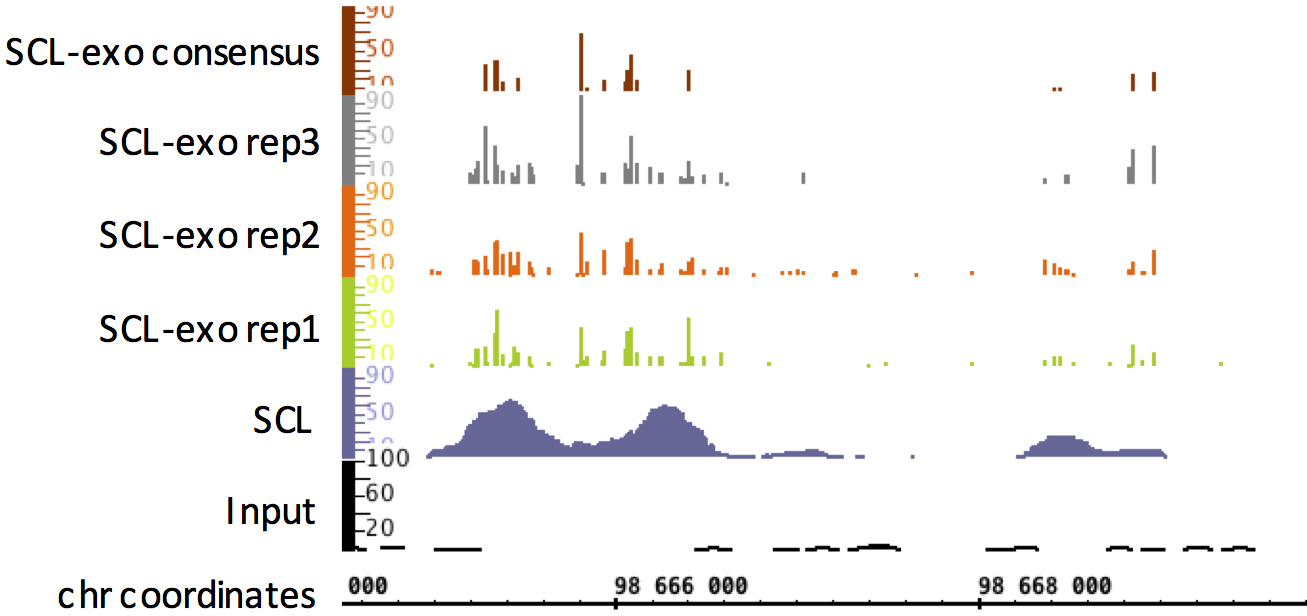
Figure 3. Integrated Genome Browser view of SCL-exo signal in a region of mm8 chr11 from P19 embryonal carcinoma cells. For comparison, the Input-seq (genomic DNA of P19 cells) and SCL-seq (no exonuclease step) are shown. - Determining significantly enriched hydroxymethylated CpGs within SCL-exo wig files
A peak-calling algorithm is used to determine the CpGs that are significantly enriched in 5hmC within an SCL-exo signal file. The algorithm looks for adjacent genomic positions, within the SCL-exo wig file, that exhibit signal values for both CpG coordinates above a predefined threshold (Sérandour et al., 2016).
The program can be used either on the consensus SCL-exo signal file or on the SCL-exo wig replicates separately.
The python program generate-SCL-exo peaks takes an SCL-exo-wig file together with the predefined threshold, and generates a bed file gathering all CpG positions that satisfy the above constraints.
Peak-calling on an SCL-exo-wig file at CpG or base-pair resolution is launched using the Linux command:
python generate-SCL-exo.py peaks SCL-exo-wig threshold.
- Trimming and filtering the sequenced reads
Data analysis
Information about data processing and analysis can be found in the original research article at: https://doi.org/10.1186/s13059-016-0919-y.
Notes
- Notes concerning Steps A15 to A24:
- Beads can be less magnetic in the 10 mM Tris-HCl, pH 8. Keep the tube on the magnetic stand during the removal of the second Tris wash, to avoid the loss of beads. Then spin the tube briefly, put it back on the magnetic stand and remove the residual Tris buffer. Tris washes should be done carefully to eliminate any trace of detergent that can be detrimental for the subsequent enzymatic reaction.
- Do not let the streptavidin beads dry out. Prepare the enzymatic mixes few minutes before the washes.
- Beads can be less magnetic in the 10 mM Tris-HCl, pH 8. Keep the tube on the magnetic stand during the removal of the second Tris wash, to avoid the loss of beads. Then spin the tube briefly, put it back on the magnetic stand and remove the residual Tris buffer. Tris washes should be done carefully to eliminate any trace of detergent that can be detrimental for the subsequent enzymatic reaction.
- Concerning the Ampure beads purification in Steps A30, A32 and A34:
Be aware that any remaining trace of EtOH would inhibit the next enzymatic reaction.
Recipes
- Annealing buffer
10 mM Tris, pH 8
50 mM NaCl
1 mM EDTA - RIPA buffer
50 mM HEPES, pH 7.6
1 mM EDTA
0.7% Na deoxycholate
1% NP-40
0.5 M LiCl - Nick Repair buffer low DTT (10x)
100 mM MgCl2
500 mM Tris-HCl, pH 7.5
100 mM (NH4)2SO4
10 mM DTT - TE buffer (pH 7.4)
10 mM Tris
1 mM EDTA
HCl - Elution buffer
95% formamide
10 mM EDTA, pH 8 - Binding & Washing (B&W) buffer (2x)
10 mM Tris-HCl (pH 7.5)
1 mM EDTA
2 M NaCl
Acknowledgments
We thank M. Bizot and G. Palierne for technical assistance. This work was funded by La Ligue Contre le Cancer, Cancéropole Grand Ouest, The CNRS and the University of Rennes 1. The authors declare no competing interests.
References
- Cox, M. P., Peterson, D. A. and Biggs, P. J. (2010). SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11: 485.
- Langmead, B., Trapnell, C., Pop, M. and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10(3): R25.
- Nicol, J. W., Helt, G. A., Blanchard, S. G., Jr., Raja, A. and Loraine, A. E. (2009). The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25(20): 2730-2731.
- Sérandour, A. A., Avner, S., Mahe, E. A., Madigou, T., Guibert, S., Weber, M. and Salbert, G. (2016). Single-CpG resolution mapping of 5-hydroxymethylcytosine by chemical labeling and exonuclease digestion identifies evolutionarily unconserved CpGs as TET targets. Genome Biol 17: 56.
- Szulwach, K. E., Song, C. X., He, C. and Jin, P. (2012). 5-hydroxymethylcytosine (5-hmC) specific enrichment. Bio-protocol 2(15).
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sérandour, A. A., Avner, S. and Salbert, G. (2018). Coupling Exonuclease Digestion with Selective Chemical Labeling for Base-resolution Mapping of 5-Hydroxymethylcytosine in Genomic DNA. Bio-protocol 8(5): e2747. DOI: 10.21769/BioProtoc.2747.
Category
Systems Biology > Epigenomics > 5-hydroxymethylcytosine
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.