- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fluorescent Measurement of Synaptic Activity Using FM Dyes in Dissociated Hippocampal Cultured Neurons
Published: Vol 8, Iss 2, Jan 20, 2018 DOI: 10.21769/BioProtoc.2690 Views: 11693
Reviewed by: Xi FengLu HanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Release and recycling of synaptic vesicles are essential for neurotransmission and synaptic plasticity. To gain mechanistic understanding of these processes, direct measurements of vesicle release and retrieval is indispensable. Styryl dyes like FM1-43 and FM4-64 have been widely used for this purpose and their loading and unloading are reliable measurements for synaptic vesicle release and retrieval in cultured neurons. This protocol describes in detail the procedure of using styryl dyes to label and measure synaptic vesicle uptake and release in cultured rat hippocampal neurons. We also include a brief description of hippocampal culture. In the end, we briefly discuss the commonality and difference among FM dye, pH-sensitive fluorescent proteins and quantum dots in terms of measuring synaptic vesicle behavior.
Keywords: FM1-43Background
Synaptic vesicles are indispensable for neurotransmission since they are the only organelle responsible for neurotransmitter release in chemical synapses. Their amount, release probability, fusion kinetics and recycling routes define synaptic transmission and neuronal communication. Various tools have been developed to probe synaptic vesicles, including electrophysiological recording of postsynaptic neurons, capacitance measurement of membrane trafficking, amperometry of oxidizable transmitters, electron microscope imaging of fixed synapses, and fluorescence imaging of vesicular labels in live neurons. Among all existing methods, the last is the only one that not only yields both spatial and temporal information about individual synapses but also provides high throughput (i.e., more data points from single synapses of different neurons). Various fluorescent probes based on different targeting and reporting mechanisms have been developed. Styryl dye (i.e., FM dyes including FM1-43, FM4-64, FM5-95), invented more than twenty years ago, remains a reliable and convenient tool. Due to its moderate affinity to lipid membrane and its lipid-sensitive emission, it can be readily loaded into recycled synaptic vesicles and released when those vesicles are exocytosed. Using more sensitive photodetectors like EMCCD, FM dyes can report single vesicle release events. Here, we provide a relatively complete description of FM-based imaging of synaptic vesicle release in primary cultures of rodent hippocampal neurons. In addition, we also discuss the commonality and the distinction between FM dyes and other fluorescent vesicle labels.
Materials and Reagents
- Pasteur pipette 9 in, cotton-plugged (Fisher Scientific, catalog number: 13-678-8B )
- Pasteur pipette 5.75 in (Fisher Scientific, catalog number: 13-678-20A )
- 24-well plates (Corning, Costar®, catalog number: 3524 )
- Kimwipes (KCWW, Kimberly-Clark, catalog number: 34155 )
- Round 12 mm-Ø glass coverslips #0 (Thermo Fisher Scientific, special order, 0.085 mm thick)
- Aluminum foil (WebstaurantStore Food Service Equipment and Supply Company, Choice, catalog number: 12224X1HD )
- Platinum wires (Alfa Aesar, catalog number: 10286 )
- 24 x 40 coverslips (Fisher Scientific, special order), all 0.085 mm thick (i.e., size 0)
- Parafilm PM-996 (Bemis, catalog number: PM996 )
- 30-mm Ø Petri dish (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 153066 )
- 15 ml conical tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339650 )
- 0.2 µm filter (Corning, catalog number: 431218 )
- 50 ml conical tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339652 )
- 1.7 ml tubes (Corning, Axygen®, catalog number: MCT-175-C )
- Neonatal rat hippocampal neurons (P0-P3)
- Trypsin-EDTA (trypsin: 0.025%/EDTA: 0.01%) (Thermo Fisher Scientific, GibcoTM, catalog number: R001100 )
- Minimum Essential Medium (MEM, 1x) (Thermo Fisher Scientific, GibcoTM, catalog number: 51200038 )
- Glucose (Thermo Fisher Scientific, GibcoTM, catalog number: 15023021 )
- Sodium bicarbonate (NaHCO3) (Sigma-Aldrich, catalog number: S5761 )
- Transferrin (Sigma-Aldrich, catalog number: T1428 )
- L-glutamine (Thermo Fisher Scientific, catalog number: 25030081 )
- Insulin (Sigma-Aldrich, catalog number: I5500 )
- Fetal bovine serum (Thermo Fisher Scientific, GibcoTM, catalog number: 26140079 )
- Matrigel (Corning, catalog number: 354234 )
- 70% EtOH (Decon Labs, catalog number: 2401 )
- Ara-C (Cytarabine) (Sigma-Aldrich, catalog number: C1768 )
- B27 supplement (Thermo Fisher Scientific, GibcoTM, catalog number: 17504044 )
- Vacuum grease (Dow Corning, catalog number: 1597418 )
- FM1-43 (Biotium, catalog number: 70022 )
- 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX) (Abcam, catalog number: ab120046 )
- D-(-)-2-Amino-5-phosphonopentanoic acid (D-AP5) (Abcam, catalog number: ab120003 )
- N-2-hydroxyethyl piperazine-n-2 ethanesulphonic acid (HEPES) (Sigma-Aldrich, catalog number: H4034 )
- DNase (Sigma-Aldrich, catalog number: D5025 )
- 10 mM HCl (diluted with ddi water 1:100 from 1 N solution) (Sigma-Aldrich, catalog number: H9892 )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: S641-212 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P5405 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (Fisher Scientific, catalog number: BP214 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Acros Organics, catalog number: 207780010 )
- Matrigel stock solution (see Recipes)
- Hanks solution (see Recipes)
- Hanks + 20 (H + 20) (see Recipes)
- Dissociation solution (see Recipes)
- DNase solution (see Recipes)
- 2 mM Ara-C stock (see Recipes)
- Plating medium (see Recipes)
- Ara-C solution (see Recipes)
- Extracellular bath Tyrode’s saline (see Recipes)
- 90 K (High Potassium Tyrode’s) (see Recipes)
Equipment
- Bunsen burner (Sigma-Aldrich, catalog number: Z270326 )
- 250 ml beaker (Fisher Scientific, Fisherbrand, catalog number: FB100250 )
- Pyrex beaker (Corning, PYREX®, catalog number: 1000-100 )
- Inverted microscope (20x lens, Mercury arc lamp, filters for EGFP) (Nikon Instruments, model: Eclipse Ti-E )
- Electric field stimulation chamber (two platinum wires glued to the sides of RC-26G perfusion chamber) (Warner Instruments, model: RC-26G )
- PH-1 platform (Warner Instruments, model: PH-1 )
- Six Channel Perfusion Control Valve System (Warner Instruments, model: VC-6 )
- Inline heater (e.g., Warner Instruments, catalog number: 64-0104 )
- TC-344B Dual Channel Temperature Controller (Warner Instruments, model: TC-344B )
- Prior Lumen 200 Illuminator (Prior Scientific, model: Lumen 200 )
- Cell culture hood, CO2 incubator
- Set of dissection tools (Fine Science Tools)
- Stimulus Isolator SD9 (Grass Instruments, model: SD9 )
- Digidata 1440A (Molecular Devices, model: Digidata 1440A )
- Dissection microscope (Nikon Instruments)
- Centrifuge (Eppendorf, model: 5702 R )
- Computer with time-lapse imaging system
- Computer with Clampex software
- Andor iXon Ultra EMCCD (Andor)
- Vibration Isolation table (Newport)
Software
- Micro-Manager (Schneider et al., 2012) or Fiji (Schindelin et al., 2012)
- Clampex (Molecular Devices)
- Microsoft Excel (Microsoft)
Procedure
- Primary culture of rat hippocampal neurons
Note: Preparation of neonatal neuronal culture is technically easier than embryonic culture.
Primary cultures of dissociated postnatal rat hippocampal cells are prepared as previously described (Liu and Tsien, 1995). Briefly, rat hippocampi (CA1-CA3) are normally dissected from P0 to P1 Sprague-Dawley rats and dissociated into a single-cell suspension with a 10-min incubation in trypsin-EDTA (trypsin: 0.025%/EDTA: 0.01%, Thermo Fisher Scientific) followed by gentle trituration using three glass pipettes of decreasing diameters (~1 mm, 0.5 mm, and 0.2 mm), sequentially (Figure 1). Pipettes were obtained by fire-polishing the tips on a Bunsen burner. Soft borosilicate glass melts and shrinks easily if exposed to fire. For the best results, pipette tips are positioned vertically to the flame and are being twisted clockwise and counterclockwise for even heating and melting. Longer flaming makes the smaller diameter tips. It is also important to make sure that the smallest tips are not fully sealed, which sometimes happens. Ideally, the smallest diameter pipette lets cell suspension through slowly and ensures single cell dissociation.
Figure 1. Example of fire-polished Pasteur pipette tips of different diameters used for dissociation of hippocampi- Dissociated cells are recovered by centrifugation (200 x g, 5 min) at 4 °C and re-suspended in plating medium composed of Minimal Essential Medium (MEM, Thermo Fisher Scientific) with (in mM) 27 glucose, 2.4 NaHCO3, 0.00125 transferrin (Sigma-Aldrich), 2 L-glutamine (Thermo Fisher Scientific), 0.0043 insulin (Sigma-Aldrich) and 10%/vol fetal bovine serum (FBS, Gibco).
- 100 μl of cell suspension is seeded onto round 12 mm-Ø glass coverslips (200-300 cells/mm2) pre-coated with Matrigel (Corning) placed in 24-well plates (Fisher Scientific).
Note: Use of Matrigel as a neuronal substrate vs. commonly used poly-L-lysine substantially improves cell culture quality. 100 μl of Matrigel is dispensed onto the surface of coverslip 1-24 h before use and is aspirated before the cell suspension is plated. Also, no antibiotics should be used in order to avoid unwanted effects on neuronal properties. - Dissection tools and glassware used for cell culture are being cleaned with no detergents in order to prevent their detrimental impact on neuronal membranes. After use, tools are washed with deionized water; blood remains are removed with Kimwipes. For sterilization before the dissection, tools are incubated with 70% EtOH in a 250 ml beaker (Fisherbrand) for 30 min. To protect fine dissection tools from mechanical damage due to bumping to the bottom of the beaker–we cover the bottom with 2-3 Kimwipes. The coverslips we use are 12 mm #0 (Thermo Fisher Scientific, special order, 0.085 mm thick). Coverslips are autoclaved in a Pyrex beaker covered with 3 layers of aluminum foil for 30 min before use.
Note: No any other special treatment of coverslips is needed. - Cells are allowed to adhere to the substrate for 30-60 min before the addition of 1 ml plating medium. After 1-2 days in culture, an additional 1 ml medium containing (in mM) 27 glucose, 2.4 NaHCO3, 0.00125 transferrin, 0.5 L-glutamine, 0.002 Ara-C, 1%/vol B27 supplement (Thermo Fisher Scientific) and 5%/vol FBS is added. Ara-C (1 µM) in the culture media efficiently prevents astroglial proliferation.
- Experiments are performed between DIV 12 and 18 (when synaptic transmission is well established).
- Dissociated cells are recovered by centrifugation (200 x g, 5 min) at 4 °C and re-suspended in plating medium composed of Minimal Essential Medium (MEM, Thermo Fisher Scientific) with (in mM) 27 glucose, 2.4 NaHCO3, 0.00125 transferrin (Sigma-Aldrich), 2 L-glutamine (Thermo Fisher Scientific), 0.0043 insulin (Sigma-Aldrich) and 10%/vol fetal bovine serum (FBS, Gibco).
- Imaging setup
- For cell culture on coverslips, an inverted microscope is preferred due to the ability to use an oil-immersion objective with high N.A. high magnification. An XYZ motorized stage is preferred but not necessary.
- An RC-26G chamber (Warner Instruments) is modified with 2 platinum wires attached to the sides of the chamber for delivering electric field stimulation. The chamber is bottom-sealed with a 24 x 40 mm size 0 cover glass (0.085 mm thick) using vacuum grease and clamped on a PH-1 platform (Warner Instruments) placed on a microscope stage (Scientifica). We use a Nikon Eclipse Ti inverted microscope with a 20x Plan Apo VC objective (N.A. 0.75) for large areas or a 100x Apo VC objective (N.A. 1.40) for a detailed view of synaptic boutons.
Note: The summative thickness of two #0 coverslips (one 24 x 40 coverslip, Fisher Scientific special order) which seals the bottom of the chamber and one 12 mm circular coverslip with cells) is equal to the thickness of one #1.5 standard coverslip (0.17 mm), for which most of the objective lenses are optimized and corrected. This eliminates the need to seal coverslips with cells directly to the bottom of the imaging chamber with vacuum grease. - Solution exchange is achieved via gravity perfusion controlled by a VC-6 valve control system and a 6-channel manifold (Warner Instruments) with a constant rate of ~50 μl/sec which allows a complete exchange of the bath solution in the recording chamber within 30 sec.
- All experiments are performed at room temperature, but physiological temperature can also easily be achieved by using an inline heater (e.g., 64-0104, Warner Instruments) and a heated stage (e.g., PH-1 stage, Warner Instruments). Both can be set to 34-37 °C (e.g., controlled through TC-344B Dual Channel Temperature Controller, Warner Instruments). To avoid heating induced stage drift, preheating for at least 1 h is recommended. Autofocus such as Nikon Perfect Focus is also helpful.
- An RC-26G with attached stimulation wires is clamped in PH-1 and a home-made water-tight insert for the Scientifica stage.
Note: It is advisable to have a leak-proof system for live imaging to minimize possible damage to the microscope. PH-1 is bolted to the stage insert. Laboratory Parafilm PM-996 is used as a seal. - Excitation light sources can be an arc lamp or a laser launched either through a liquid light guide or a laser launcher box. We use a Prior Lumen 200 Illuminator connected to the microscope via a liquid light guide.
- Fluorescence excitation/dichroic/emission filter combination corresponding to selected FM dyes can be found online (e.g., http://www.chroma.com). Filters are either installed in the filter cubes inside the microscope housing, or separately within the light path. For example, FM1-43 imaging is done using a fluorescence filter set: Ex. 460/50; DIC: 495LP; Em: 510/25BP. All optical filters and dichroic mirrors are purchased from Chroma or Semrock.
- Image acquisition and synchronized perfusion are controlled via Micro-Manager and Clampex software. The acquisition settings including excitation power, fluorescence filter set (excitation, dichroic and emission filters), exposure time, camera gain and frame rate are all kept the same among different samples. Images are taken at 0.1 Hz rate. Baseline fluorescence is captured during 1 min before electrical/chemical stimulation/depolarization of neurons or for at least 3 frames.
- Imaging with Andor EMCCD. 1) Set Andor temperature to -80 °C; 2) Set Andor readout mode to 5 MHz (instead of 17 MHz); 3) Set Andor vertical speed to 0.5, instead of 3.3: these settings help to increase the dynamic range for imaging fluorescence intensity.
- For cell culture on coverslips, an inverted microscope is preferred due to the ability to use an oil-immersion objective with high N.A. high magnification. An XYZ motorized stage is preferred but not necessary.
- Imaging procedure
- Synaptically mature primary rat hippocampal neurons (DIV 12-18) are incubated with 10 µM FM1-43 (i.e., SynaptoGreen C4, Biotium) for 0.5 h at 37 °C in a 5% CO2 incubator to load FM1-43 into synaptic vesicles through spontaneous exo-/endocytosis. For dye loading, 10 μl sterile 1 mM stock solution in ddiH2O mixed thoroughly with 990 μl of the conditioned plating medium from the neuronal culture will be added to and dispensed into a new well of the 24-well plate. Then, a coverslip with neurons is transferred to the well containing 10 μM FM1-43 by means of burner sterilized forceps.
- A more common loading procedure is acute loading by stimulation. For example, FM1-43 dye is loaded in the presence of high K+ saline, which promotes massive evoked exo-/endocytosis.
- For that, in a laminal flow culture hood, use burner sterilized forceps to transfer one coverslip of primary rat hippocampal culture from the 24-well plates to a 30-mm Ø Petri dish containing 3 ml of normal Tyrode’s solution. Bring the coverslips to the microscope. To prevent dye loss due to spontaneous neuronal activity, Glutamate receptor blockers (10 μM NBQX and 20 μM D-AP5) are added at least 1 min before imaging and are present in the bath throughout the whole imaging experiment.
- Transfer the coverslips to the imaging chamber preloaded with normal Tyrode’s solution with perfusion speed of 0.05 ml/sec.
- Stop perfusion by closing both inlet and outlet (i.e., vacuum), and remove ~90-95% solution from the chamber.
- Dropwise add 500 μl high K+ solution containing 10 µM FM1-43 (i.e., SynaptoGreen C4, Biotium) to the imaging chamber and mix it gently.
- Incubate the cells for 2 min with the dye and turn on the perfusion for 5-min washout of surface FM dyes with normal Tyrode’s solution. Healthy looking neurons with moderate FM1-43 labeling are identified at 20x magnification (as in Figure 2).

Figure 2. Sample zoom-out images which show optimal cell density in DIC and overlay with FM1-43 fluorescence- To induce synaptic vesicle release, either electric field stimulation or high K+ perfusion is used. The rate of evoked FM1-43 loss from presynaptic terminals is measured.
- The neuronal culture is exposed to electrical field stimulation at 10 Hz with 1 msec 70 V pulses generated by Grass SD9 stimulus isolator for 2 min or high-potassium (90 K) 1 min perfusions.
- Stimulation control is interfaced through Clampex software (Molecular Devices). An example of Clampex protocol that controls timing of electric field stimulation and switch between perfusion channels is shown in Figure 3.
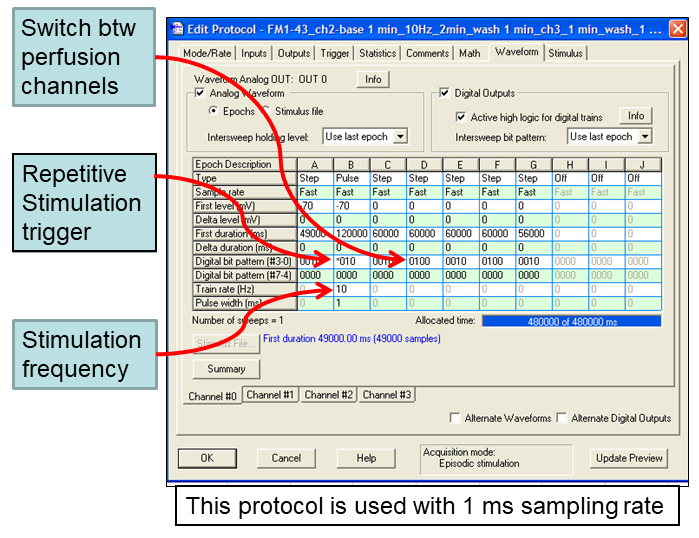
Figure 3. Example of Clampex protocol that can be used to control timing of electric field stimulation and to switch between solution perfusion channels. Sampling rate is set at 1 msec. - SD9 is triggered with a 5 V TTL pulse from #4 digital output port on 1440A digidata (Molecular Devices). SD9 settings are shown in Figure 4. 70 V output pulses are delivered from positive and negative ports on SD9 to the platinum wires (Alfa Aesar 0.5 mm dia), super-glued to the sides of the RC-26G chamber. Stimulation conditions were optimized in whole-cell patch clamp recordings on current-clamped neurons and are sufficient to reliably evoke action potentials without obvious detrimental effects. In the example Clampex protocol (Figure 2), electric field stimulation is encoded as a 10 Hz, pulse train (Epoch B), which is 120,000 msec (120 sec = 2 min) long. Repetitive triggering of the stimulator through digital output #4 is encoded by the star (*) in digital bit pattern.

Figure 4. Grass instruments SD9 stimulus isolator settings. Frequency–0, Delay–0, duration–10 x 0.1, Volts–7 x 10, output–mono, polarity–normal. - Digital control of perfusion is achieved through a VC-6 valve controller. Individual perfusion channels are set to external trigger mode, and open or close in response to TTL pulse sent from the Digidata 1440A digital output.
- The Andor iXon Ultra EMCCD camera’s External Input/Output ‘Fire’ port is connected to the Start port on the Digidata 1440A, which allows it to trigger Clampex protocols via the Digitizer Start Input immediately when the imaging acquisition begins. The schematic diagram of controller/devices interconnections is depicted in Figure 5.
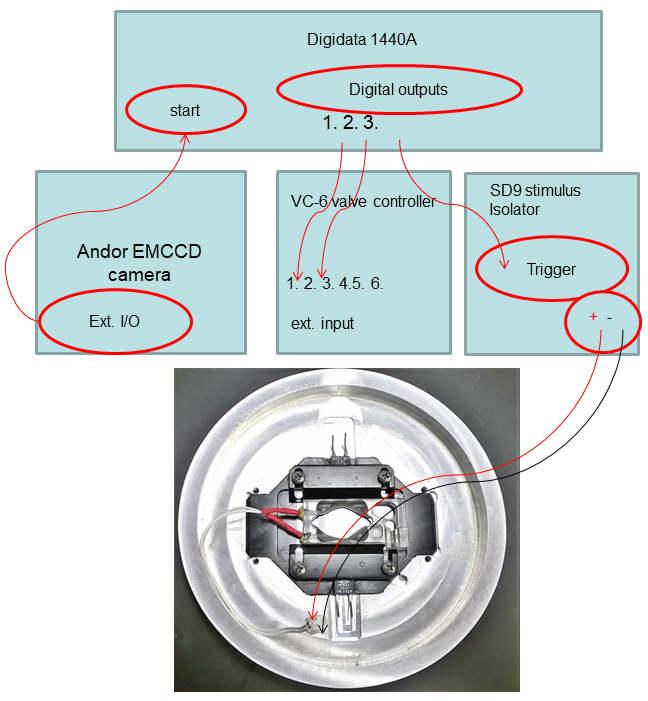
Figure 5. Diagram of controller/devices interconnections - Orientation of coverslips on the microscope is shown in Figure 6.
- Baseline fluorescence is captured for 1 min (or at least 3 frames) before electrical/chemical stimulation/depolarization of neurons.
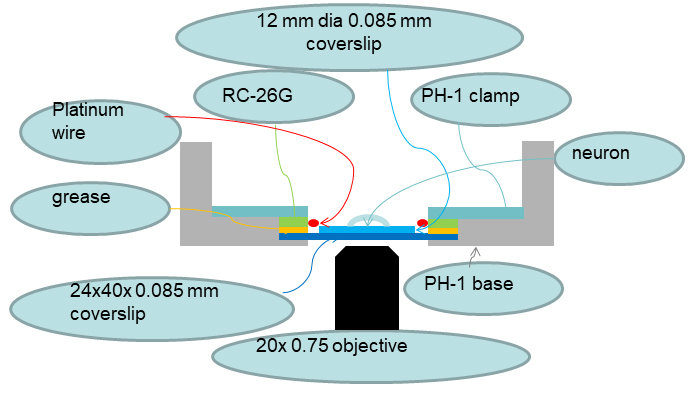
Figure 6. Diagram of coverslips’ orientation on the microscope
- Synaptically mature primary rat hippocampal neurons (DIV 12-18) are incubated with 10 µM FM1-43 (i.e., SynaptoGreen C4, Biotium) for 0.5 h at 37 °C in a 5% CO2 incubator to load FM1-43 into synaptic vesicles through spontaneous exo-/endocytosis. For dye loading, 10 μl sterile 1 mM stock solution in ddiH2O mixed thoroughly with 990 μl of the conditioned plating medium from the neuronal culture will be added to and dispensed into a new well of the 24-well plate. Then, a coverslip with neurons is transferred to the well containing 10 μM FM1-43 by means of burner sterilized forceps.
- Imaging analysis
Imaging analysis in ImageJ/Fiji:- Open ImageJ or Fiji (not Micro-Manager).
- File → Import image sequence.
- Image → Duplicate first (not a stack) and use it for ROI selection.
- If want to select/save/analyze a portion of image, e.g., area in focus: select it, right click, duplicate.
- If you want to smoothen the image: Process → Filters → Mean (e.g., average 4-5 pixel values).
- Process → FFT → Bandpass Filter, large structures–down to 40 px, small–down to 3, Suppress stripes: None, 5% tolerance of direction, Autoscale after filtering, Saturate image.
- Having duplicated the active image, open Image → Adjust → Threshold, Dark background-B&W.
- Set lower threshold level, click set, apply: the program assigns values in a range from 0 to 255 for the 8-bit image.
- Analyze → Analyze Particles: size 2-200 pixels (good range to cover synaptic structures). An example of threshold-defined and size-restricted ROIs is shown in Figure 7.

Figure 7. Example of the threshold based selection of FM1-43 labeled ROIs. Image on the right depicts outlined ROIs determined based on the threshold and particle size. - Image → Adjust → Brightness/Contrast: drag brightness to Max to be able to find background spots with almost no signal.
- Select 4 ROIs for background: One by one select background regions and click Add on the ROI manager.
- On the ROI manager, go to More → Save ROIs and save in the root folder, not in the stack.
- Go to original image stack, open ROI manager, uncheck, check back, show all.
- ROI manager, select More → Multi measure to get the intensity values for each ROI through the whole stack of images. Copy values to Excel.
Notes:- To correct for lateral drifting in a stack of images, have your stack open with the 1st frame active, to which you want to align subsequent frames, then go to: Plugins → Registration → StackReg → Translation. After correction, the new stack will have blank areas along the edges where drifting happened. To exclude those areas from analysis–just select the area you want to keep, right-click, duplicate and save the stack as a new image sequence in a new folder.
- StackReg is built into Fiji (if the Fiji installation does not have it, go to Help → Update, click ‘Manage update sites’, add http://sites.imagej.net/BIG-EPFL/ and it will be automatically installed at next update), but can be added to ImageJ as well. It requires TurboReg.
http://bigwww.epfl.ch/thevenaz/turboreg/
http://bigwww.epfl.ch/thevenaz/stackreg/ - MultiStackReg (available from http://bradbusse.net/downloads.html, requires StackReg and TurboReg) can be used to save the transformations and apply them to another stack. This is extremely useful if another channel that has a lower signal is imaged simultaneously and is difficult to automatically correct.
- To correct for lateral drifting in a stack of images, have your stack open with the 1st frame active, to which you want to align subsequent frames, then go to: Plugins → Registration → StackReg → Translation. After correction, the new stack will have blank areas along the edges where drifting happened. To exclude those areas from analysis–just select the area you want to keep, right-click, duplicate and save the stack as a new image sequence in a new folder.
Data analysis in Excel:- Insert 1 column at the beginning of the data set to be able to calculate average background values (from the last 4 columns that represent background ROIs-regions of interest).
Note: Columns of data contain fluorescence intensity values of individual ROIs over frame number, plotted in rows. Frame numbers can easily be translated into time points from the imaging frequency if need be. If the frequency is 0.1 Hz, interval between frames is 10 sec. - Make a copy of the entire spreadsheet and subtract averaged background values (in column 1) from individual intensity values.
- Make a second copy of the spreadsheet with background values subtracted. Insert 2 rows on top of the data set. Calculate average values of the first 3 rows (baseline, Fmax) and last 3 rows (after 2-nd bout in 90 K, Fmin).
- Copy the spreadsheet one more time and normalize data as (F - Fmin)/(Fmax - Fmin) for individual ROI. Visual representation of Excel analysis can be found in Figure 8.
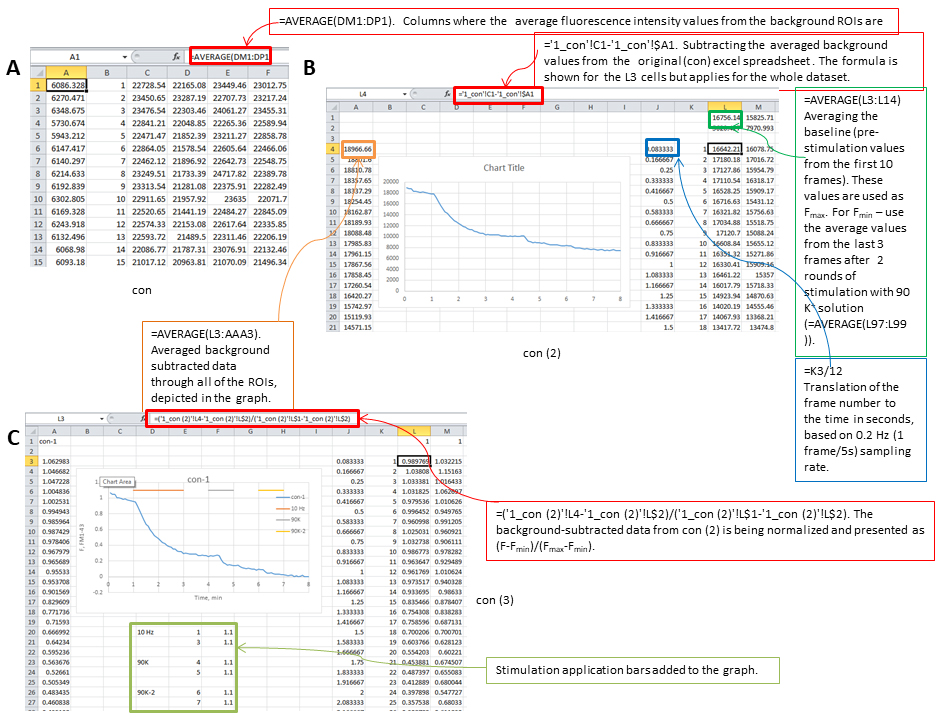
Figure 8. Screenshots from Excel analysis. A. Background averaging column is added to the dataset, then a copy of the spreadsheet is made (con (2)). B. In the con (2) spreadsheet background is subtracted, frame number is translated to time, Fmax and Fmin values are calculated, graph panel is added for visual control of the FM1-43 fluorescence changes, then the spreadsheet is copied one more time (con (3)). C. In con (3) spreadsheet the data from con (2) is being normalized and presented as (F - Fmin)/(Fmax - Fmin) and application bars are being added to the graph. - Calculate average values across multiple ROIs over time. An example of the resultant graph is shown in Figure 9. Sample images at baseline, end of electric field stimulation and end of second bout in 90 K+ are shown in Figure 10.
Note: Different Fields of View (FOVs) contain different numbers of threshold-detected ROIs. To eliminate over-influence of FOVs with high ROI number on the averaged data, a set of randomly selected ROIs which is equal or smaller than the number of ROIs of the least populated FOV can be used for averaging. To randomize ROIs in excel–insert a new row on top of the ROI data set. In this new row, select cells, aligned to the columns with ROIs. Type in =rand(), then press Ctrl+Enter. Having the row of randomized values selected, go to Menu-Data-Sort-Expand the selection-Options-Sort left to right. Sort data by row1 (randomized values).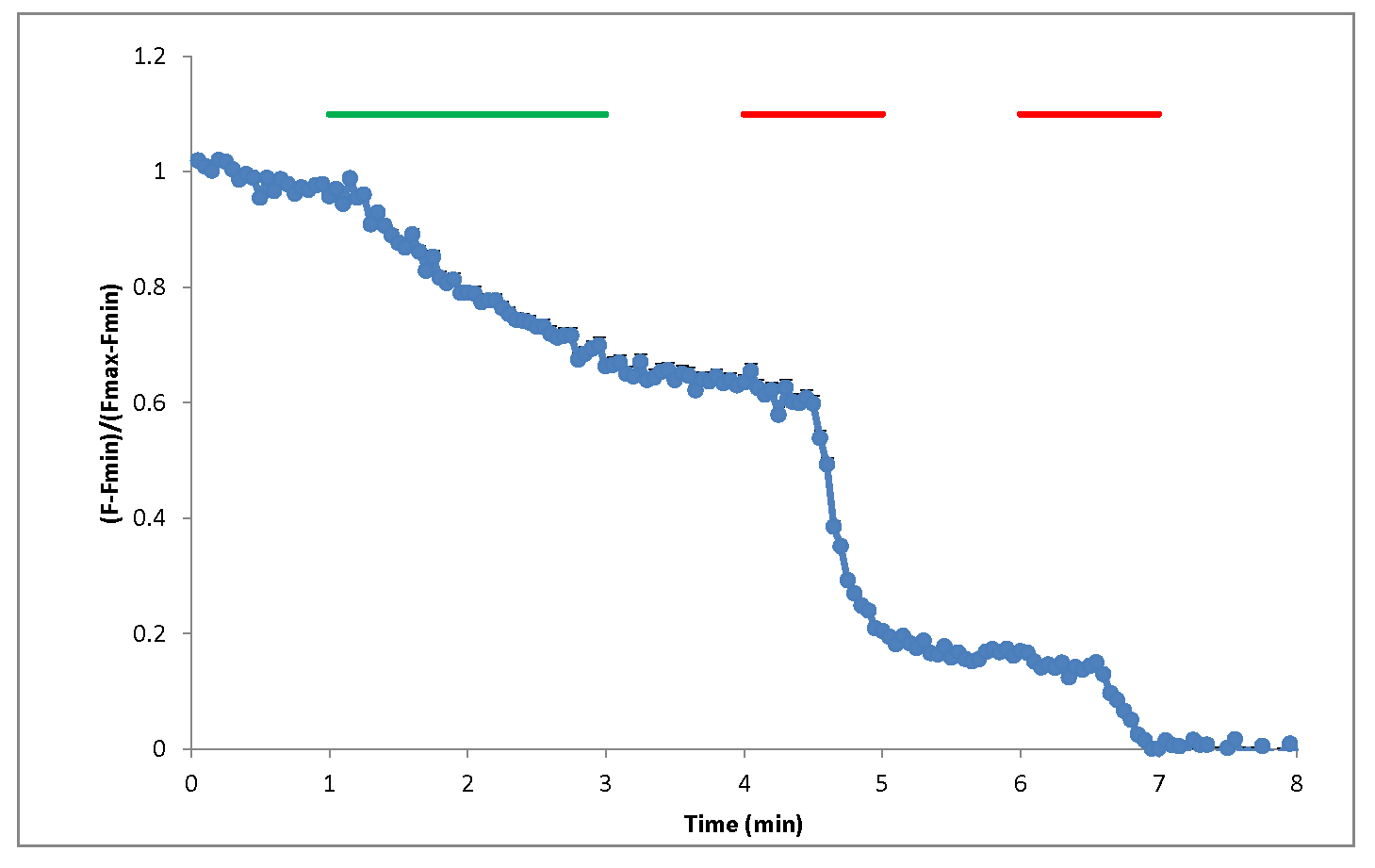
Figure 9. Example of normalized FM1-43 fluorescence in response to 2 min 10 Hz electric field stimulation (green bar) or 1 min high potassium (90 K) chemical stimulation (red)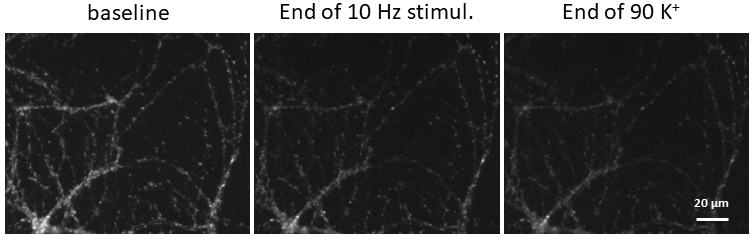
Figure 10. Example images at baseline, end of electric field stimulation and end of second bout in 90 K+
- Open ImageJ or Fiji (not Micro-Manager).
Data analysis
Initial image analysis is done in ImageJ. Regions of interest (ROIs) are selected by using the same fluorescence intensity threshold across different samples. Average intensity from every ROI and average background intensities from four cell-free regions in every image stack are exported to Excel. The FM1-43 signal in every ROI is calculated as (F - Fmin)/(Fmax - Fmin), in which Fmax is the average of the first 3 frames at a baseline before stimulation, and Fmin is the average of the last 3 frames after multiple bouts with 90 K stimulation. All fluorescence intensity values are background subtracted.
To determine the minimum number of ROIs for FM1-43 destaining, a power analysis is performed using G*Power (Faul et al., 2007). An effect size of 25% is estimated with the error probability set to 0.05, power to 0.95 and an expected standard deviation of 40% is chosen based on FM destaining experiments performed in the lab. A sample size of 53 is needed to achieve significance with a two-tailed Student’s t-test. All image processing is performed in ImageJ. All experiments are performed in two to three different batches of cell cultures. All values presented are mean ± SEM. For calculating statistical significance, the Student’s t-test is used for 2-group comparison, and one-way analysis of variance (ANOVA) followed by the Tukey-Kramer method as post-hoc analysis is used for comparing three or more groups.
Notes
- Reproducibility and variability
In our tests, we observed a moderate variability in FM loading and unloading in untreated control. This variation mostly originates from the variations of cell culture such as days in vitro and cell density. Therefore, reducing such variables can significantly improve experimental consistency. In addition, we notice that unhealthy neurons often have excessive FM staining, which we have used as a criterion to exclude damaged synapses. - The comparison between FM and other synaptic vesicle labels
Synaptic vesicles are so essential in neurobiology that many methodologies such as electrophysiology, amperometry, electron microscopy and fluorescence imaging have been developed over the past fifty years to study them. Fluorescence imaging of fluorophores that selectively target synaptic vesicles has the advantage of directness, ability to dynamically monitor synaptic events in real time with high throughput and high spatial resolution (Kavalali and Jorgensen, 2014). FM dyes are the most popular fluorescent labels (Gaffield and Betz, 2007). Other probes include fluorescently tagged antibodies recognizing vesicle luminal epitopes, vesicular proteins whose luminal domain is tagged with pH-sensitive fluorescent protein (Syn-pHFP) (Afuwape and Kavalali, 2016) and quantum dots (Qdots) with surface affinity to vesicular membranes (Zhang et al., 2009) or conjugated to antibodies recognizing vesicle luminal epitopes (Park et al., 2012). Below, we will make a concise comparison among those fluorescent labels in terms of operational difference.- Targeting difference
FMs are styryl dyes that randomly insert into cell membranes. With stimulation-induced vesicle release and compensatory retrieval, FM dyes are believed to end up mostly in recycled synaptic vesicles, partially in endosomes and lysosomes, and a little in Golgi and endoplasmic reticulum (ER). Qdots with vesicular membrane affinity and the vesicle-targeting antibodies conjugated to fluorophores including Qdots have similar targeting outcomes with a lower possibility of being targeted to Golgi and ER. Shortening loading time is the most effective way to reduce non-synaptic vesicle targeting. Differently, Syn-pHFPs are transgenically expressed and distributed to all cell membranes the original proteins reside in, which includes vesicles and the cell surface. Generally, it is believed that Syn-pHFPs label all synaptic vesicles, releasable and non-releasable. - Signal difference
Since FM dyes are prone to washout when surfaced, the signal reporting vesicle release is a decrease of their fluorescence. In contrast, Syn-pHFPs report exocytosis as an increase of fluorescence because exocytosis causes deacidification of the vesicular lumen, and their fluorescence decrease reports endocytosis and vesicle re-acidification. - Normalization difference
Different synaptic boutons contain different numbers of vesicles; therefore, normalization is important to obtain a measurement of population vesicle behavior. For FM dyes and other externally added probes, the most common and simplest way to normalize the response is to define the fluorescence intensity prior to stimulation as 100% with the intensity after exhaustive stimulation as 0. For genetically expressed Syn-pHFP, the most common normalization is to use a high concentration of NH4Cl solution to de-acidify all cytoplasmic compartments to completely unquench Syn-pHFP and achieve the maximum 100% fluorescence intensity. To determine minimal Syn-pHFP intensity (0%), cells are normally perfused with low pH-solution (e.g., pH 5.5 Tyrode’s). Clearly, both pH manipulations have a possible impact on synaptic vesicle behavior. Notably, our recent paper demonstrated that even a moderate concentration of NH4Cl (i.e., 5 mM) can change synaptic vesicle release and retrieval over an extended period of time (Lazarenko et al., 2017).
- Targeting difference
Recipes
- Matrigel stock solution
- Make Matrigel stock solution
Thaw 10 ml bottle on ice in a cold room for 24-48 h, then aliquot in 1 ml aliquots and freeze. If you try to thaw it at a higher temperature, it will turn into sludge - Make working Matrigel (i.e., what is called Matrigel in the protocol)
- Thaw 1 ml aliquot on ice for several hours and mix with 49 ml MEM
- Matrigel is thick and viscous, so you will probably need to flush the 1 ml aliquot with MEM to get the whole thing out
- Shake the MEM + Matrigel to mix and let it sit overnight in a cold room to dissolve completely before using
- Aliquot it into four 15 ml conical tubes with 12.5 ml in each
- Cover all tubes with aluminum foil to protect from light
- Do not use an aliquot older than one month, and do not filter Matrigel
- Make Matrigel stock solution
- Hanks solution
Hanks solution is directly made from Sigma-Aldrich H2837 powdered HBSS, or directly ordered from Invitrogen
0.2 µm filter sterilize and aliquot in 50 ml tubes - Hanks + 20 (H + 20)
400 ml 1x Hanks + 100 ml fetal bovine serum (20% FBS)
0.2 µm filter sterilized and aliquoted in 50 ml tubes - Dissociation solution (500 ml)
Hank’s salt solution +12 mM MgSO4·6H2O - DNase solution
Dissolve entire 375 kU in 5 ml sterilized Milli-Q water and aliquot in 40 µl aliquots - 2 mM Ara-C stock (500 μl aliquots)
- 9.7 mg Ara-C (usually stored in refrigerator or cold room) to 20 ml of deionized water in a 50-ml conical tube
- Vortex well
- Filter (0.2 μm) into 50 ml conical tube
- Aliquot into 1.7 ml tubes, 500 μl each
- Store at -20 °C
- 9.7 mg Ara-C (usually stored in refrigerator or cold room) to 20 ml of deionized water in a 50-ml conical tube
- Plating medium
500 ml MEM
2.5 g glucose
100 mg NaHCO3
50 mg transferrin
Mix well with stir bar on lab bench
Note: You can also make 1 L solution and divide it for plating and Ara-C.
50 ml FBS (~10%)
5 ml 0.2 M L-glutamine
1 ml insulin stock (12.5 mg/ml insulin in 10 mM HCl)
0.2 µm filter sterilize and aliquot in 50 ml tubes in culture hood - Ara-C solution
500 ml MEM
2.5 g glucose
100 mg NaHCO3
50 mg transferrin
Mix well with stir bar on bench
1.25 ml of 0.2 M L-glutamine
10 ml B27 supplement
500 µl Ara-C stock (2 mM, in Milli-Q water), for the final concentration 1 µM
25 ml FBS (5%)
0.2 µm filter sterilize and aliquot in 50 ml tubes - Extracellular bath Tyrode’s saline
150 mM NaCl
4 mM KCl
2 mM MgCl2
2 mM CaCl2
10 mM N-2 hydroxyethyl piperazine-n-2 ethanesulfphonic acid (HEPES)
10 mM glucose
pH 7.35 - 90 K (High Potassium Tyrode’s)
64 mM NaCl
90 mM KCl
2 mM MgCl2
2 mM CaCl2
10 mM N-2 hydroxyethyl piperazine-n-2 ethanesulphonic acid (HEPES)
10 mM glucose
pH 7.35
Acknowledgments
The methods were adapted from (Lazarenko et al., 2017). Techniques were also adapted from all of the references cited. This work was supported by NIH Grant OD008761, NS094738 and DA025143 to Q.Z. We have no conflicts of interest or competing interests to declare.
References
- Afuwape, O. A. and Kavalali, E. T. (2016). Imaging synaptic vesicle exocytosis-endocytosis with pH-sensitive fluorescent proteins. Methods Mol Biol 1474: 187-200.
- Faul, F., Erdfelder, E., Lang, A. G. and Buchner, A. (2007). G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39(2): 175-191.
- Gaffield, M. A. and Betz, W. J. (2006). Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc 1(6): 2916-2921.
- Kavalali, E. T. and Jorgensen, E. M. (2014). Visualizing presynaptic function. Nat Neurosci 17(1): 10-16.
- Lazarenko, R. M., DelBove, C. E., Strothman, C. E. and Zhang, Q. (2017). Ammonium chloride alters neuronal excitability and synaptic vesicle release. Sci Rep 7(1): 5061.
- Liu, G. and Tsien, R. W. (1995). Synaptic transmission at single visualized hippocampal boutons. Neuropharmacology 34(11): 1407-1421.
- Park, H., Li, Y. and Tsien, R. W. (2012). Influence of synaptic vesicle position on release probability and exocytotic fusion mode. Science 335(6074): 1362-1366.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Schneider, C. A., Rasband, W. S. and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9(7): 671-675.
- Zhang, Q., Li, Y. and Tsien, R. W. (2009). The dynamic control of kiss-and-run and vesicular reuse probed with single nanoparticles. Science 323(5920): 1448-1453.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lazarenko, R. M., DelBove, C. E. and Zhang, Q. (2018). Fluorescent Measurement of Synaptic Activity Using FM Dyes in Dissociated Hippocampal Cultured Neurons. Bio-protocol 8(2): e2690. DOI: 10.21769/BioProtoc.2690.
Category
Neuroscience > Cellular mechanisms > Synaptic physiology
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.