- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunoprecipitation of Tri-methylated Capped RNA

Published: Vol 8, Iss 3, Feb 5, 2018 DOI: 10.21769/BioProtoc.2717 Views: 9849
Reviewed by: Gal HaimovichSavita NairAnca Flavia Savulescu
Original research article
The authors used this protocol in:

Jun 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Cellular quiescence (also known as G0 arrest) is characterized by reduced DNA replication, increased autophagy, and increased expression of cyclin-dependent kinase p27Kip1. Quiescence is essential for wound healing, organ regeneration, and preventing neoplasia. Previous findings indicate that microRNAs (miRNAs) play an important role in regulating cellular quiescence. Our recent publication demonstrated the existence of an alternative miRNA biogenesis pathway in primary human foreskin fibroblast (HFF) cells during quiescence. Indeed, we have identified a group of pri-miRNAs (whose mature miRNAs were found induced during quiescence) modified with a 2,2,7-trimethylguanosine (TMG)-cap by the trimethylguanosine synthase 1 (TGS1) protein and transported to the cytoplasm by the Exportin-1 (XPO1) protein. We used an antibody against (TMG)-caps (which does not cross-react with the (m7G)-caps that most pri-miRNAs or mRNAs contain [Luhrmann et al., 1982]) to perform RNA immunoprecipitations from total RNA extracts of proliferating or quiescent HFFs. The novelty of this assay is the specific isolation of pri-miRNAs as well as other non-coding RNAs containing a TMG-cap modification.
Keywords: m2,2,7G-cap RNABackground
Cellular quiescence, a type of reversible growth arrest, is an important cellular state involved in wound healing, organ regeneration, and preventing neoplasia (Coller, 2011; Valcourt et al., 2012). Small non-coding RNAs such as miRNAs have been found involved in the regulation of cellular quiescence. miRNAs are small non-coding RNAs ~22-nucleotides long that regulate the expression of protein-coding genes by base-pairing with the 3’ untranslated region (3’UTR) of messenger RNAs (mRNAs) (Esteller, 2011). The canonical miRNA biogenesis pathway is based on a stepwise processing machinery (Ha and Kim, 2014; Kim et al., 2016). miRNAs are transcribed to produce a primary miRNA (pri-miRNA) with an imperfect loop structure that is recognized by the enzyme Drosha and its binding partner DGCR8 in the nucleus. Cleavage of the pri-miRNA generates a precursor miRNA (pre-miRNA) that is recognized and transported to the cytoplasm by the Exportin-5 (XPO5) protein. The pre-miRNA is cleaved by the enzyme Dicer (mature miRNA) and loaded into the RNA-induced silencing complex (RISC). On the other hand, precursors of small nuclear RNAs (snRNAs) involved in mRNA processing such as U1, U2, U4, and U5 have a (m7G)-cap, which is recognized by cap-binding complex (CBC) and the phosphorylated adaptor for RNA export (PHAX) in the nucleus to enable their export to the cytoplasm by XPO1 (Ohno et al., 2000). These snRNAs are then recognized by Sec1/Munc18 (Sm) proteins (by binding to Sm binding site sequences) in the cytoplasm and TGS1 is recruited to hypermethylate the (m7G)-cap into a (m2,2,7G, TMG)-cap. This modification is recognized by Snuportin-1 in association with Importin-β and other factors to import the snRNAs back into the nucleus (Palacios et al., 1997; Kiss, 2004). Interestingly, XPO1 also has high affinity for the (TMG)-capped small nucleolar RNA (snoRNA) U3 in the nucleus and transports it from Cajal bodies to the nucleoli (Boulon et al., 2004). A previous study showed that TGS1 enhances Rev-dependent HIV-1 RNA expression by (TMG)-capping viral mRNAs in the nucleus, thereby increasing recognition by XPO1 for transport to the cytoplasm (Yedavalli and Jeang, 2010). These findings suggest that TMG-capping of RNAs gives plasticity to different types of RNA molecules in order to regulate their processing and cellular localization. Our recent findings demonstrated the existence of a group of pri-miRNAs modified with a 2,2,7-trimethylguanosine (TMG)-cap by TGS1 protein and transported to the cytoplasm by XPO1 during quiescence. Previous publications have shown the ability to pull-down (TMG)-cap RNAs, such as snRNAs and snoRNAs, with specific antibodies against (TMG)-cap RNAs (Luhrmann et al., 1982). Our previous publications demonstrated for the first time the pull-down of (TMG)-cap pri-miRNAs in human cells (Martinez et al., 2017). Understanding which RNAs could be modified with a TMG-cap will provide new important insights into RNA biogenesis in normal or disease-related conditions.
Materials and Reagents
- 1.5 ml microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-129 )
- 15 ml conical centrifuge tubes (DNase-/RNase-free) (Corning, catalog number: 430052 )
- GilsonTM EXPERTTM University Fit pipette filter tips (Gilson, catalog numbers: F1731031 , F1733031 , F1735031 , F1737031 )
- Gel-loading pipet tips (Fisher Scientific, catalog number: 02-707-139 )
- Large-orifice pipet tips (Fisher Scientific, catalog number: 02-707-134 )
- Sterile polystyrene disposable serological pipettes (Greiner Bio One International, catalog number: 710180 )
- Serological pipettes
2 ml serological pipettes (Fisher Scientific, catalog number: 13-678-11C )
5 ml serological pipettes (Fisher Scientific, catalog number: 13-678-11D )
10 ml serological pipettes (Fisher Scientific, catalog number: 13-678-11E )
25 ml serological pipettes (Fisher Scientific, catalog number: 13-678-11 ) - 150 cm2 vented tissue culture treated flasks (Corning, Falcon®, catalog number: 355001 )
- 100 mm TC-treated cell culture dish (Corning, Falcon®, catalog number: 353033 )
- Cell scrapers (Fisher Scientific, catalog number: 08-100-242 )
- HFF cells (obtained from the Yale Skin Disease Research Center) (Alternative source of HFF cells from ATCC: Hs27 (ATCC, catalog number: CRL-1634 )
- DMEM (Sigma-Aldrich, catalog number: D7777 )
- Minimum essential medium (MEM) non-essential amino acids, 100x (Thermo Fisher Scientific, catalog number: 11140076 )
- 0.05% trypsin-EDTA with phenol red (Thermo Fisher Scientific, catalog number: 25300054 )
- 10x PBS (Sigma-Aldrich, catalog number: P5493 )
- Nuclease-free water (not DEPC-Treated) (Thermo Fisher Scientific, catalog number: AM9937 )
- TRIzolTM Reagent (Thermo Fisher Scientific, catalog number: 15596026 )
- RNase AWAYTM Surface Decontaminant (Thermo Fisher Scientific, catalog number: 7002 )
- Chloroform (Sigma-Aldrich, catalog number: C2432 )
- Ethanol, molecular biology grade (Fisher Scientific, catalog number: BP2818-500 )
- Isopropanol, molecular biology grade (Fisher Scientific, catalog number: BP26184 )
- GlycoBlueTM Coprecipitant (15 mg/ml) (Thermo Fisher Scientific, catalog number: AM9516 )
- TURBO DNA-freeTM Kit (Thermo Fisher Scientific, catalog number: AM1907 )
- Anti-m3G-cap, rabbit polyclonal, antiserum (Synaptic Systems)*
Note: *Synaptic Systems discontinued the production of the Anti-m3G-cap, rabbit polyclonal, antiserum. Creative Diagnostics has a rabbit anti-TMG antibody (Anti-m3G-cap polyclonal antibody, Creative Diagnostics, catalog number: DPAB29202 ) that would be similar to the one we previously used but the experimental conditions have to be re-evaluated. - Protein G Sepharose® 4 Fast Flow Beads (GE Healthcare, catalog number: 17061801 )
- Normal rabbit serum (control, EMD Millipore, catalog number: NS01L-1ML )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: S671-3 )
- NP-40 (Thermo Fisher Scientific, catalog number: 28324 )
- Tris base (Fisher Scientific, catalog number: BP152-5 )
- Hydrochloric acid (HCl) (VWR, catalog number: BDH7204-1 )
- RNasinTM Plus RNase inhibitor (Promega, catalog number: N2611 )
- NaOAc (AMRESCO, catalog number: 0602 )
- Ethylenediaminetetraacetate acid (EDTA), pH 8 (Thermo Fisher Scientific, catalog number: AM9260G )
- Ethylenediaminetetraacetate acid (EDTA) (AMRESCO, catalog number: 0105 )
- Sodium dodecyl sulfate (SDS) (Thermo Fisher Scientific, catalog number: AM9822 )
- Phenol/Chloroform/Isoamyl Alcohol; 125:24:1 mixture, pH 4.5 (Thermo Fisher Scientific, catalog number: AM9720 )
- Agarose LE (Denville Scientific, catalog number: CA3510-8 )
- iScriptTM cDNA Synthesis Kit (Bio-Rad Laboratories, catalog number: 1708891 )
- Sso AdvancedTM Universal SYBR® Green Supermix (Bio-Rad Laboratories, catalog number: 1725274 )
- PARISTM Kit (Thermo Fisher Scientific, catalog number: AM1921 )
- Boric acid (Fisher Scientific, catalog number: A73-1 )
- NET-2 Buffer (see Recipes)
- G-50 Buffer (see Recipes)
- 1x TBE (see Recipes)
Equipment
- Micropipettes (Gilson, model: Pipetman® L, catalog number: F167370 )
- FormaTM Steri-CycleTM CO2 Incubator (Thermo Scientific, model: FormaTM Steri-CycleTM CO2 Incubators, catalog number: 370)
- -80 °C freeze
- SorvallTM LegendTM Micro 21R Microcentrifuge (Thermo Fisher Scientific, model: SorvallTM LegendTM Micro 21R , catalog number: 75002490)
- EppendorfTM ThermomixerTM R (Eppendorf, model: Thermomixer R , catalog number: 05-412-401)
- LabquakeTM Tube Shaker/Rotator (Thermo Fisher Scientific, catalog number: C4152110Q )
- SorvallTM ST 40R Centrifuge (Thermo Fisher Scientific, model: SorvallTM ST 40R , catalog number: 75004525)
- NanoDropTM 2000 Spectrophotometer (Thermo Fisher Scientific, model: NanoDropTM 2000 , catalog number: ND-2000)
- T100TM Thermal Cycler (Bio-Rad Laboratories, catalog number: 1861096 )
- CFX ConnectTM Real-Time PCR Detection System (Bio-Rad Laboratories, catalog number: 1855200 )
- UV transilluminator
Procedure
- Plate HFF cells in serum-free DMEM supplemented with MEM non-essential amino acids at low density (approx. 1 x 106 cells/150 cm2 flask). Incubate HFF cells in humidified atmosphere of 5% CO2 at 37 °C for 48 h (Figure 1).
Notes:- In order to obtain enough RNA for each experiment, use 4 to 6 150 cm2 flasks.
- To plate HFF cells in serum-free DMEM; wash 70-80% confluent HFF cells (100 mm TC-treated cell culture dish) three times each with 10 ml phosphate buffered saline, add 1 ml 0.05% trypsin with phenol red to detach cells, add 10 ml serum-free DMEM media and collect cells in 15 ml conical tube, pellet cells by centrifuging at 300 x g at room temperature, and re-suspend pellet in serum-free DMEM supplemented medium.
- It is important to remove trypsin from HFF cells before re-plating since medium doesn’t contain FBS to inactive the trypsin.
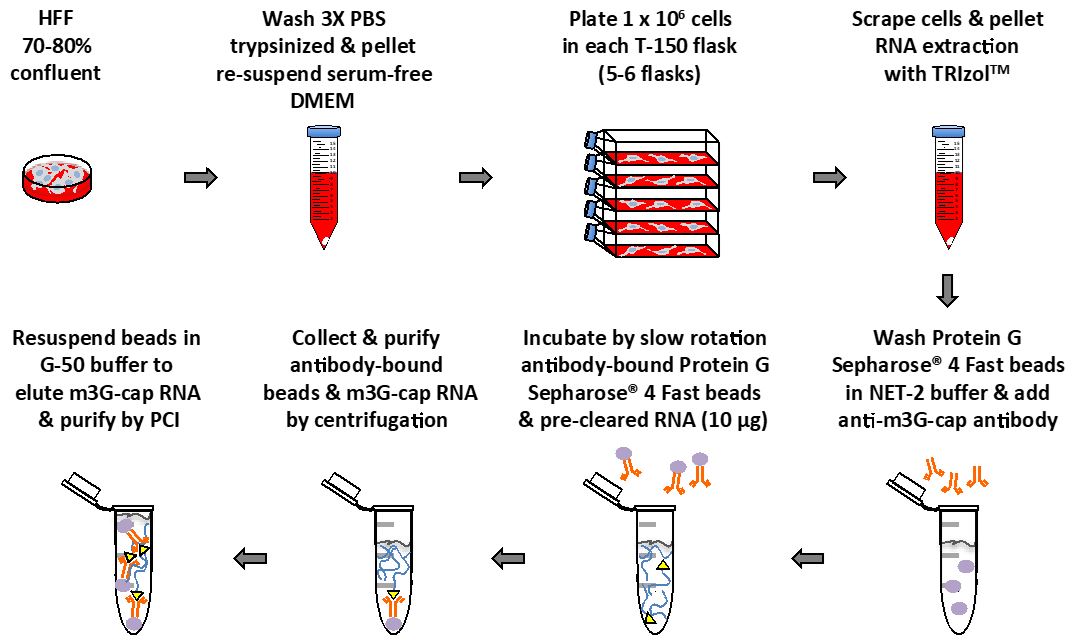
Figure 1. Main steps diagram of this protocol - In order to obtain enough RNA for each experiment, use 4 to 6 150 cm2 flasks.
- Collect HFF cells by scraping flask on ice, pellet at 300 x g for 5 min at 4 °C, and wash two times each with 2 ml of phosphate buffered saline. Cell pellets can be frozen at -80 °C until RNA extraction (cell pellets must be kept on ice until lysed with TRIzolTM) (Figure 1).
Notes:- To pellet HFF cells, remove old serum-free DMEM from cells and add 3 ml of fresh serum-free DMEM. Use the blade of cell scraper to collect cells in one corner of flask taking care to not splash the cells in flask. Add 1-2% of volume of FBS or BSA to cells on ice to assist in pelleting. Extract RNA from pelleted cells using TRIzolTM Reagent per manufacturer’s instructions.
- All steps in this protocol should be performed in an RNase free bench area by using RNase AWAYTM Surface Decontaminant to spray and wipe most surface areas including micropipettes and ice bucket.
- Resuspend cell pellets thawed on ice in 1 ml of TRIzolTM Reagent for 5 min to lyse.
- Add chloroform (20% of TRIzolTM Reagent volume), mix samples well, and incubate at room temperature for 2-3 min.
- Centrifuge samples at 12,000 x g for 15 min at 4 °C to separate red phenol-chloroform (red/lower layer), interphase, and aqueous phase (clear/upper layer).
- Collect aqueous layer and precipitate RNA for 10 min at room temperature with isopropanol (0.5 volume of original TRIzolTM Reagent); GlycoBlueTM Coprecipitant (15 μg) can be added at this time to aid RNA precipitation.
- Pellet precipitated RNA by centrifugation (12,000 x g for 10 min at 4 °C).
- Wash RNA pellets with 75% ethanol (add an equal volume of 75% ethanol to TRIzolTM Reagent and briefly vortex) and re-pellet by centrifuging at 7,500 x g for 5 min at 4 °C.
- Air-dry RNA pellets for a few minutes and re-suspend in nuclease-free water.
Note: Do not completely dry the RNA pellet because it will be difficult to re-suspend. Incubation at 55 °C for 10 min can help solubilize the RNA.
- To pellet HFF cells, remove old serum-free DMEM from cells and add 3 ml of fresh serum-free DMEM. Use the blade of cell scraper to collect cells in one corner of flask taking care to not splash the cells in flask. Add 1-2% of volume of FBS or BSA to cells on ice to assist in pelleting. Extract RNA from pelleted cells using TRIzolTM Reagent per manufacturer’s instructions.
- Use DNase from TURBO DNA-freeTM Kit to digest DNA contamination in RNA samples according to manufacturer’s protocol:
- Mix RNA with 10x TURBO DNase Buffer (0.1 volume) and 1 μl of TURBO DNase.
- Incubate RNA at 37 °C for 20 min and halt the reaction by adding DNase Inactivation Reagent (0.1 volume).
- Incubate RNA samples for 5 min with intermittent mixing by flicking tube then centrifuge at 10,000 x g at room temperature for 2 min.
- Collect RNA (supernatant) and quantitate with NanoDropTM 2000 Spectrophotometer.
- Mix RNA with 10x TURBO DNase Buffer (0.1 volume) and 1 μl of TURBO DNase.
- Pre-load Protein G Sepharose® 4 Fast beads with rabbit serum control or anti-m3G-cap antibody (Figure 1).
Note: Use large-orifice pipet tips or clip the tip off of a pipet tip to aid the transfer of Protein G Sepharose® 4 Fast beads.- Wash 4 times the Protein G Sepharose® 4 Fast beads (40 μl slurry/IP sample) each with 1 ml NET-2 Buffer (see Recipes) using short 30 sec spins at 1,000 x g to pellet beads.
Note: Non-filtered gel-loading pipet tips can be used during ‘wash’ steps to help prevent accidental loss of beads. - Dilute washed Protein G Sepharose beads in 500 μl of NET-2 Buffer and add rabbit anti-m3G-cap antibody (15 μl/IP sample, approximately 150 μg) or rabbit serum control (15 μl/IP sample, approximately 150 μg). To achieve equal loading of beads with antibodies only use one microcentrifuge tube/antibody; aliquot the beads into separate tubes after they are pre-loaded.
- Incubate beads/antibody for 1.5 h at room temperature on tube rotator; make sure samples are actually mixing.
- Remove excess antibody by washing Protein G Sepharose Beads 5 times each with 1 ml NET-2 Buffer (short 30 sec spins at 1,000 x g); Resuspend beads in NET-2 Buffer (50 μl/sample).
- Aliquot antibody-bound Protein G Sepharose Beads into 1.5 microcentrifuge tubes; use enough tubes to have one of each antibody (anti-m3G-cap and control) per sample.
- Wash 4 times the Protein G Sepharose® 4 Fast beads (40 μl slurry/IP sample) each with 1 ml NET-2 Buffer (see Recipes) using short 30 sec spins at 1,000 x g to pellet beads.
- Pre-clear RNA with Protein G Sepharose Beads to reduce non-specific binding:
- Add RNasin Plus RNase inhibitor (1 μl) to RNA (10 μg/500 μl diluted with NET-2 buffer) and incubate for 3 to 5 min at 85 °C.
Note: Plunge RNA into ice immediately after heating to avoid refolding of RNA. - After heating, add an additional 1 μl of RNasin Plus RNase inhibitor to RNA.
Note: Remember to wipe tubes with RNase AWAYTM Surface Decontaminant before opening to prevent possible contamination and degradation of RNA. - Rotate RNA (10 μg/sample for each antibody) slowly (without vibration) with 40 μl of NET 2-washed Protein G Sepharose 4 Fast Flow beads (without antibody) for 2 h at 4 °C.
- Centrifuge RNA at 1,000 x g for 2 min to remove pre-clearing beads.
- Add RNasin Plus RNase inhibitor (1 μl) to RNA (10 μg/500 μl diluted with NET-2 buffer) and incubate for 3 to 5 min at 85 °C.
- Rotate pre-cleared RNA slowly (without vibration) with anti-m3G-cap or control pre-loaded Protein G Sepharose® 4 Fast beads for 4 to 16 h at 4 °C. Confirm that samples are mixing (Figure 1).
- Collect the beads by centrifuging at 1,000 x g for 2 min and remove supernatant (save 250 μl for RNA extraction).
- Wash beads 5 times with 1 ml of NET-2 Buffer using short 30 sec spins at 1,000 x g.
Note: RNasin Plus RNase inhibitor can be added to NET-2 Buffer (see Recipes) to prevent degradation of RNA. - Resuspend beads in 250 μl of G-50 Buffer (see Recipes) to elute TMG-capped RNA (Figure 1).
- Purify RNA by phenol-chloroform-isoamyl alcohol (PCI) extraction (also extract RNA from TMG-capped depleted supernatant)
- Add 250 μl of phenol-chloroform-isoamyl alcohol to RNA in G-50 Buffer.
- Vortex RNA samples for 20 sec (10 up and 10 angled) and centrifuge samples for 10 min at 12,000 x g at room temperature.
- Transfer the aqueous phase with RNA (upper layer) to a clean microcentrifuge tube containing 50 μl of 3 M sodium acetate (NaOAc), pH 5.2.
- Add 1 ml of 100% ethanol (precooled to -20 °C) to precipitate RNA; 1 μl of GlycoBlue can be added to help precipitate RNA and visualize pellets.
- Incubate RNA at -20 °C for 48 h.
- Pellet RNA by centrifuging sample at 12,000 x g for 10 min at 4 °C.
- Aspirate ethanol and wash RNA pellet with 500 μl of 75% ethanol (precooled to -20 °C).
- Re-pellet purified RNA by short spin for 1 min at 12,000 x g at 4 °C and aspirate 75% ethanol.
- Dissolve the dried m3G-capped RNA pellet in nuclease-free water (20 to 30 μl).
- Add 250 μl of phenol-chloroform-isoamyl alcohol to RNA in G-50 Buffer.
- RT-PCR of m3G-capped RNAs by using iScriptTM cDNA Synthesis Kit and 2x SsoAdvancedTM SYBR® Green Supermix PCR according to manufacturer’s protocols:
Note: Perform RT-PCR using un-processed RNA (input) and RNA extracted from supernatant to determine expression levels of RNA of interest (m3G-capped and uncapped).- Mix 5 μl of m3G-capped RNA with 5x iScript Reaction Mix (0.2 volume) and 1 μl of iScript Reverse Transcriptase.
- Process RNA to cDNA in a thermocycler by undergoing the following conditions: 25 °C for 5 min, 46 °C for 20 min, 95 °C for 1 min, and held at 4 °C. The reaction can be stored at 4 °C for short-term storage or -20 °C for long-term storage.
- Mix the resulting cDNA (around ¼ of the total cDNA reaction mix) with 2x SsoAdvancedTM SYBR® Green Supermix (0.5 volume) and the desired forward and reverse primers (12.5 μM stock; 0.04 volume).
- cDNA then undergoes the following conditions to produce a PCR product: 95 °C for 1.5 min; 40 cycles: 95 °C for 30 sec, 60 °C for 30 sec.
- The PCR product is ran on a 1% agarose gel (made with 1x TBE [see Recipes] and ethidium bromide) at 100 V for 2 h, and bands are imaged with a UV transilluminator.
- Mix 5 μl of m3G-capped RNA with 5x iScript Reaction Mix (0.2 volume) and 1 μl of iScript Reverse Transcriptase.
Data analysis
The recovery of (TMG)-capped RNAs was measured by RT-PCR amplification of well-known hypermethylated RNAs such as small nuclear RNA (snRNA) U7 (Figure 2) or small nucleolar RNA (snoRNA) U3 (Martinez et al., 2017).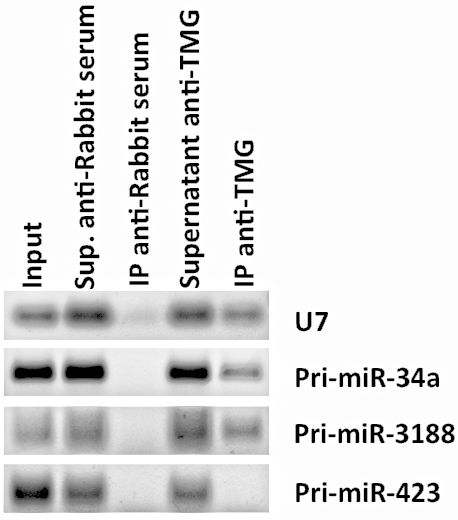
Figure 2. RNA immunoprecipitation of (TMG)-capped primary miRNAs (Pri-miRNAs) in quiescent human foreskin fibroblasts (HFFs). RT-PCR data shows the RNA immunoprecipitation of Pri-miR-34a and Pri-miR-3188 in quiescent HFFs (as well as the positive control snRNA U7), but not Pri-miR-423 using an antibody against (TMG)-capped RNAs. Total RNA was extracted using TRIzol Reagent, and 10 μg of RNA was diluted in NET-2 buffer, precleared and incubated with Protein G Sepharose 4 Fast Flow beads loaded with 15 μl of control antibody (Normal Rabbit Serum, EMD-Millipore) or with antibody recognizing the (TMG)-cap (Anti-m3G-cap, rabbit polyclonal, Synaptic Systems). Beads were rinsed five times with NET-2 buffer and were resuspended in G-50 buffer. RNA was extracted from the beads by phenol-chloroform-isoamyl alcohol extraction and resuspended in 20 μl of nuclease-free water. Immunoprecipitated tri-methylated capped RNA was converted to cDNA using iScript cDNA synthesis kit (Bio-Rad), followed by RT-PCR, and visualized after gel electrophoresis.
Notes
This protocol could be modified to determine the location of TMG-capped RNA by separation of nuclear and cytoplasmic RNA fractions from fresh cultured cells (PARISTM Kit).
Recipes
Note: All reagents should be made with nuclease-free water and autoclaved.
- NET-2 Buffer
150 mM NaCl
0.05% NP-40
50 mM Tris-HCl, pH 7.4 - G-50 buffer
20 mM Tris, pH 7.5
300 mM NaOAc
2 mM EDTA, pH 8
0.25% SDS - 1x TBE
216 g Tris base
110 g boric acid
80 ml 0.5 M EDTA, pH 8
1.5 L water
(Bring solution to pH 8.3)
Acknowledgments
We thank Dr. Joan Steitz’s laboratory for sharing part of this protocol. I.M., J.A.B., and K.E.H. were supported in part by a WVU Foundation Fund (2V882). K.E.H. was supported in part by funding from the Ladies Auxiliary to the VFW of the United States (CK 003229). M.X. is supported by NIH grant R00 CA190886. J.A.S. is an Investigator of the Howard Hughes Medical Institute. This protocol was modified from previous work (Yu et al., 1998). We do not have any conflict of interest or competing interests.
References
- Boulon, S., Verheggen, C., Jady, B. E., Girard, C., Pescia, C., Paul, C., Ospina, J. K., Kiss, T., Matera, A. G., Bordonne, R. and Bertrand, E. (2004). PHAX and CRM1 are required sequentially to transport U3 snoRNA to nucleoli. Mol Cell 16(5): 777-787.
- Coller, H. A. (2011). Cell biology. The essence of quiescence. Science 334(6059): 1074-1075.
- Esteller, M. (2011). Non-coding RNAs in human disease. Nat Rev Genet 12(12): 861-874.
- Ha, M. and Kim, V. N. (2014). Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15(8): 509-524.
- Kim, Y. K., Kim, B. and Kim, V. N. (2016). Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc Natl Acad Sci U S A 113(13): E1881-1889.
- Kiss, T. (2004). Biogenesis of small nuclear RNPs. J Cell Sci 117(Pt 25): 5949-5951.
- Luhrmann, R., Appel, B., Bringmann, P., Rinke, J., Reuter, R., Rothe, S. and Bald, R. (1982). Isolation and characterization of rabbit anti-m3 2,2,7G antibodies. Nucleic Acids Res 10(22): 7103-7113.
- Martinez, I., Hayes, K. E., Barr, J. A., Harold, A. D., Xie, M., Bukhari, S. I. A., Vasudevan, S., Steitz, J. A. and DiMaio, D. (2017). An Exportin-1-dependent microRNA biogenesis pathway during human cell quiescence. Proc Natl Acad Sci U S A 114(25): E4961-E4970.
- Ohno, M., Segref, A., Bachi, A., Wilm, M. and Mattaj, I. W. (2000). PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell 101(2): 187-198.
- Palacios, I., Hetzer, M., Adam, S. A. and Mattaj, I. W. (1997). Nuclear import of U snRNPs requires importin β. EMBO J 16(22): 6783-6792.
- Valcourt, J. R., Lemons, J. M., Haley, E. M., Kojima, M., Demuren, O. O. and Coller, H. A. (2012). Staying alive: metabolic adaptations to quiescence. Cell Cycle 11(9): 1680-1696.
- Yedavalli, V. S. and Jeang, K. T. (2010). Trimethylguanosine capping selectively promotes expression of Rev-dependent HIV-1 RNAs. Proc Natl Acad Sci U S A 107(33): 14787-14792.
- Yu, Y. T., Shu, M. D. and Steitz, J. A. (1998). Modifications of U2 snRNA are required for snRNP assembly and pre-mRNA splicing. EMBO J 17(19): 5783-5795.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hayes, K. E., Barr, J. A., Xie, M., Steitz, J. A. and Martinez, I. (2018). Immunoprecipitation of Tri-methylated Capped RNA. Bio-protocol 8(3): e2717. DOI: 10.21769/BioProtoc.2717.
Category
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.