- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Electron Tomography to Study the Three-dimensional Structure of Plasmodesmata in Plant Tissues–from High Pressure Freezing Preparation to Ultrathin Section Collection

Published: Vol 8, Iss 1, Jan 5, 2018 DOI: 10.21769/BioProtoc.2681 Views: 10248
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Plasmodesmata (PD) are nanometric (~20 nm wide) membrane lined pores encased in the cell walls of the adjacent plant cells. They allow the cells to exchange all types of molecules ranging from nutrients like sugar, hormones, to RNAs and various proteins. Unfortunately, they are also hijacked by phyto-viruses, enabling them to spread from cell-to-cell and then systematically throughout the whole plant. Their central position in plant biology makes it crucial to understand their physiology and especially link their function to their structure. Over the past 50 years, electron microscopists have observed them and attempted to ultrastructurally characterize them. They laid the foundation of what is known about these pores (Tilney et al., 1991; Ding et al., 1992; Oparka and Roberts, 2001; Nicolas et al., 2017a).
Despite the explosion of three-dimensional electron microscopy (3D-EM), PD ultrastructure remained recalcitrant to such technique. The first technical difficulty is to process them in such a way where they are as close to their native state as possible. Secondly, plant samples reveal themselves as being difficult to process due to the poor staining/fixating reagents penetration rates, their increased size, their high water content and the presence of an acidic vacuole. On top of this, their very unique position in the cell wall and their nanometric size make them difficult to conveniently stain in order to see the inner-workings of these pores.
Here we describe in detail the protocol used in Nicolas et al. (2017b) to image PD in fine detail and produce high-resolution tomograms.
Background
High Pressure Freezing (HPF) relies on the vitrification of the water present in the sample. By cooling down the sample at a high enough freezing rate (104-105 °C/sec), its contained water molecules cannot reorganize in a crystal-fashion and remain vitrified in an amorphous state (see Dubochet [2007] for further reading on the physics behind water crystallization). The sample is then said to be ‘cryoimmobilized’ or ‘vitrified’. This is generally achieved by the use of liquid nitrogen (-195 °C) or liquid ethane (-188 °C). At ambient pressure, this phenomenon can only be achieved on a few microns (< 5 microns), however by raising the pressure to approximately 2,000 bars (~2,000 atmospheres), this depth can reach at least 200 microns (up to 500 microns in certain conditions). This allows the vitrification of thick biological tissues without any osmotic artefacts.
Classically, HPF is followed by Freeze Substitution (FS). This step comprises staining, dehydration and resin embedding of the sample. The good success of the FS is critical for the final sub-cellular preservation state of the sample and will be dependent upon the staining agents used, their concentration and also the temperature kinetic followed during the different stages of FS. These parameters are key for adequate fixation and cross-linking of cellular components, the proper staining (not too little nor too much) and embedding, and heavily impact the final resolution reached. Although this protocol is a good starting point, depending on the samples (tissues, developmental stage, plant species) and organelles of interest, it will need to be adapted to the reader’s specific task.
Despite published FS protocols custom tailored for plant samples have yielded spectacular results over the past decades (Donohoe et al., 2006; Kang et al., 2011), in our hands, it was not suitable for the study of PD ultrastructure for two reasons: i) The epoxy resin used in such protocols is highly electron-scattering and generates a bad signal to noise ratio when imaged with a 120 kV electron microscope, subsequently preventing us from clearly imaging the internal details of the nanometric pores (Figure 1A). ii) The elements inside the pores are so tightly packed in such a small space (< 10 nm) that heavy staining activity during FS prevents clear imaging of these elements (Figure 1A).
In this context, we opted for HPF followed by a modified FS course to prepare our samples (Figure 1C and Table 1). This led to the production of high resolution tomograms, enabling the appreciation of PD ultrastructure in all dimensions of space. We hope our readers find this protocol useful and will eventually improve it for better visualization of nanometric membrane details in thick plant samples.
Table 1. Comparative cryofixation procedures between Donohoe et al. (2006) and Nicolas et al. (2017b). Left table recapitulates dehydration + fixation steps and right table the resin-embedding step. Cells marked in blue represent temperatures below 0 °C and cells marked in orange represent temperatures above 0 °C.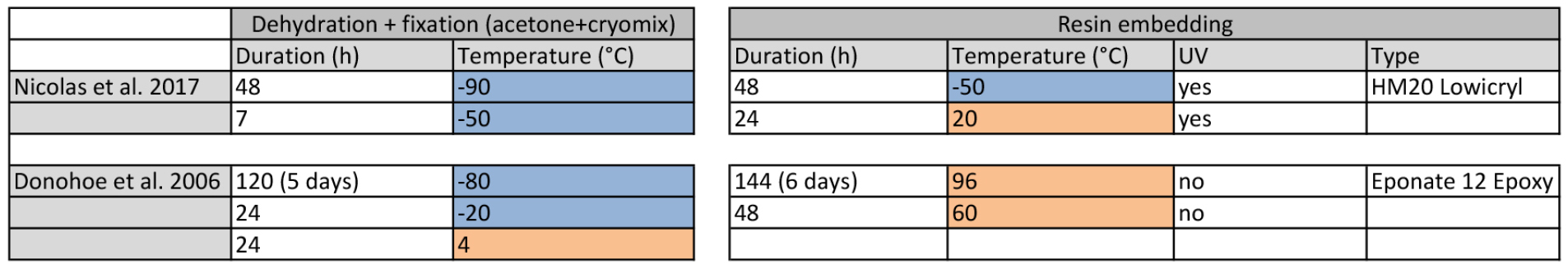
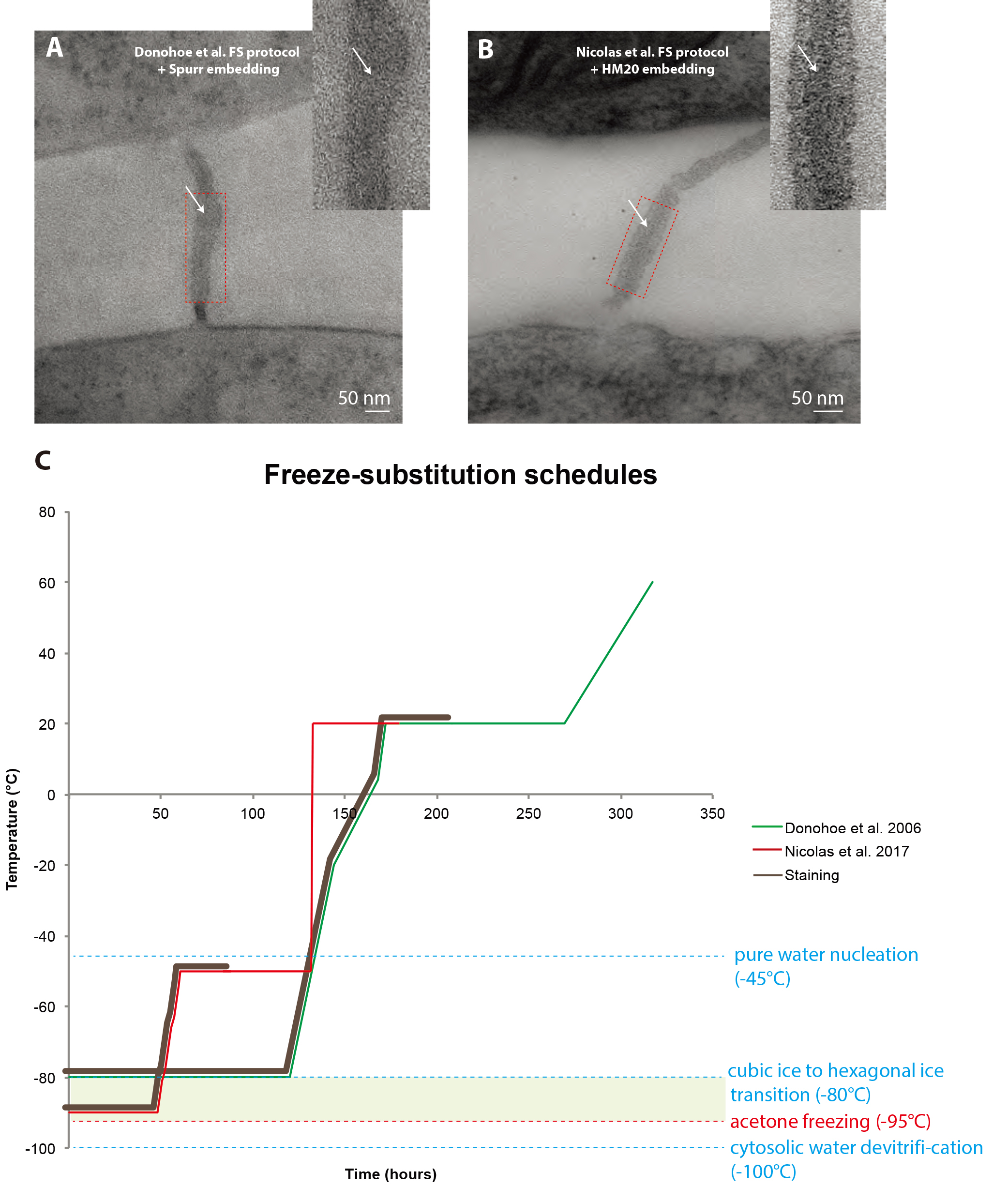
Figure 1. Comparison between freeze-substitution protocols. A. Root tip plasmodesma micrograph acquired from a 90 nm thick section prepared with the regular cryosubstitution protocol (Donohoe et al., 2006). The central element (white arrows) is barely visible, the membrane bilayers are not as lightly stained as in (B) and the overall signal-to-noise ratio is low. B. 2D micrograph of a plasmodesma situated in the root tip acquired at a 0° tilt from a 180 nm thick section prepared with our improved cryosubstitution protocol. Despite the thickness of the section, details in the vicinity of the pore are discernible, notably the central element (white arrows). Dense material can be seen in the cytoplasmic sleeve space. Insets show a close-up view of the red-boxed region ns. C. This temperature versus time curve depicts the commonly used freeze substitution schedule and the one we developed. Brown sections of the curve correspond to when the sample is submerged in the cryosubstitution cocktail containing the highly reactive staining agents. Green zone corresponds to the ideal area for the frozen hydrated samples, below which hexagonal ice cannot form and acetone won’t freeze at atmospheric pressure (between -95 °C and -80 °C). In Donohoe et al., 2006 (green curve) the samples remain in the cocktail for a prolonged amount of time, until the temperature reaches 20 °C. In our protocol (red curve, Nicolas et al., 2017b) the cocktail is removed from the sample very early on in the process when the temperature is at -50 °C.
Materials and Reagents
- Plant material
- Square plastic culture plates for Arabidopsis seedlings vertical culture (VWR, catalog number: 391-0444 )
- MS medium + vitamins (Duchefa Biochemie, catalog number: M0222.0050 ) for seedlings and cell cultures
- Sucrose (Sigma-Aldrich, catalog number: 84100 )
- 2-(N-morpholino)ethanesulfonic acid (MES) (Euromedex, catalog number: EU0033-A )
- Naphthaleneacetic acid (NAA) stock solution (10 mg/ml aliquoted at -20 °C) (Sigma-Aldrich, catalog number: N064025G )
- Kinetin hormone stock solution (1 mg/ml aliquoted at -20 °C) (Sigma-Aldrich, catalog number: K3253 )
- 1 N KOH (for pH)
- MES (Euromedex)
- Plant agar (Duchefa Biochemie, catalog number: P1001.1000 )
- Culture mediums for cells and seedlings (see Recipe 1)
- Murashige and Skoog medium for liquid cultured cells
- Murashige and Skoog medium for seedlings
- Murashige and Skoog medium for liquid cultured cells
- Square plastic culture plates for Arabidopsis seedlings vertical culture (VWR, catalog number: 391-0444 )
- High pressure freezing (Figure 2A)
- Aclar sheet, 51 µm thick (Electron Microscopy Sciences, catalog number: 50426 )
- Cryotubes (VWR, catalog number: 479-1207 )
- Eppendorf 1.5 ml tubes (SARSTEDT, catalog number: 72.708 )
- 15 ml Falcon tube (SARSTEDT, catalog number: 62.553.542 )
- Tooth picks
- BSA fraction V (Sigma-Aldrich, old catalog number: 85040C, now catalog number: 05482 )
- Liquid MS medium
- MethylCycloHexane (MCH) (Merck Schuchardt OHG 85662 Hohenbrunn, Germany)
- BSA solution for cryoprotection during HPF (see Recipe 2)

Figure 2. High Pressure Freezing procedure. A. 1. Insulated tweezers for manipulation in liquid nitrogen bath. 2. Leica loading system (see Video 1 for loading procedure) with its associated platelet holder rod. 3. 200 µm deep, 1.5 mm diameter, copper platelets. 4. 100 µm deep, 1.5 mm membrane carriers, copper membrane carriers. 5. Sample transfer metal containers. 6. Pod holder. 7. Pods. 8. Harris puncher for production of the 1.5 mm diameter aclar disks to be optionally laid at the bottom of the 200 µm deep platelets to facilitate the later separation of the sample. 9. Torque screwdriver for tightening the membrane carrier in the pods (#3, 4 and 5 respectively). B. View of the EMPACT1 touchscreen displaying the status of the machine. (10) is the valve controlling liquid nitrogen flux from the main tank (11) to the liquid nitrogen bath (12). C. View of the display after processing a sample. Curve dropping down is temperature and curve rising up is pressure.
- Aclar sheet, 51 µm thick (Electron Microscopy Sciences, catalog number: 50426 )
- Freeze substitution (Figure 3)
- Screw top 2 ml tubes (SARDTEDT, catalog number: 72.693 )
- Disposable regular pipets 1.5, 2 and 3 ml and FS specific 1 ml with thin tips (Ratiolab, catalog number: 2600155 )
- Wheaton glass sample vials with snap-cap for cryomix preparation (DWK Life Sciences, WHEATON, catalog number: 225536 .)
- Personal cartridge half mask 6100 (Honeywell International, catalog number: 1029471 )
- Plastic coffins or pills Leica molds (AFS2 consumable, Leica, catalog numbers: 16707155 and 16707157 respectively)
- Plastic solvent containers with screw tops (Leica, catalog number: 16707158 )
- Tinfoil
- Uranyl acetate powder (Merck, catalog number: 8473 )
- Pure methanol
- Glutaraldehyde 10% EM-grade anhydrous solutions in acetone (Electron Microscopy Sciences, catalog number: 16530 )
- Ultra pure 100% ethanol and acetone (VWR, catalog numbers: 83813.440 and 20066.558 , respectively)
- Osmium tetroxide vials 0.1 g (Electron Microscopy Sciences, catalog number: 19134 )
- Nail polish (color does not matter)
- Cryosubstitution Uranyl-acetate stock solution (20%) (see Recipe 3)
- Cryosubstitution mix (see Recipe 4)

Figure 3. Freeze substitution procedure. 1. AFS tube holders. The one on the right being made according to Heinz Schwarzes blueprints (heinz.schwarz@tuebingen.mpg.de). 2. Metal cups for holding cryomix tubes. 3. Metal socket for mould containers for resin polymerization. 4. Screwdriver used to transfer tube holders 1 and 2 and sample transfer containers shown in Figure 1 (#5). 5. EMS micro-needle and Ted Pella Inc. ultra-micro-needle tools. 6. Ted Pella Inc. micro-tool adapter. 7. Hand-made tool holder. 8. Leica plastic ‘pill’ mould. 9. Leica plastic ‘coffin’ mould. 10. Plastic solvent container with screw top. 11. Leica plastic mould container.
- Screw top 2 ml tubes (SARDTEDT, catalog number: 72.693 )
- Resin embedding
- HM20 resin (Electron Microscopy Sciences, catalog number: 14345 )
- HM20% solutions (see Recipe 5)
- HM20 resin (Electron Microscopy Sciences, catalog number: 14345 )
- Ultramicrotomy
- Crystalizing dish (Fisher Scientific, catalog number: 11766582 )
Manufacturer: DWK Life Sciences, DURAN, catalog number: 213133408 . - Fast absorbent paper filters (Whatman, Filter papers 41) (GE Healthcare, catalog number: 1441-070 )
- Glass wand (Fisher Scientific, catalog number: 12441627 )
Manufacturer: MBL, catalog number: SRF380 . - Grids with different meshes (200 L/inch and 300 L/inch Delta Microscopy) and slot grids (EMS Formvar Carbon Film 2 x 0.5 mm copper grids lot, Electron Microscopy Sciences, catalog number: EMS200-Cu )
- Paraffin films (Bemis, Parafilm ‘M’)
- Pasteur pipet (VWR, catalog number: 612-1720 )
- 0.2 µm filter
- Syringe
- 0.5 ml Eppendorf tubes
- 5 nm colloidal gold solution (BBI solution, catalog number: EM-GC5 , www.bbisolutions.com)
- Solid parlodion (Electron Microscopy Sciences, catalog number: 19220 )
- Isoamyl-acetate (Sigma-Aldrich, catalog number: W205532 )
- Toluidine blue powder (Sigma-Aldrich, catalog number: T3260 )
- Sodium borate (Sigma-Aldrich, catalog number: B3545 )
- Ultrapure (MilliQ) water
- 2% Parlodion solution for grid filming (see Recipe 6)
- Toluidine blue solution (see Recipe 7) for screening block sections on table top microscope during resin block milling
- Fiducial marker solution (see Recipe 8)
- Crystalizing dish (Fisher Scientific, catalog number: 11766582 )
Equipment
- Plant material
- 200 ml glass flasks for liquid cell culture (Dutscher, catalog number: 232522 )
- Flask shaker to keep liquid cultured cells moving (Digital Shaker, Southwest Science)
- Green room for in vitro cultivation of seedlings and liquid cultured cells (see Recipe 1 for growth conditions)
- 200 ml glass flasks for liquid cell culture (Dutscher, catalog number: 232522 )
- High pressure freezing (Figure 2A)
- Liquid nitrogen, liquid nitrogen container and adapted personal protection gear
- Air compressor (JUN-AIR)
- Leica EMPACT1 machine (Leica Microsystems)
- Leica loading system
- Pod holders
- Pods
- Puncher for aclar disks (Harri’s Micro Punch 1.20 mm)
- Regular biomolecular pipets from 2 µl to 1,000 µl range
- 200 ml flask
- Binocular for root dissection (Nikon, model: SMZ-10A )
- Heating surface for quick drying of Pods and pod holders during the session
- Insulated tweezers for manipulation in N2 (VOMM Germany, 22 SA ESD)
- Little metal scooper for sampling liquid cultured cells
- Membrane carriers (100 µm or 200 µm deep) (Leica Microsystems, catalog numbers: 16707898 and 16706898 respectively)
- Metal containers for frozen sample transfer and associated screw driver for lifting procedures (provided with EMPACT1)
- Torque screwdriver (TOHNICHI, Torque driver RTD60CN) set to 30Ncm
- Liquid nitrogen, liquid nitrogen container and adapted personal protection gear
- Freeze Substitution and resin embedding (Figure 3)
- Leica EM Freeze Substitution Processor (FSP) equipped with UV light (Leica Microsystems)
- Leica Automatic Freeze Substitution 2 (EM AFS2) machine (Leica Microsystems, model: Leica EM AFS2 )
- Hand made tool holders (out of paper clips) for better disposal of the various tools in AFS2 well
- AFS tube holders Metal cups
- Metal socket for mould containers
- Microtools for separating the membrane carriers from frozen samples and disposing samples in the moulds correctly (micro-needle #1 0.025 mm, Electron Microscopy Sciences, catalog number: 62091-01 and tungsten 5 µm ultra-micro needle, Ted Pella, catalog number: 13625 )
- Ventilated hood
- Leica EM Freeze Substitution Processor (FSP) equipped with UV light (Leica Microsystems)
- Ultramicrotomy (Figure 4)
- Petri dishes of various sizes (Dutscher, catalog numbers: 068515 , 068516 , 068517 )
- Various EM grade precision tweezers (EMS style 2, 5, 5X, style 7 https://www.emsdiasum.com/ https://www.dumonttweezers.com)
- Block holders
- Carbon coater (optional) (SPI-MODULE Carbon Coater)
- Diamond knives (DIATOME Trim20 dry, Ultra wet and HISTO wet)
- ELMO Glow discharger (optional) (CORDOUAN Technologies)
- Glass knife maker (LKB BROMMA, model: 7800 Knifemaker )
- Glass knives for first milling steps to reach the sample
- Grid carriers for grid storage
- Leica Ultracut UC7 (Leica Microsystems, model: Leica Ultracut UC7 )
- Razor blades for coarse block preparation
- Table top microscope (Olympus, model: CX-41 )

Figure 4. Ultramicrotomy procedure. 1. Sharp razor blades. 2. Tweezers (shapes n°3, 7, 5 and 5X, EMS). 3. Grid holders. 4. Block holders (sold with ultramicrotome, Leica Microsystems). 5. Diamond knives Histo, ultra 45° for section collection and trim 20 dry for precise trimming (from left to right). 6. Glass knife for coarse trimming. 7. Cat whisker mounted on a wooden stick for section gathering during on-grid collection step. Circular inset shows an enlarged view of the cat whisker mounted on the stick using nail polish.
- Petri dishes of various sizes (Dutscher, catalog numbers: 068515 , 068516 , 068517 )
Software
- Tecnai imaging and analysis (TIA) software (https://www.fei.com/software/)
TEM User Interface was used in conjunction with Tecnai Imaging and Analysis (TIA) software to control and acquire micrographies with the EM (https://www.fei.com/software/) - Xplore3D (FEI) was used for automated tilt series acquisitions (https://www.fei.com/software/)
- ImageJ (https://imagej.nih.gov/ij/download.html) with the Input/Output plugin (http://www.cmib.fr/en/download/softwares/input-output.html) were used to readout the .mrc tilt series files and tomograms
- IMOD suite (http://bio3d.colorado.edu/imod/)
Note: All processing was done using the IMOD suite (Kremer et al., 1996) (http://bio3d.colorado.edu/imod/), from the alignment of the raw tilt series to tomogram reconstruction, segmentation and data analysis.
Procedure
Note: This procedure involves the manipulation of dangerous compounds, namely liquid nitrogen, acetone, osmium tetroxide, glutaraldehyde, uranyl-acetate, Lowicryl resin. If you are not familiar with how to handle such compounds, please refer to the official guidelines and our Notes 1 and 2.
- High pressure freezing
Steps A1 through A13 should be carried out as fast as possible to limit deterioration of sampled live tissue. In our case, this period is less than a minute. A thorough description on how to use the EMPACT1 is provided in user manual and also in the proof of concept paper (Studer et al., 2001).- Turn on Leica EMPACT and air compressor hooked up to it.
- Fill up the chamber with MCH and evacuate air in the circuit by gently pushing on the vertical rod.
- Fill in liquid nitrogen tank up to ¾ (keep liquid nitrogen source handy for eventual mid-way top-ups during session).
- Switch on the EMPACT1 and wait until working pressure reaches between 4.8 and 5 bars.
- On the touchscreen tap on the corresponding valve to allow filling of the bath with liquid nitrogen (Figure 2B).
- Load an empty pod and tap on prepare → lock → start to shoot in order to clear out all the air out of the system.
- Charge membrane carrier on the Leica loading system (Figure 2A, #2 and Video 1).
 Video 1. Loading the biological sample in the HPF pods. (1) 200 µm deep membrane carriers (Leica Microsystems); (2) Vertically grown Arabidopsis seedlings; (3) Becker filled liquid MS medium; (4) 20% BSA diluted in liquid MS medium; (5) 0.5-2 µl and 20-200 µl range pipets (Sartorius); (6) Torque screwdriver set to 30Ncm (TOHNICHI, Torque driver RTD60CN); (7) Disposable razor blades; (8) Style 7 EMS tweezers; (9) Disposable tooth picks; (10) Leica loading system (with rod), pods, pod holder.
Video 1. Loading the biological sample in the HPF pods. (1) 200 µm deep membrane carriers (Leica Microsystems); (2) Vertically grown Arabidopsis seedlings; (3) Becker filled liquid MS medium; (4) 20% BSA diluted in liquid MS medium; (5) 0.5-2 µl and 20-200 µl range pipets (Sartorius); (6) Torque screwdriver set to 30Ncm (TOHNICHI, Torque driver RTD60CN); (7) Disposable razor blades; (8) Style 7 EMS tweezers; (9) Disposable tooth picks; (10) Leica loading system (with rod), pods, pod holder. - If using the 200 µm membrane carriers with a hole on the bottom, wedge an aclar disk generated with the puncher. Disks can be generated in batches the day before cryofixation, incubated in 95% ethanol and let to evaporate.
- With a 0.5-2 µl pipet, fill the carrier with 20% BSA (Recipe 2) in MS (Murashige and Skoog medium). It must form a clean dome in the socket without bleeding on the edges and with no air bubbles (Video 1).
- Handling of biological material: i) Roots are quickly dissected under binocular in MS medium on a glass slide with a sharp razor blade and then carefully picked up with a toothpick. ii) Cells are aliquoted in a 1.5 ml Eppendorf tube from the main 200 ml flask and are let to sediment down for a few minutes. Then, the cells are picked up by scratching from the green cell pellet visible at the bottom with a toothpick or a little metal spoon shaped scooper, Cells are then carefully placed in a membrane carrier. Every 2 to 3 HPF trials (~3 min time frame when two experimenters are implicated), replace the cells in the Eppendorf tube by fresh cells.
- Dispose the sample in the BSA bubble in the membrane carrier. It is best when the sample is completely submerged by the BSA (Video 1).
- Push the membrane carrier in the pod positioned in its special socket on the Leica loading system (Video 1).
- Screw the sapphire tight on the carrier using the TORX screwdriver (set on 2.5 cN m-1) (Video 1).
- Screw the white pod holder (Video 1).
- Load it in the Leica EMPACT machine and the loading stage and push the piston towards the left (Video 2). Video 2. Loading the pod containing the sample in the HPF machine
- On the touch screen tap on prepare → lock→ start. The system automatically flushes out the pod holder, making the pod loaded with the sample fall in a liquid N2 bath.
At this stage the sample must never come out of the liquid N2, risking sudden devitrification and the waste of the sample (Video 2). - Check the freezing rate and pressure rise on the touchscreen. It should look like in Figure 2C, where maximum pressure is reached ~20 msec before reaching final temperature.
- Place pod holder in the specially designed socket bathing in the liquid nitrogen (Video 2).
- Unscrew the pod holder (Video 2).
- Carefully release the clamping from the sapphire by unscrewing and then gently tug out the membrane carrier by tapping the pod against the bottom of the bath. Use insulated tweezers to manipulate the frozen membrane carrier (Video 2).
- Place the membrane carrier in the sample carrier dedicated to this usage and keep in liquid N2 (Video 2).
- When all samples are ready to be transferred for subsequent steps, place metal lids on the metal containers and quickly transfer the containers from the EMPACT bath to an intermediate liquid nitrogen container. Then transfer to AFS2 machine, already running, filled up with liquid N2 and pre-cooled to -90 °C.
- Turn on Leica EMPACT and air compressor hooked up to it.
- Freeze substitution
During the freeze substitution, it is important to always maintain the frozen hydrated samples in the cold, below -80 °C to avoid too much hexagonal ice formation (Dubochet, 2007). Therefore, never pick up the tubes out of the well and always use instruments, containers and FS solutions that have been precooled in the well for a good 15-20 min.
Because Steps B7 to B9 are tedious, we would recommend at the beginning to devote a full day to it.- Under a vented hood, dispatch cryomix (see Recipe 4) immediately after having added osmium into the screw top tubes (numbered with a Sharpie and wrapped in scotch tape to avoid washing out of the ink) and then dip the tubes in liquid nitrogen to freeze the cryomix (less volatile when frozen).
- Insert frozen cryomix containing tubes in the AFS well, inside the metal cups.
- While cryomix is still solid, carefully dispatch the frozen sample containing membrane carriers inside the cryomix tubes (maximum of 2 carriers/tube).
- During the thawing process of the cryomix from the -195 °C to -90 °C transition, the carriers will sink in the mix and freeze-substitution will start.
- Set the AFS as following:
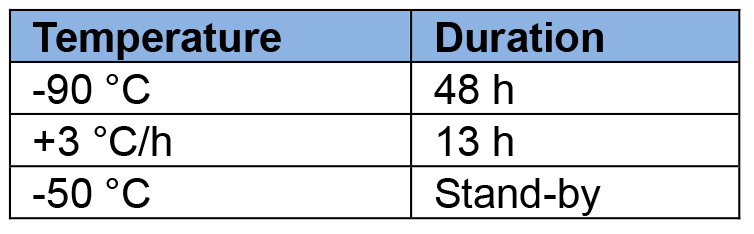
- (OPTIONAL but highly encouraged) At day +1 after the start of the FS, open the cryomix tubes and redispose the carriers at the bottom in such a way that the carriers do not lay on top of each other in order to maximise the exchanges between the mix and the samples. Also, a gentle shake can be given to the tubes.
- After completion of the -90 °C to -50 °C transition, the machine is on hold at -50 °C.
- Wash thoroughly every tube 3 times with ultra-pure acetone and then 3 times with ultrapure ethanol (Note 3).
- During the last wash, transfer the samples into a plastic mould container containing 100% ethanol.
- Carefully separate the samples from the membrane carriers in a plastic mould container using the microtools (Figure 3, #5 and #6). Use them to scrape the rim of the carriers to loosen the frozen BSA. If using 200 µm deep carriers (Leica Microsystems), the hole at the bottom can be used to push out the frozen sample by fitting the ultra-micro needle tool (Figure 3, #5, right one).
- Prior to the Transfer of the detached sample in the moulds with a 3 ml pipette, fill the moulds with cold ultrapure ethanol and let them cool down to -50 °C (Note 4).
- Under a vented hood, dispatch cryomix (see Recipe 4) immediately after having added osmium into the screw top tubes (numbered with a Sharpie and wrapped in scotch tape to avoid washing out of the ink) and then dip the tubes in liquid nitrogen to freeze the cryomix (less volatile when frozen).
- Resin embedding
- When all samples are in place in the adequate moulds, gently pipet out the excess ethanol from the moulds (see Note 3 for pipetting technique) and replace with 25% HM20 (see Recipe 5).
- The following incubation kinetic is just a guideline. Adapt according to your work days:
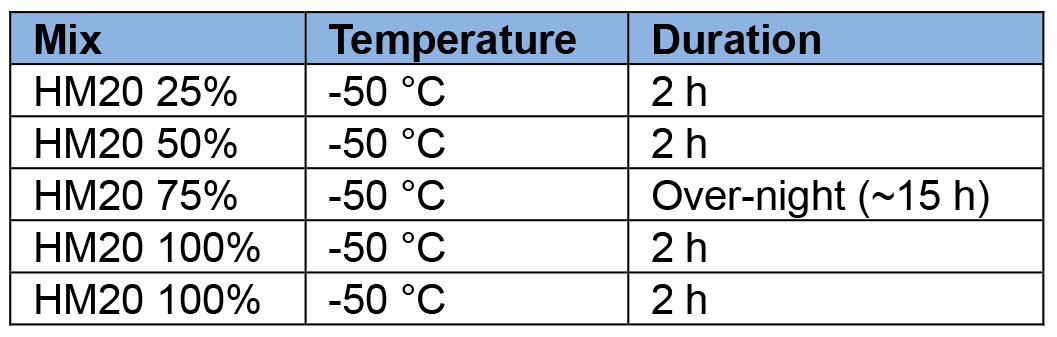
- On the last 100% HM20 bath, place the mould containers in the metal piece (Figure 3, #3) that has been cooled down to -50 °C before hand.
- Mount the FSP module and set the AFS as following:
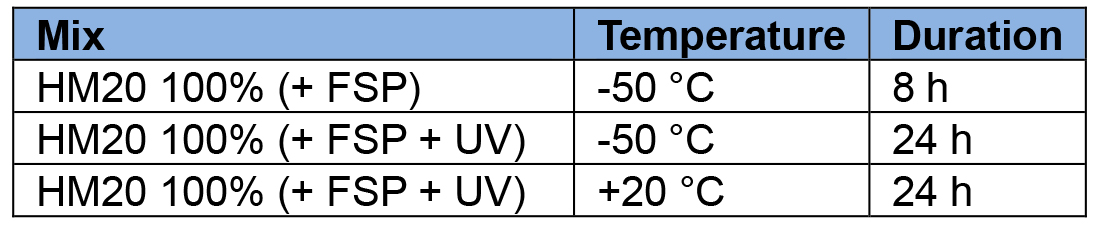
- Take out hardened resin blocks from their casts by either slicing the casts carefully with razor blades or, if using the pill-shape moulds, the blocks can be punched out using a hexagonal screwdriver placed on the bottom (Video 3). Video 3. Puncturing out the pill-shaped blocks. It requires a Wiha 353/SW 3.0x75 screwdriver for puncturing.
- When all samples are in place in the adequate moulds, gently pipet out the excess ethanol from the moulds (see Note 3 for pipetting technique) and replace with 25% HM20 (see Recipe 5).
- Ultramicrotomy
- Grid filming
Because we embed our samples in HM20 Lowicryl, a more fragile resin than EPON, the sections need to be deposited on grids that have been coated with parlodion or formvar.
Formvar-coated slot grids were purchased and useful because no grid bars can potentially block the imaging in high tilts. However, they are more fragile and very sensitive to shifts induced by resin retraction. For parlodion-filmed mesh grids, they are home-made as follows (Video 4):- Fill a crystalizing dish, placed in a large Petri dish, with distilled water to the top.
- Rack the top of the crystalizing dish with a glass wand to flatten out the surface of the water.
- Pipet the parlodion solution with a Pasteur pipet and place the pipet vertically over the crystalizing dish. Let a drop fall on the surface of the water.
- Let the parlodion spread at the surface of the water.
- With clamps, remove this first film (used to eliminate potential dust and particles on the water surface).
- Repeat Steps D1b and D1c.
- Carefully place the grids on the floating parlodion film (opaque side face down on the film). Use the mesh desired (200 mesh/µm2 is optimal for better overviews and tilt series acquisition).
- With tweezers, gently place a Whatman type 41 absorbent paper (or equivalent absorption speed) on the floating grids and let it soak.
- Carefully remove excess parlodion film on the sides of the absorbent paper, either by tearing it away or folding it on top of the floating paper. This is to prevent excess film to fold onto the grids, resulting in a double layer of film on the filmed grids.
- To avoid rippling and/or sliding of the film, firmly grip the absorbent paper by the sides with 2 tweezers type 5X or like (which close automatically) and transfer the absorbent paper by flipping it over into a glass/plastic Petri dish.
- Let the Petri dish open for a day under ventilated hood, in order to let the filmed grids dry out.
Video 4. Grid filming procedure. 1- Glass Pasteur pipet and its pump; 2- Petri dish; 3- Glass wand; 4- Large Petri dish lid; 5- Whatman filter paper type 41; 6- Crystallizer; 7- EM grids. 8- 2% parlodion solution; 9- Type 5X tweezers. - Fill a crystalizing dish, placed in a large Petri dish, with distilled water to the top.
- Section collection
- Since the aim of this protocol is more focused on how to process the samples as this is the critical factor for the final images, we will not go into details on how to properly use an ultramicrotome, prepare the block-face for cutting etc. Instead please refer to Hagler (2007). The following is some comments for properly performing these steps.
- For the study of PD, 90 nm or 180 nm thick sections were collected with the Leica Ultracut UC7 (Leica Microsystems).
- Producing serial sections can be useful for two main reasons: i) with one grid, more objects of interest, ii) serial tomography can be performed where the same structure is reconstructed on serial sections, making it possible to recover 3D volumes of structures a lot bigger than the section thickness (Kang et al., 2011).
- To do so, the block face needs to be reasonably small enough so that multiple sections can be placed on a single grid. Carving the block face in a trapeze shape will allow the sections to stick together as they are generated, creating a contiguous ribbon of serial sections that can be easily deposited on a single grid simultaneously.
- Although it is usually not a problem when using non-carbonated parlodion-filmed copper grids, if sections are repelled by the grids due to static charge during the collection step, grids can be glow-discharged prior to section collection.
- Depending on the EM voltage (more voltage equals more kinetic energy of the electrons, and therefore a better ability to image thick samples well) and the size of the structures of interest, thickness of the sections can vary from 90 nm to 180 nm. Finer sections, closer to 90 nm in thickness will yield slightly better x, y resolution, especially in medium range EM voltages as in our case. 180 nm and greater will allow better volume recovery at the expense of x, y resolution when imaged at 120 kV. In the case of PD study, simple ones have a diameter range from 20 to 40 nm thick, so 90 nm thick sections are appropriate. However, it has to be noted that PD can be of various lengths and sizes, especially when it comes to branched complex PD. Therefore they can extend to several hundreds of nm across the cell wall in all directions. In this case, increasing the section thickness grants better chances to fully recover the volume of the pores.
- Since the aim of this protocol is more focused on how to process the samples as this is the critical factor for the final images, we will not go into details on how to properly use an ultramicrotome, prepare the block-face for cutting etc. Instead please refer to Hagler (2007). The following is some comments for properly performing these steps.
- Coating grids with fiducial markers (gold particles)
Gold particles are used for the subsequent alignment steps required prior to tomogram reconstruction. Because pure colloidal gold tends to aggregate due to its inherent negative charge, it is often used diluted with BSA, making the spread of the gold more homogenous across the section.- On a sheet of paraffin film, lay drops of gold solution (see Recipe 8) and water (Figure 5).
- Gently lay the grids on the gold drops for 20 sec approximately.
- (Optional) Gently pipet up and down the mixture to favor good contact between the grid and the gold.
- Sequentially lay the grids for a few seconds on two water drops to remove excess gold solution.
- Absorb the remaining liquid by approaching absorbent paper (Whatman paper Grade 5) on the side of the grid.
- Repeat Steps D3a to D3e on the other side of the grid in order to have gold particles on both sides.
- Let the grids dry prior to introduction in the EM (damages the ionic pump system otherwise).

Figure 5. Laying the fiducial markers on the grids. Typical setting for fiducial marker deposition on grid prior to tomography. Grids are first laid on top of the red drops (fiducial marker + BSA solution) for 15-20 sec then dragged consecutively in the water drops and carefully dried by the rim of the grid with absorbent paper. This process needs to be repeated on both sides of the grids.
- On a sheet of paraffin film, lay drops of gold solution (see Recipe 8) and water (Figure 5).
- Grid filming
Data analysis
- With this high-pressure freezing–freeze substitution protocol, morphological study of nanometric details inside the cells is rendered possible. Combined with the use of Lowicryl HM-20 resin, more fragile but more electro-lucent and able to polymerize at low temperature (with UV light), this protocol allows efficient tomographic reconstruction of details around 10 nm in dimension. Low magnification images show well-preserved clumps of cells (Figure 6A). At the interfaces between these cells, PD are seen bridging the cells (Figure 6B). Tilt series acquisition of a typical type I pore (Video 5) renders tomograms where the whole volume of the pore can be appreciated (Figures 6C-6E and Video 6). Video 5. Tilt series of type I PD. Full range tilt series (-65° to +65°) of the type I plasmodesma (in the centre) found in 4 days old cultured cells, processed with this protocol and shown in Figure 5. Dense black spots are the fiducial markers used for alignment with Etomo. Video 6. Tomogram of type I PD. Reconstructed tomogram and its manual segmentation of the type I plasmodesma showed in Figure 5.
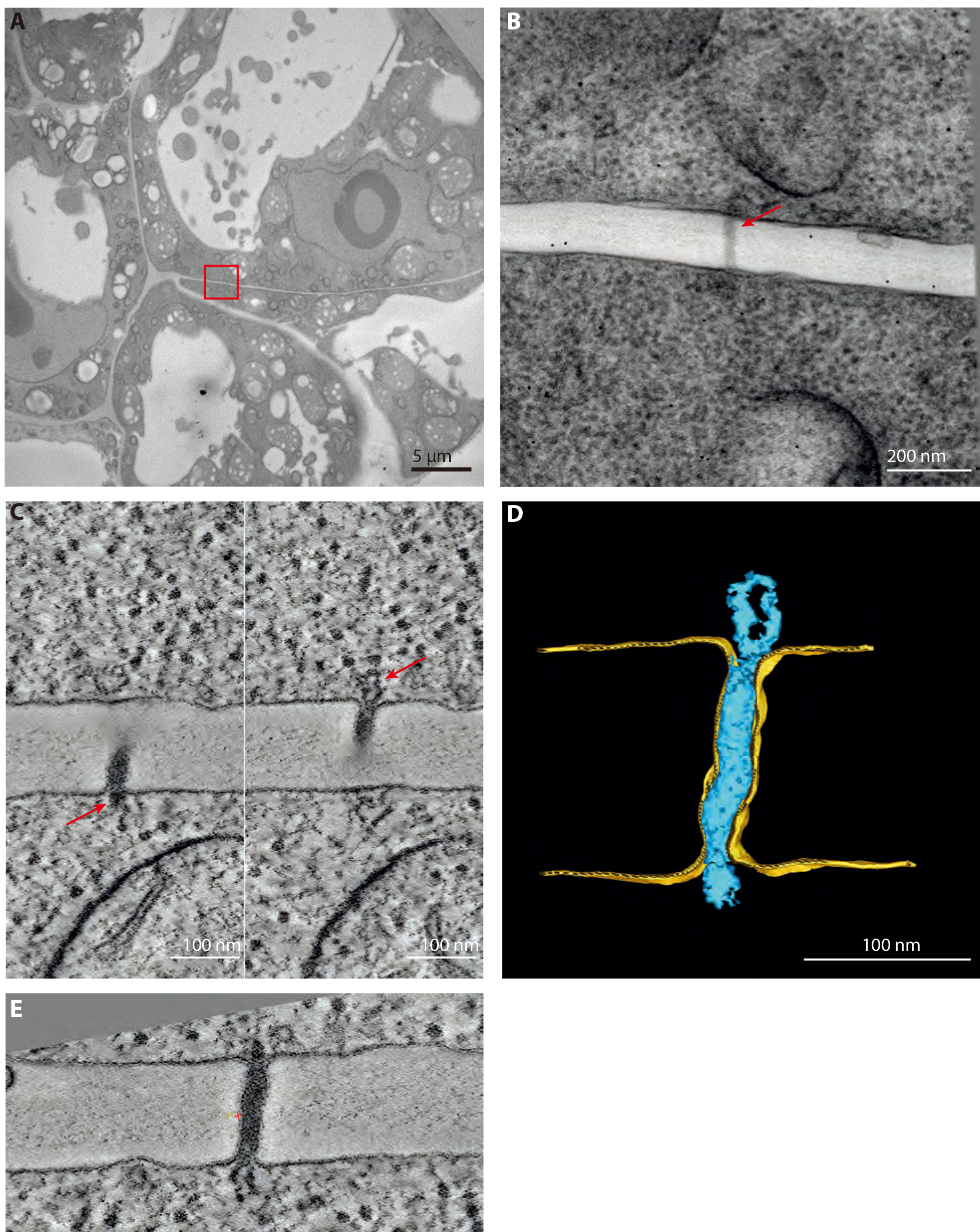
Figure 6. Representative data obtained after HPF and FS. A. Overview of 4 days old cells processed with the procedure described in this article and used in (Nicolas et al., 2017b). B. 0° tilt of the tilt series of a type I plasmodesma (red arrow) found in the red boxed region in (A). Section thickness: 180 nm. C. 0.56 nm thick tomographic slices of the plasmodesma showed in (B). Left and right panel show the two extremities of the pore where the ER can be seen entering the pore (red arrows). D. Segmentation, realized with IMOD, of the same plasmodesma. E. Resliced tomographic slice of the plasmodesma in (C) to have the whole length on one plane. Done with the slicer tool of 3DMOD. - This protocol has also succeeded in delivering clear snapshots of plant organelles such as lipid droplets and Golgi apparatuses (Wattelet-Boyer et al., 2016; Brocard et al., 2017)
- Biological replicates and independent experiments:
PD counting and PD tomography were done on at least two biological replicates, three if possible. For cultured cells, one biological replicate constituted one independent high pressure freezing session on a particular date. Indeed, because the cultured cells were usually sampled in the same flask, we did not consider different blocks from the same HPF as biological replicates.
For roots, each seedling was considered an individual, therefore were considered biological replicates. Nevertheless, data from roots was also collected on multiple HPF session. - PD screening method:
In (Nicolas et al., 2017b), counting type I versus type II pores followed basic principles of stereology (Lucocq, 1993). These are: i) avoid double counting the pores and ii) establishing a precise rule on when a PD can be included in data or not. It is important to stress out that this general method can be applied to counting other features on PD (simple versus branched for example) and also other organelles.
i) When counting on serial sections, there is an inherent risk of double counting structures that span on multiple sections. This risk increases with the size of the structures of interest. Accordingly, a precise counting protocol adapted to the size of the object of interest is required.
First, the ‘chance’ of encountering a given structure of interest on multiple contiguous sections is assessed. The structures are spotted on a ‘reference’ section (n), then we assess if these same structures are still present on a ‘look-up’ section (n + 1). If the structure is not there anymore, it is said to be ‘resolved’ (Figure 6). The fraction of resolved features can then be computed for section n + 1, n + 2 etc., depending on the size of object of interest relative to the section thickness.
In the case of PD, which are at the most 40 nm wide, they are largely inferior to section thickness. Hence, at the n + 1 section in cultured cells (Nicolas et al., 2017b Figure 3g), 80% of the pores screened were resolved at section n + 1. Therefore, by counting every 2 sections (n, n + 2, n + 4 etc.), we can reasonably avoid the double counting of PD.
ii) PD were counted as ‘valid’ and included in the data when both of the plasma membranes were visible and the PD connected at least one of the two adjacent cells (Figure 7).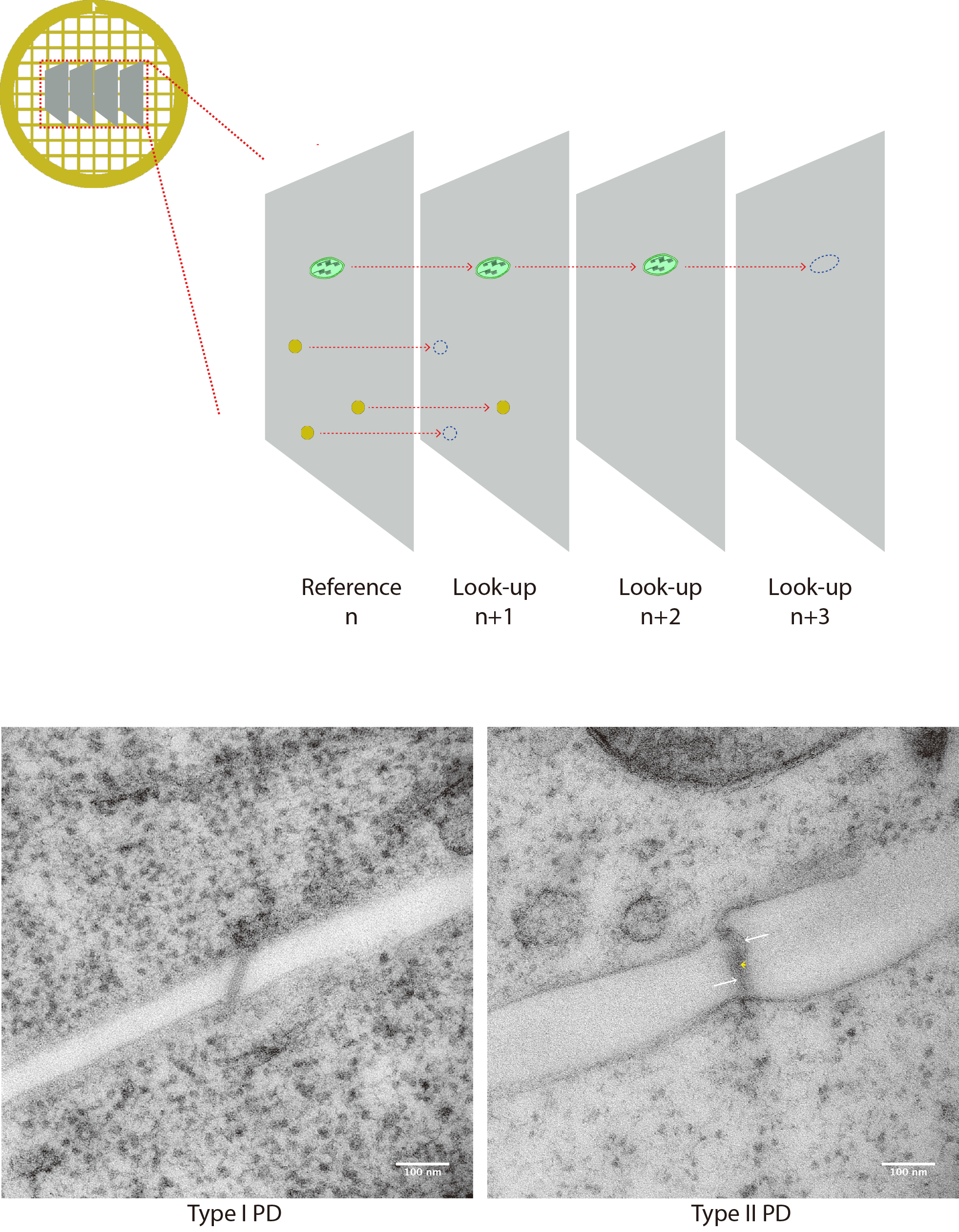
Figure 7. PD counting on serial sections. A. Cartoon depicts 4 serial sections on a grid. Structures of interest are spotted on the reference section n and then followed on look-up sections n + 1, n + 2 etc. When structure cannot be seen anymore, it is said to be resolved. (B-C) Micrographies of 90 nm thick sections of a type I PD (left) and type II (PD) typically collected for quantification. Membranes of the pores are visible, and they connect with both sides. In the latter, heterogeneous densities inside the pore allow the visualization of cytoplasmic sleeve space (white arrows). Although not as resolutive as tomography, the central element (desmotubule) can be seen spanning the pore in the center.
Notes
- Liquid nitrogen manipulation
Because liquid nitrogen can burn, always wear adapted eye/face protection, hand protection and a lab coat during manipulation of liquid nitrogen containers.
Because this gas can cause asphyxiation, always ensure the room is properly ventilated. - Cryosubsitution reagents and resin
Glutaraldehyde and uranyl acetate solutions must be stored at -20 °C in a separate container.
Osmium tetroxide should be stored apart, preferably under a vented hood and always manipulated with special lab-gloves (SHIELDskin Orange nitrile 250). When cryomix is being manipulated outside a ventilated hood, i.e., in the AFS during extraction of the cryomix, experimenter (and others present in the same room) should have complete protection on, including lab-gloves, lab-coat, cartridge mask (SPERIAN half mask and T48-ABEK1 P3 dual cartridges) and protective eyewear to avoid any contact with osmium tetroxide fumes.
HM20 and any other kind of resin should also be manipulated with gloves and cartridge mask as well. For convenience, we wear complete protective gear during all steps from starting of the FS to resin polymerization.
Overall, all these compounds, including acetone, should be manipulated using lab-gloves at all time. Gloves should be changed every 20 min and changed immediately when contaminated. - Bath changing advice during cryosubstitution
Changing baths during FS must be done with care and patience. Gently pipet out the previous medium with the thin tips 1 ml pipets (prevents accidental sucking in of the sample). During this step avoid pipetting in and out; turbulences may detach the samples from the carriers. Always leave remaining medium at the bottom to prevent the samples from air-drying. Then add the next medium gently with a regular plastic pipet (either size) by letting it drip on the plastic tube wall. - Unambiguous tracing of samples in moulds
Keep precise track of what sample/condition is in which mould to avoid any ambiguity. The moulds can be marked with colored nail polish with multiple shapes/dots to distinguish them one-another.
Recipes
- Culture mediums for cells and seedlings
- Murashige and Skoog medium for liquid cultured cells (1 L)
Murashige and Skoog medium + vitamins (Duchefa Biochemi): 4.41g
Sucrose: 30 g
2-(N-morpholino)ethanesulfonic acid (MES) (Euromedex): 0.5 g
NAA: 50 µl
Kinetin stock solution: 50 µl
Adjust pH with 1 N KOH solution to 5.8
Autoclave flasks 110 °C for 30 min
Note: Cells are cultivated under constant light (20 µE/m/sec) and constant agitation at 22 °C. - Murashige and Skoog medium for seedlings (1 L)
Murashige and Skoog medium + vits (Duchefa Biochemi): 4.4 g
MES (Euromedex): 0.5 g
Plant agar (Duchefa Biochemi): 7 g
Adjust pH with 1 N KOH solution to 5.8
Autoclave flasks 110 °C for 30 min
Note: Seedlings are cultivated in vertically placed plates in order for the root to stay on the surface of medium in a greenhouse at 22 °C set on a long day photoperiod (16 h, 100 µE/m/sec).
- Murashige and Skoog medium for liquid cultured cells (1 L)
- BSA solution for cryoprotection during HPF (2 ml)
Weigh 0.4 g of BSA and put it in a 15 ml Falcon tube
Complete at the 2 ml mark with liquid MS medium for cells (see Recipe 1)
Vortex thoroughly until BSA is dissolved entirely
Store at -20 °C - Cryosubstitution Uranyl-acetate stock solution (20%, 500 µl)
Weigh 0.1g of uranyl acetate powder (gloves and dust mask required) and put in 2 ml screwtop tube
Add pure methanol (under ventilated hood) up to 500 µl
Wrap the tube in tinfoil and store in the dark, at -20 °C in dedicated cryosubstitution box - Cryosubstitution mix

- HM20% solutions at different concentrations (4 ml)
Make HM20 solutions in the FS Leica plastic solvent containers (Figure 3, #10). Make the solutions in advance and pre-cool in AFS well at -50 °C before use
- 2% Parlodion solution for grid filming (20 ml)
Weigh 0.4 g of solid parlodion (Electron Microscopy Sciences)
Add isoamyl-acetate (store in a vented solvent cupboard and keep away from humidity) up to 20 ml
Agitate until the solid parlodion is dissolved
Wrap the flask in tinfoil and seal the lid with Parafilm - Toluidine blue solution
Make mother solution: 1 g of Toluidine blue powder (Sigma-Aldrich) and 1 g of sodium borate (Sigma-Aldrich) in 20 ml of distilled water
Working solution is diluted 25 times in distilled water - Fiducial marker solution
Make a solution of 0.5% BSA in MilliQ water
Filtrate solution with a 0.2 µm filter connected to a syringe
Mix colloidal gold solution in 0.5% BSA (1:1 ratio)
Aliquot in 0.5 ml Eppendorf tubes and store at -20 °C
Acknowledgments
This work was supported by the Region Aquitaine (to E.M.B) and PEPS (Initial Support for Exploratory Projects to E.M.B) and the National Agency of Research (Grant ANR-14-CE19-0006-01 to E.M.B). All sample preparation and imaging was done on the Pôle Imagerie du Végétale, appended to the Bordeaux Imaging Centre (http://www.bic.u-bordeaux.fr/). The Region Aquitaine also supported the acquisition of the electron microscope (grant No. 2011 13 04 007 PFM). Thanks to Clément Chambaud that assisted in making the explanatory videos. The authors declare having no conflicts of interests.
References
- Brocard, L., Immel, F., Coulon, D., Esnay, N., Tuphile, K., Pascal, S., Claverol, S., Fouillen, L., Bessoule, J. J. and Brehelin, C. (2017). Proteomic analysis of lipid droplets from Arabidopsis aging leaves brings new insight into their biogenesis and functions. Front Plant Sci 8: 894.
- Ding, B., Turgeon, R. and Parthasarathy, M. V. (1992). Substructure of freeze-substituted plasmodesmata. Protoplasma 169: 28-41.
- Donohoe, B. S., Mogelsvang, S. and Staehelin, L. A. (2006). Electron tomography of ER, Golgi and related membrane systems. Methods 39(2): 154-162.
- Dubochet, J. (2007). The physics of rapid cooling and its implications for cryoimmobilization of cells. Methods Cell Biol 79: 7-21.
- Hagler, H. K. (2007). Ultramicrotomy for biological electron microscopy. In: Kuo, J. (Ed.). Electron Microscopy: Methods and Protocols. Humana Press pp: 67-96.
- Kang, B. H., Nielsen, E., Preuss, M. L., Mastronarde, D. and Staehelin, L. A. (2011). Electron tomography of RabA4b- and PI-4Kβ1-labeled trans Golgi network compartments in Arabidopsis. Traffic 12(3): 313-329.
- Kremer, J. R., Mastronarde, D. N. and McIntosh, J. R. (1996). Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116(1): 71-76.
- Lucocq, J. (1993). Unbiased 3-D quantitation of ultrastructure in cell biology. Trends Cell Biol 3(10): 354-358.
- Nicolas, W. J., Grison, M. S. and Bayer, E. M. (2017a). Shaping intercellular channels of plasmodesmata: the structure-to-function missing link. J Exp Bot.
- Nicolas, W. J., Grison, M. S., Trepout, S., Gaston, A., Fouche, M., Cordelieres, F. P., Oparka, K., Tilsner, J., Brocard, L. and Bayer, E. M. (2017b). Architecture and permeability of post-cytokinesis plasmodesmata lacking cytoplasmic sleeves. Nat Plants 3: 17082.
- Oparka, K. J. and Roberts, A. G. (2001). Plasmodesmata. A not so open-and-shut case. Plant Physiol 125(1): 123-126.
- Studer, D., Graber, W., Al-Amoudi, A. and Eggli, P. (2001). A new approach for cryofixation by high-pressure freezing. J Microsc 203(Pt 3): 285-294.
- Tilney, L. G., Cooke, T. J., Connelly, P. S. and Tilney, M. S. (1991). The structure of plasmodesmata as revealed by plasmolysis, detergent extraction, and protease digestion. J Cell Biol 112(4): 739-747.
- Wattelet-Boyer, V., Brocard, L., Jonsson, K., Esnay, N., Joubes, J., Domergue, F., Mongrand, S., Raikhel, N., Bhalerao, R. P., Moreau, P. and Boutte, Y. (2016). Enrichment of hydroxylated C24- and C26-acyl-chain sphingolipids mediates PIN2 apical sorting at trans-Golgi network subdomains. Nat Commun 7: 12788.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nicolas, W. J., Bayer, E. and Brocard, L. (2018). Electron Tomography to Study the Three-dimensional Structure of Plasmodesmata in Plant Tissues–from High Pressure Freezing Preparation to Ultrathin Section Collection. Bio-protocol 8(1): e2681. DOI: 10.21769/BioProtoc.2681.
Category
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Electron microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.