- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Extraction and Analysis of Pan-metabolome Polar Metabolites by Ultra Performance Liquid Chromatography–Tandem Mass Spectrometry (UPLC-MS/MS)
Published: Vol 8, Iss 3, Feb 5, 2018 DOI: 10.21769/BioProtoc.2715 Views: 10026
Reviewed by: Neelanjan BoseAnca Flavia SavulescuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

![Automated Sequential Derivatization for Gas Chromatography-[Orbitrap] Mass Spectrometry-based Metabolite Profiling of Human Blood-based Samples](https://en-cdn.bio-protocol.org/imageup/arcimg/20250107224024942.jpg?t=1770522920)
Abstract
Modern triple quadrupole mass spectrometers provide the ability to detect and quantify a large number of metabolites using tandem mass spectrometry (MS/MS). Liquid chromatography (LC) is advantageous, as it does not require derivatization procedures and a large diversity in physiochemical characteristics of analytes can be accommodated through a variety of column chemistries. Recently, the comprehensive optimization of LC-MS metabolomics using design of experiments (COLMeD) approach has been described and used by our group to develop robust LC-MS workflows (Rhoades and Weljie, 2016). The optimized LC-MS/MS method described here has been utilized extensively for metabolomics analysis of polar metabolites. Typically, tissue or biofluid samples are extracted using a modified Bligh-Dyer protocol (Bligh and Dyer, 1959; Tambellini et al., 2013). The protocol described herein describes this workflow using targeted polar metabolite multiple reaction monitoring (MRM) from tissues and biofluids via ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). This workflow has been utilized extensively for chronometabolic analysis (Krishnaiah et al., 2017), with applications generalized to other types of analyses as well (Sengupta et al., 2017; Sivanand et al., 2017).
Keywords: UPLC-MSBackground
Metabolomics is a field of study aiming to comprehensively analyze metabolites through the use of various analytical detection methods, namely mass spectrometry (MS) and nuclear magnetic resonance (NMR) (Liu and Locasale, 2017). Although both NMR and MS are essential tools in metabolomics, mass spectrometry can analyze samples with greater sensitivity (Liu and Locasale, 2017). Within mass spectrometry, various approaches are available, however a rapid approach to profile the global metabolome is needed (Lv et al., 2011; Rhoades and Weljie, 2016). Advancements in triple quadrupole mass spectrometers have made them well suited for ion-switching methods (scanning in both positive and negative ion modes within a single analysis) in addition to enabling reproducible and sensitive targeted profiling of numerous metabolites (Lv et al., 2011; Gika et al., 2012; Yuan et al., 2012; Rhoades and Weljie, 2016). Additionally, LC typically does not require extensive sample preparation, nor derivatization, which allows for the detection of a broader range of metabolites (Gika et al., 2012; Liu and Locasale, 2017). Nonetheless, development of LC-MS methods to comprehensively analyze small polar metabolites is nontrivial, and requires advanced modeling to optimize both LC and MS factors simultaneously. Thus, the COLMeD approach aimed to address this challenge and enabled a more comprehensive metabolite analysis across platforms including on a triple quadrupole mass spectrometer (Rhoades and Weljie, 2016). The LC-MS workflow on a triple quadrupole is described here and has been used to successfully study metabolomics in a circadian context in which 179 metabolites were successfully profiled and analyzed (Krishnaiah et al., 2017).
Materials and Reagents
- Pipette tips 1,000 μl, 200 μl, 10 μl (Gilson, catalog numbers: F1735001 , F1733001 , F1732001 )
- 1.7 ml PosiClick tubes (Denville Scientific, catalog number: C2170 )
- 2.0 ml Safe-Lock tubes (Eppendorf, catalog number: 022363352 )
- Mass spectrometry vials and caps–Verex Vial Kit, 9 mm, PP, 300 μl + PTFE/Silicone, pre slit (Phenomenex, catalog number: AR0-9992-13 )
- Stainless steel beads 5 mm (QIAGEN, catalog number: 69989 )
- Gloves (Denville Scientific, catalog number: G4161 )
- Sample trays
- VanGuard cartridge holder (WATERS, catalog number: 186007949 )
- XBridge BEH Amide 2.5 μm XP Vanguard Cartridge, 2.1 x 5 mm (WATERS, catalog number: 186007763 )
- XBridge BEH Amide 2.5 μm, 2.1 x 100 mm column XP (WATERS, catalog number: 186006091 )
- Acquity UPLC column In-Line Filter Kit (WATERS, catalog number: 205000343 )
- MilliQ water–18 mΩ, 0.22 μm filter (Merck, catalog number: MPGP04001 )
- Acetonitrile–Optima LC/MS (Fisher Scientific, catalog number: A955-4 )
- Ammonium acetate (Sigma-Aldrich, catalog number: 73594-25G-F )
- Ammonium hydroxide TraceMetal grade (Fisher Scientific, catalog number: A512-P500 )
- Argon compressed gas (Airgas)
- Chloroform (Fisher Scientific, catalog number: C298-1 )
- Methanol optima LCMS (Fisher Scientific, catalog number: A456-4 )
- Nitrogen gas (Airgas, catalog number: NI230LT230RB )
- Solvent A (see Recipes)
- Solvent B (see Recipes)
- Seal wash (see Recipes)
Equipment
- Pipettes (P1000, P200, P20, P10)
- Centrifuge (Eppendorf, model: 5430 R , catalog number: 022620511)
- Vortex
- ACQUITY H-Class UPLC (WATERS, model: ACQUITY UPLC H-Class )
- Bath Sonicator (VWR, catalog number: 97043-976 )
- Speed Vacuum–Vacufuge Plus (Eppendorf, model: Vacufuge plus , catalog number: 022822993)
- TissueLyser II (QIAGEN, catalog number: 85300 )
- Xevo TQ-S Micro (WATERS, model: Xevo TQ-S Micro )
Software
- MassLynx Version 4.1
- TargetLynx XS
- R (version 3.3)
Procedure
- Extracting polar metabolites
Polar metabolites were extracted using a modified Bligh-Dyer protocol as described below (Bligh and Dyer, 1959; Tambellini et al., 2013). Extractions were performed on ice unless otherwise indicated.- One set of 1.5 ml microfuge tubes for the collection of polar metabolites should be labeled for each sample. (A second set can be labeled if collection of the nonpolar metabolites is desired as well.) The number of biological replicates required for analysis is dependent upon the study and can range from as few as 5 for highly controlled models to several hundred to thousands for human studies.
- The following solvents should be prepared and cooled prior to beginning the extraction protocol:
- 2:1 Methanol:Chloroform mixture (300 μl per sample)
- Pure methanol (100 μl per sample)
- Water (18 mΩ, 0.22 μm filtered) (100 μl per sample)
- 2:1 Methanol:Chloroform mixture (300 μl per sample)
- Centrifuge should also be cooled to 4 °C.
- Samples (i.e., U2OS and primary hepatocyte cell pellets [number of cells equivalent to a 50 mg cell pellet] or 50 mg of liver tissue) should be placed on ice to thaw.
Notes:- Smaller sample sizes can be successfully used. E.g., 1 x 106 U2OS cells were utilized in Krishnaiah et al., 2017.
- If extracting tissue, 2 ml safe-lock microfuge tubes should be utilized.
- Smaller sample sizes can be successfully used. E.g., 1 x 106 U2OS cells were utilized in Krishnaiah et al., 2017.
- (Optional) If quantitation is required, stable-isotope labeled internal standards at a concentration determined on a per metabolite basis should be added to thawed samples at this point.
- Once samples are thawed, follow Step A6a if extracting cell pellets or A6b if extracting tissue.
- Extraction of cell pellets:
- To each sample, 300 μl of the cold 2:1 Methanol:Chloroform mixture is added.
- Each microfuge tube containing the sample is vortexed until a homogenous mixture is formed. Figure 1A shows a representative endpoint.
Note: The cell pellet may not be homogenized upon vortexing and thus it is essential to sonicate the sample. - Next, the samples are sonicated in a bath sonicator for 15 min at 35 kHz. Figure 1B shows a representative endpoint.
- At the end of sonication, samples are placed back on ice.

Figure 1. Examples of representative endpoints during an extraction. A. An example of a cell pellet extraction to which 300 μl of 2:1 Methanol:Chloroform was added and vortexed. B. An example of a cell extraction after sonication (Step A6a.iii). C. The aqueous (1) and organic (2) layers are shown at the end of centrifugation (Steps A10 to A11).
- To each sample, 300 μl of the cold 2:1 Methanol:Chloroform mixture is added.
- Extraction of tissue using the TissueLyser II:
- Pre-cool tissue homogenizer blocks by placing at -80 °C.
- To each tissue sample, carefully add a stainless steel bead followed by 300 μl of the cold 2:1 Methanol:Chloroform mixture.
- Next, using the pre-cooled blocks (handle using gloves), homogenize the tissue samples at 25 Hz for 4 min using the TissueLyser II.
- At the end of homogenization, place the samples back on ice.
- Pre-cool tissue homogenizer blocks by placing at -80 °C.
- Extraction of cell pellets:
- Next, 100 μl each of water and chloroform are added to each microfuge tube containing the samples (cell pellets or tissue).
- The samples are then vortexed again until a homogenous mixture is formed.
- In order to separate the polar and non-polar metabolites, microfuge tubes containing the samples are centrifuged at 18,787 x g (RCF) for 7 min at 4 °C.
- Samples are then placed back on ice carefully in order to avoid disruption of the separated layers.
- Next, carefully collect the upper layer (indicated as layer 1 in Figure 1C) containing the polar metabolites into the pre-labeled set of microfuge tubes taking care not to disturb the interface and/or collect insoluble particulates. (The lower fraction [indicated as layer 2 in Figure 1C] containing nonpolar metabolites can be collected in a similar manner if desired.)
Note: It is recommended to perform the extraction procedure on test samples prior to actual samples. - The microfuge tubes containing the aqueous layer metabolites are then dried in a speed vacuum for 4 h or until dry.
- Upon completion of drying, metabolites can either be re-suspended as indicated below or stored at -80 °C.
Note: Storing dried samples at -80 °C impacts the stability of metabolites in a differential manner dependent on various compound classes, therefore, it is recommended to perform LC-MS analysis soon after extraction.
- One set of 1.5 ml microfuge tubes for the collection of polar metabolites should be labeled for each sample. (A second set can be labeled if collection of the nonpolar metabolites is desired as well.) The number of biological replicates required for analysis is dependent upon the study and can range from as few as 5 for highly controlled models to several hundred to thousands for human studies.
- Re-suspending and preparing polar metabolites for LC-MS
- A randomized run order should be generated in order to minimize variation as a result of sample preparation order.
- A minimum of 5 blanks in addition to 10 equilibration (EQ) samples will be run at the beginning of the analysis (see Step B13 in this section for information on preparing EQ and QC samples). Next, 4 quality control (QC) samples will be run followed by the samples. Every 6 to 10 samples, a QC will be run in addition to 2 QC’s at the end of the analysis (Figure 2). Note that the spacing between QC injections can vary, and may be dependent on sample quantity. Greater QC frequency (e.g., every 6 samples) provides more robust data correction, however requires more sample.
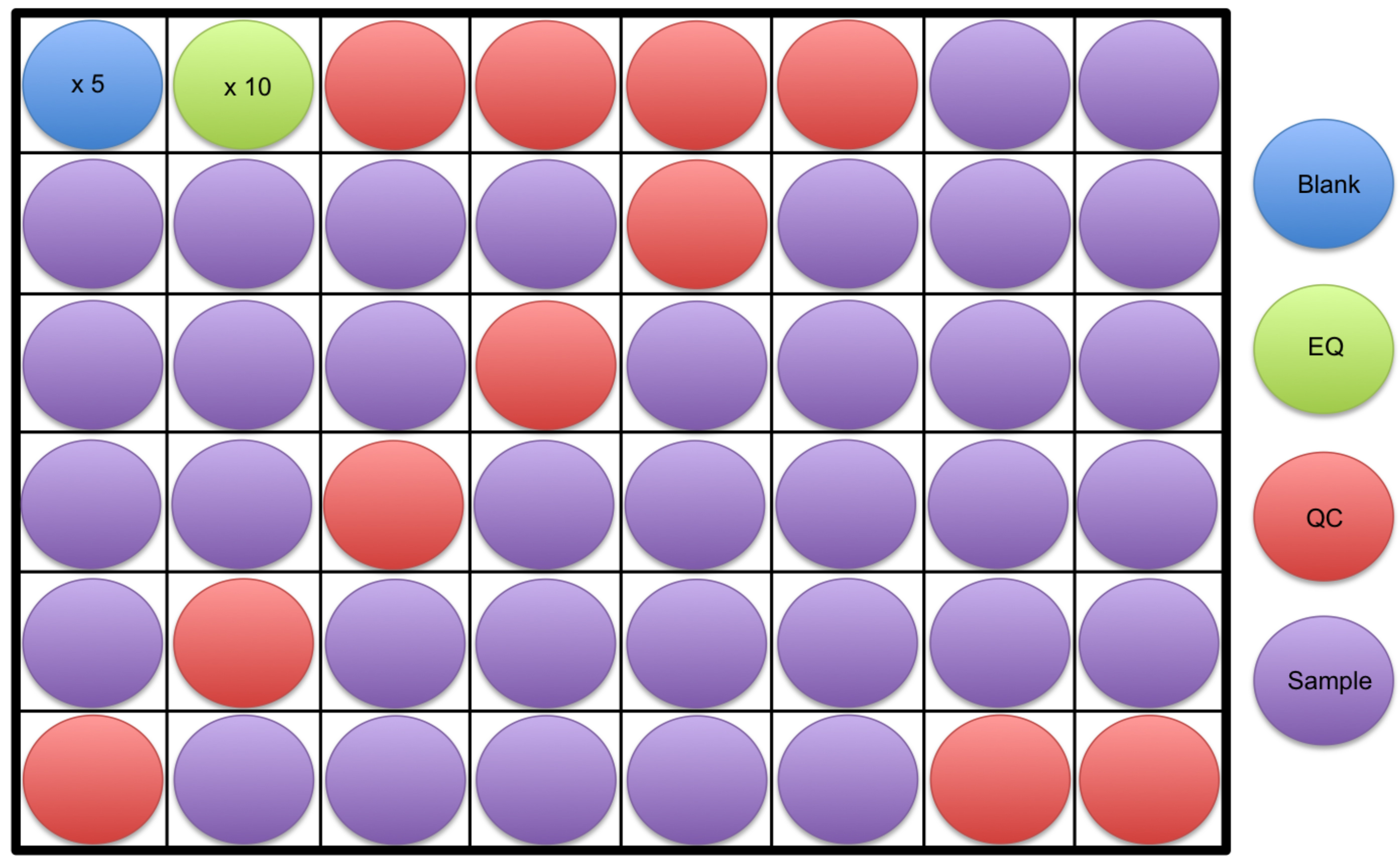
Figure 2. An example of a sample run order is shown. At least 5 blanks (blue) followed by 10 equilibration samples (green) should be run at the start of an analysis. This is followed by 4 quality control samples (red), samples to be analyzed (purple), and then 2 quality control samples again at the end. The example shows a quality control sample run every 6 samples throughout the analysis, however, this can vary from every 6 to 10 samples. - Diluent consisting of 1:1 Acetonitrile:Water should be prepared and cooled beforehand.
- In addition, the centrifuge should be cooled to 4 °C.
- Dried polar metabolites, diluent as well as sample tray(s) should be placed on ice.
- 100 μl of diluent are added to each microfuge tube containing the dried polar metabolites as well as to an empty microfuge tube to be used as a blank. Again, the volume metabolites are resuspended in is dependent on the sample concentration and is adjusted to ensure metabolites are detected within a linear range. Proper dilution of metabolites also ensures longer column life.
- Each microfuge tube is then vortexed for a minimum of 20 sec in order to re-suspend the metabolites.
- Next, centrifuge the microfuge tubes for 7 min at 18,787 x g (RCF) at 4 °C as an additional precautionary step to clean up samples.
- The supernatant from each sample is then aliquoted as follows:
- 5 μl is added to a microfuge tube to generate a pooled quality control (QC) sample. (Volume added to the pooled quality control sample is dependent on the total number of quality control samples to be run. For instance, if 10 QC samples are to be run, a minimum of 200 μl would be required if 20 μl are to be added to each vial.)
- A minimum of 20 μl is added to LC-MS vials for each replicate to be analyzed. (A minimum of 2 analytical replicates, ideally 3 is recommended.) Volume added to each vial is dependent on injection volume as injection volumes can range from 2 to 10 μl. (It has been noted that smaller injection volumes yield better chromatography.)
Note: It should be noted that MS vials with inserts were utilized and larger volumes may be required if utilizing standard 1.5 ml HPLC vials.
- 5 μl is added to a microfuge tube to generate a pooled quality control (QC) sample. (Volume added to the pooled quality control sample is dependent on the total number of quality control samples to be run. For instance, if 10 QC samples are to be run, a minimum of 200 μl would be required if 20 μl are to be added to each vial.)
- LC-MS vials are placed in sample tray(s) according to a pre-randomized run order.
- 90 μl of the blank (or volume sufficient for the number of blanks to be run) is added to an LC-MS vial and placed in the sample tray.
- Once all samples have been aliquoted, the pooled QC sample is vortexed to ensure homogeneity.
- From the pooled QC sample:
- An equilibration sample is prepared by diluting the QC sample 10 fold using the diluent (1:1 Acetonitrile:Water).
- 20 μl is added to mass spec vials and vials are placed in the appropriate positions in the sample tray(s). (The number of vials is dependent on total QC samples to be run and the volume added to vials varies based on desired injection volume.)
- An equilibration sample is prepared by diluting the QC sample 10 fold using the diluent (1:1 Acetonitrile:Water).
- 90 μl of the EQ sample is added to an LC-MS vial and placed in the sample tray. Volume can be adjusted based on the number of EQ’s to be run.
- A randomized run order should be generated in order to minimize variation as a result of sample preparation order.
- Acquiring data by UPLC-MS
Polar metabolites were then analyzed through UPLC-MS utilizing a method optimized through the COLMeD approach as described in (Rhoades and Weljie, 2016). The following procedure should be used as guidelines in acquiring data by individuals trained in using a mass spectrometer.- Solvent A is prepared fresh according to Recipe 1. In addition, ensure that there is enough of solvent B (Recipe 2) and the seal wash (Recipe 3) present. If not prepare the solvents.
- An XBridge BEH Amide column (2.5 μm 100 x 2.1 mm) with a pre-column inline filter and vanguard is utilized at a temperature of 40 °C. The column is utilized on an Acquity (H-Class) UPLC System coupled to a Waters Xevo Triple quadrupole mass spectrometer.
- The temperature for the sample manager is set to 8 °C.
- Additionally, for the duration of the analysis, nitrogen pressure should be maintained above 100 PSI. Argon gas is used as the collision gas.
- Solvent lines, seal wash, and sample manager are primed prior to the start of the analysis. In addition, the needle is washed using 100% acetonitrile. The mass spectrometer is placed in operation for a minimum of 30 min prior to the start of the analysis.
- The column is equilibrated in the solvents as well as the starting conditions prior to beginning the analysis according to Water’s XP 2.5 micron Columns Care and Use Manual.
- The following parameters as indicated in Table 1 are utilized for the LC Inlet method for each injection. The run time for each injection is 30 min followed by a 1 min seal wash.
Table 1. LC parameters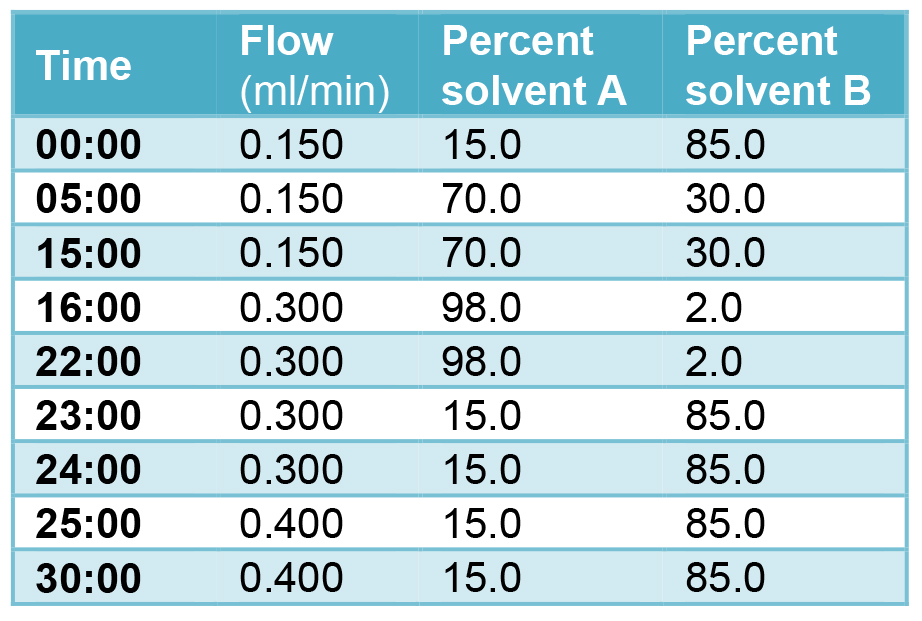
- Parameters utilized for the mass spectrometer in MSMS mode are indicated in Table 2 and are utilized for both electrospray positive and negative modes.
Table 2. MS parameters for both positive and negative modes
- 2 μl of the blanks and equilibration samples are injected while a 2 to 10 μl injection volume is utilized for the quality control samples and samples.
- Samples are analyzed utilizing a multiple reaction monitoring (MRMs) mode in which 338 metabolites are targeted through their mass over charge ratios however the number of metabolites detected varies based on sample concentration, sample type, as well as sample handling. MRMs were generated utilizing the databases METLIN (Smith et al., 2005) and HMDB (Wishart et al., 2013) in addition to Waters’ IntelliStart software and standards as well as prior established protocols (Basu and Blair, 2011; Yuan et al., 2012). Metabolites scanned for and their settings (transitions and cone and collision voltages) are detailed in Table S1.
- Solvent A is prepared fresh according to Recipe 1. In addition, ensure that there is enough of solvent B (Recipe 2) and the seal wash (Recipe 3) present. If not prepare the solvents.
Data analysis
- Data acquired through LC-MS as indicated above is processed through the use of the TargetLynx software. This software is utilized to identify peaks from Total Intensity Chromatograms, integrate them and to obtain ion counts (Figure 3).
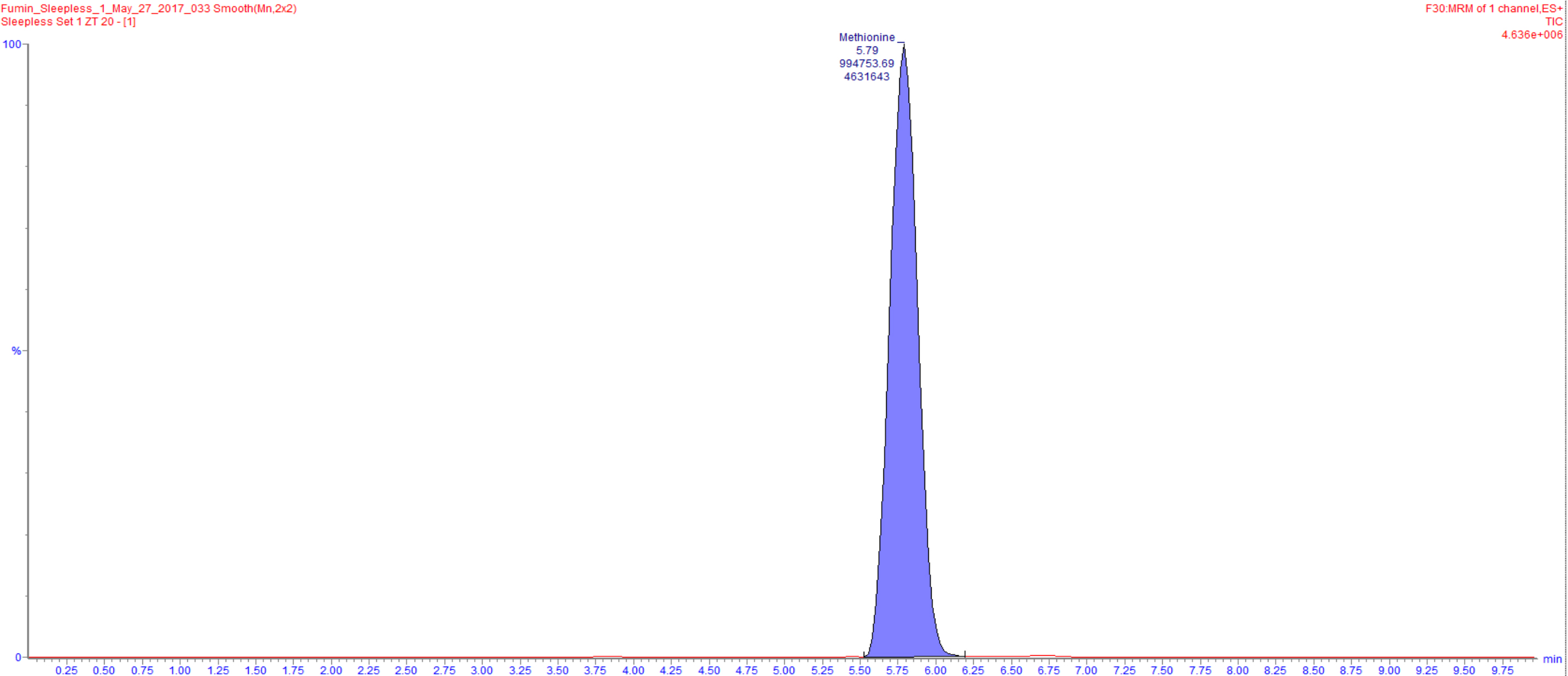
Figure 3. An example of a total intensity chromatogram is shown. Here, the peak for methionine was identified and integrated using TargetLynx resulting in an ion count of 994753.69. - Ion counts obtained from TargetLynx are then exported to a text file for further processing in R.
- In R, a customized script is utilized to remove metabolites with metabolic features appearing in less than 50% of QC samples or those that have a coefficient of variation greater than 30%. The QC samples are also used to fit a cross-validated locally estimated scatterplot smoothing function (LOESS) to each metabolite (Dunn et al., 2011) in order to account for instrumental drift over the course of the analysis to be used for normalization of ion counts (Figure 4). (The R script is available upon request however it should be noted that it is specific for data obtained using Waters’ TargetLynx software.)
- The normalized ion counts are then utilized for further statistical and multivariate analyses.
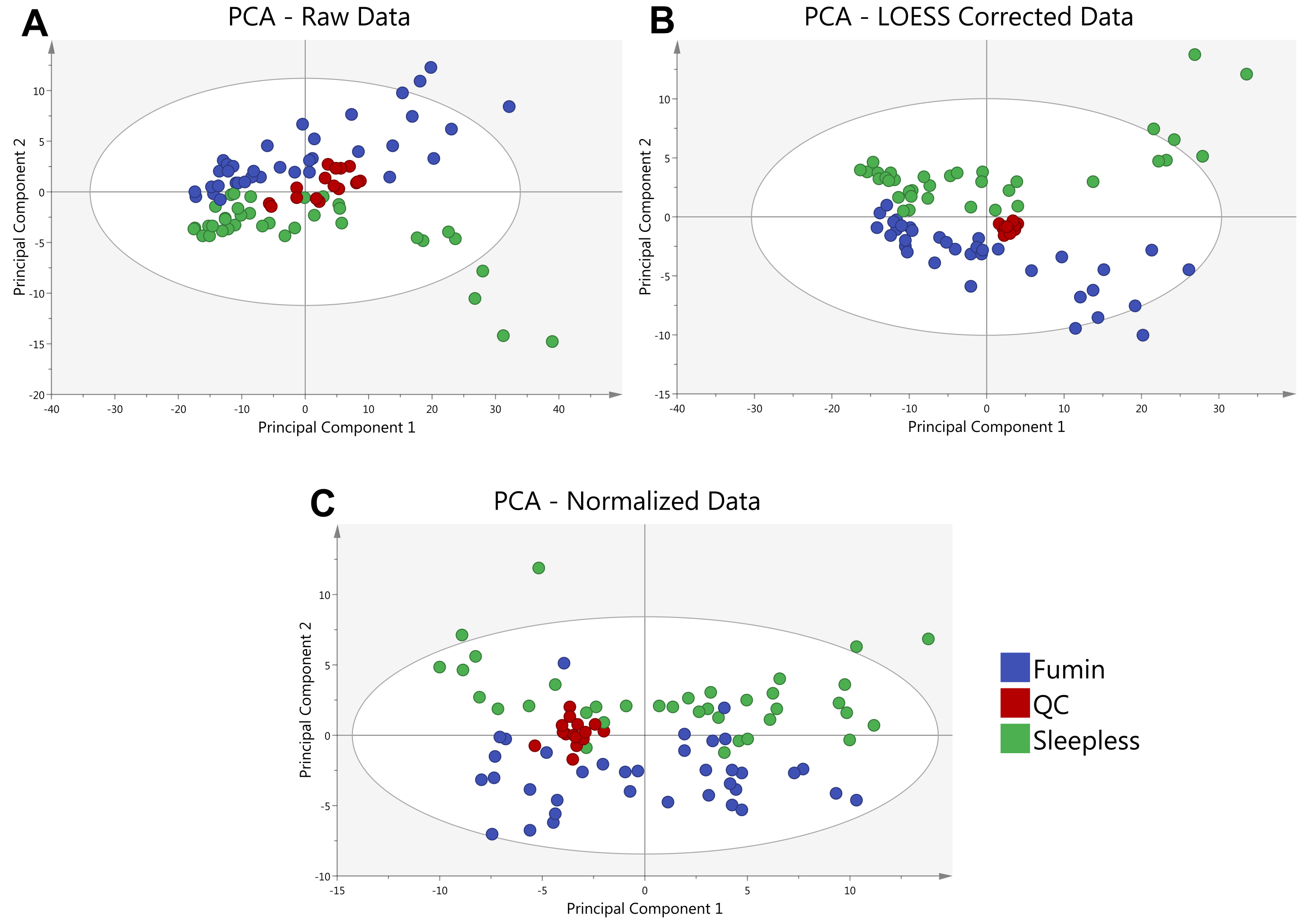
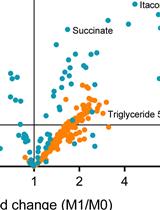
Figure 4. Principal Component Analysis (PCA) of a sample data set is shown. A PCA of the average of 2 analytical replicates for 2 Drosophila genotypes (Fumin and Sleepless) shows the clustering of quality control (QC) samples at various steps in the data analysis process. QC samples (red) are shown prior to LOESS correction and Normalization (A) to exhibit a greater spread as compared to after LOESS Correction (B). QC samples remain tightly clustered after median fold change normalization of the LOESS corrected Data (C).
Notes
The above described extraction protocol has been utilized in the analysis of cell pellets (Krishnaiah et al., 2017), liver (Krishnaiah et al., 2017), whole Drosophila (Maguire et al., 2015), and brain tissues (McGrath et al., 2008) as well as other sample types such as plasma (Banoei et al., 2017), serum (Beaudry et al., 2016 and Hao et al., 2016), cereberal spinal fluid biofluids, yeast (Tambellini et al., 2013 and 2017) etc. We also strongly recommend that any results be validated by inclusion of a stable-isotope labeled internal standard prior to extraction or unlabeled standard analyzed in a similar matrix to confirm retention times and fragmentation patterns.
Recipes
- Solvent A
95:5 Water:Acetonitrile
20 mM ammonium acetate
Ammonium hydroxide, pH of 9
Sonicate at 35 kHz for 15 min - Solvent B
100% acetonitrile
Sonicate for 5 min - Seal wash
50:50 Water:Acetonitrile
Sonicate at 35 kHz for 15 min
Acknowledgments
D.M.M and S.D.R. are supported through a Pharmacology T32 Training Grant (T32 GM008076) and the project was supported in part by the Institute for Translational Medicine and Therapeutics’ (ITMAT) Transdisciplinary Program in Translational Medicine and Therapeutics of the University of Pennsylvania via an award to AMW. The project described was supported by the National Center for Research Resources, Grant UL1RR024134, and is now at the National Center for Advancing Translational Sciences, Grant UL1TR000003. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The above-described protocol has been adapted from (Rhoades and Weljie, 2016) and (Krishnaiah et al., 2017). The authors declare no conflicts of interest or competing interests.
References
- Banoei, M. M., Vogel, H. J., Weljie, A. M., Kumar, A., Yende, S., Angus, D. C., Winston, B.W., and the Canadian Critical Care Translational Biology Group. (2017). Plasma metabolomics for the diagnosis and prognosis of H1N1 influenza pneumonia. Crit Care 21(1): 97.
- Basu, S. S. and Blair, I. A. (2011). SILEC: a protocol for generating and using isotopically labeled coenzyme A mass spectrometry standards. Nat Protoc 7(1): 1-12.
- Beaudry, P., Campbell, M., Dang, N. H., Wen, J., Blote, K., and Weljie, A. M. (2016). A pilot study on the utility of serum metabolomics in neuroblastoma patients and xenograft models. Pediatr Blood Cancer 63(2): 214-220.
- Bligh, E. G. and Dyer, W. J. (1959). A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37(8): 911-917.
- Dunn, W. B., Broadhurst, D., Begley, P., Zelena, E., Francis-McIntyre, S., Anderson, N., Brown, M., Knowles, J. D., Halsall, A., Haselden, J. N., Nicholls, A. W., Wilson, I. D., Kell, D. B., Goodacre, R. and Human Serum Metabolome, C. (2011). Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc 6(7): 1060-1083.
- Gika, H. G., Theodoridis, G. A., Vrhovsek, U. and Mattivi, F. (2012). Quantitative profiling of polar primary metabolites using hydrophilic interaction ultrahigh performance liquid chromatography-tandem mass spectrometry. J Chromatogr A 1259: 121-127.
- Hao, D., Sarfaraz, M. O., Farshidfar, F., Bebb, D. G., Lee, C. Y., Card, C. M., David, M., and Weljie, A. M. (2016). Temporal characterization of serum metabolite signatures in lung cancer patients undergoing treatment. Metabolomics 12: 58.
- Krishnaiah, S. Y., Wu, G., Altman, B. J., Growe, J., Rhoades, S. D., Coldren, F., Venkataraman, A., Olarerin-George, A. O., Francey, L. J., Mukherjee, S., Girish, S., Selby, C. P., Cal, S., Er, U., Sianati, B., Sengupta, A., Anafi, R. C., Kavakli, I. H., Sancar, A., Baur, J. A., Dang, C. V., Hogenesch, J. B. and Weljie, A. M. (2017). Clock regulation of metabolites reveals coupling between transcription and metabolism. Cell Metab 25(4): 961-974 e964.
- Liu, X. and Locasale, J. W. (2017). Metabolomics: A primer. Trends Biochem Sci 42(4): 274-284.
- Lv, H., Palacios, G., Hartil, K. and Kurland, I. J. (2011). Advantages of tandem LC-MS for the rapid assessment of tissue-specific metabolic complexity using a pentafluorophenylpropyl stationary phase. J Proteome Res 10(4): 2104-2112.
- Maguire, S. E., Rhoades, S., Chen, W. F., Sengupta, A., Yue, Z., Lim, J. C., Mitchell, C.H., Weljie, A., and Sehgal, A. (2015). Independent effects of γ-aminobutyric acid transaminase (GABAT) on metabolic and sleep homeostasis. J Biol Chem 290(33): 20407-20416.
- McGrath, B. M., McKay, R., Dave, S., Seres, P., Weljie, A. M., Slupsky, C. M., Hanstock, C. C., Greenshaw, A. J., and Silverstone, P. H. (2008). Acute dextro-amphetamine administration does not alter brain myo-inositol levels in humans and animals: MRS investigations at 3 and 18.8 T. Neurosci Res 61(4): 351-359.
- Rhoades, S. D. and Weljie, A. M. (2016). Comprehensive optimization of LC-MS metabolomics methods using design of experiments (COLMeD). Metabolomics 12(12).
- Sengupta, A., Rhoades, S. D., Kim, E. J., Nayak, S., Grant, G. R., Meerlo, P. and Weljie, A. M. (2017). Sleep restriction induced energy, methylation and lipogenesis metabolic switches in rat liver. Int J Biochem Cell Biol.
- Sivanand, S., Rhoades, S., Jiang, Q., Lee, J. V., Benci, J., Zhang, J., Yuan, S., Viney, I., Zhao, S., Carrer, A., Bennett, M. J., Minn, A. J., Weljie, A. M., Greenberg, R. A. and Wellen, K. E. (2017). Nuclear acetyl-CoA production by ACLY promotes homologous recombination. Mol Cell 67(2): 252-265 e256.
- Smith, C. A., O’Maille, G., Want, E. J., Qin, C., Trauger, S. A., Brandon, T. R., Custodio, D. E., Abagyan, R. and Siuzdak, G. (2005). METLIN: a metabolite mass spectral database. Ther Drug Monit 27(6): 747-751.
- Tambellini, N. P., Zaremberg, V., Krishnaiah, S., Turner, R. J., and Weljie, A. M. (2017). Primary metabolism and medium-chain fatty acid alterations precede Long-chain fatty acid changes impacting neutral lipid metabolism in response to an anticancer lysophosphatidylcholine analogue in yeast. J Proteome Res 16(10): 3741-3752.
- Tambellini, N. P., Zaremberg, V., Turner, R. J. and Weljie, A. M. (2013). Evaluation of extraction protocols for simultaneous polar and non-polar yeast metabolite analysis using multivariate projection methods. Metabolites 3(3): 592-605.
- Wishart, D. S., Jewison, T., Guo, A. C., Wilson, M., Knox, C., Liu, Y., Djoumbou, Y., Mandal, R., Aziat, F., Dong, E., Bouatra, S., Sinelnikov, I., Arndt, D., Xia, J., Liu, P., Yallou, F., Bjorndahl, T., Perez-Pineiro, R., Eisner, R., Allen, F., Neveu, V., Greiner, R. and Scalbert, A. (2013). HMDB 3.0--The human metabolome database in 2013. Nucleic Acids Res 41(Database issue): D801-807.
- Yuan, M., Breitkopf, S. B., Yang, X. and Asara, J. M. (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7(5): 872-881.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Malik, D. M., Rhoades, S. D. and Weljie, A. M. (2018). Extraction and Analysis of Pan-metabolome Polar Metabolites by Ultra Performance Liquid Chromatography–Tandem Mass Spectrometry (UPLC-MS/MS). Bio-protocol 8(3): e2715. DOI: 10.21769/BioProtoc.2715.
Category
Systems Biology > Metabolomics > Biofluid > Polar Metabolite
Systems Biology > Metabolomics > Tissue
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.