- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Targeted Genome Editing of Virulent Phages Using CRISPR-Cas9




Published: Vol 8, Iss 1, Jan 5, 2018 DOI: 10.21769/BioProtoc.2674 Views: 16773
Reviewed by: Modesto Redrejo-RodriguezRajesh ThippeshappaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
This protocol describes a straightforward method to generate specific mutations in the genome of strictly lytic phages. Briefly, a targeting CRISPR-Cas9 system and a repair template suited for homologous recombination are provided inside a bacterial host, here the Gram-positive model Lactococcus lactis MG1363. The CRISPR-Cas9 system is programmed to cleave a specific region present on the genome of the invading phage, but absent from the recombination template. The system either triggers the recombination event or exerts the selective pressure required to isolate recombinant phages. With this methodology, we generated multiple gene knockouts, a point mutation and an insertion in the genome of the virulent lactococcal phage p2. Considering the broad host range of the plasmids used in this protocol, the latter can be extrapolated to other phage-host pairs.
Keywords: PhageBackground
Phages are bacterial viruses found in abundance in every ecosystem (Suttle, 2005; Breitbart and Rohwer, 2005) and unsurprisingly, they are natural inhabitants of milk. Phage p2 is a model for the most prevalent group (Sk1virus) of virulent lactococcal phages found in the dairy industry (Deveau et al., 2006; Mahony et al., 2012) and it infects the Gram-positive bacterium Lactococcus lactis MG1363, also a model strain for basic research. Despite the status of p2 as a reference phage, almost half of its genes encode uncharacterized proteins. Likewise, a clear majority of phage genes identified by metagenomics have no functional assignment and no homolog in public databases (Hurwitz et al., 2016; Paez-Espino et al., 2016).
One of the ways to study genes is through their modification and subsequent observation of the resulting phenotypes. Phage genomes can only be modified inside a host, in their biologically active form. Virulent phages are strictly lytic; thus, their genome never integrates into the bacterial chromosome. This adds a time constraint for the in vivo modification of their DNA, which can be manipulated only during their short infection cycle. The emergence of CRISPR-Cas research in the last decade lead to the adaptation of this natural prokaryotic defense mechanism into a powerful tool to edit the genome of a plethora of organisms and viruses, including virulent phages (Kiro et al., 2014; Martel and Moineau, 2014; Box et al., 2015; Pires et al., 2016; Bari et al., 2017; Lemay et al., 2017; Manor and Qimron, 2017; Tao et al., 2017).
Here, we detail a simple and reproducible protocol to edit the genome of phage p2 using the well-known Streptococcus pyogenes Cas9 (SpCas9) cloned into the lactic acid bacterium L. lactis MG1363 (Lemay et al., 2017). Within our laboratory, this protocol has also been successfully applied to edit the genome of a virulent phage infecting the Gram-negative E. coli (unpublished), illustrating its broad applicability.
Materials and Reagents
- Materials
- Disposable Pasteur pipettes (VWR, catalog number: 14672-200 )
- 3 mm glass beads (VWR, catalog number: 26396-508 )
Manufacturer: Walter Stern, catalog number: 100C . - 100 mm glass tubes with caps (Fisher Scientific, catalog numbers: 14-961-27 and 05-888-1A )
Manufacturer: Bal Supply, catalog number: 13144UL . - 150 mm glass tubes with caps (Fisher Scientific, catalog numbers: 14-961-32 and 05-888C )
Manufacturer: Bal Supply, catalog number: 18144CL . - 0.5 ml micro-tubes (SARSTEDT, catalog number: 72.699 )
- 1.5 ml micro-tubes (SARSTEDT, catalog number: 72.690 )
- Sterile 100 x 15 mm plastic Petri dishes (‘plates’) (VWR, catalog number: 25384-302 )
- 10 ml sterile BD Luer-LokTM Tip syringe (BD, catalog number: 309604 )
- 0.2 µm sterile PES syringe filter (SARSTEDT, catalog number: 83.1826.001 )
- 0.45 µm sterile PES syringe filter (SARSTEDT, catalog number: 83.1826 )
- Disposable Pasteur pipettes (VWR, catalog number: 14672-200 )
- Phage and bacterial strain
- Lactococcus lactis MG1363 (Félix d’Hérelle Reference Center for Bacterial Viruses, catalog number: HER1439 )
Note: L. lactis is generally recognized as a safe bacterium and all the experiments can be performed in a biosafety level 1 laboratory. - Phage p2 (Félix d’Hérelle Reference Center for Bacterial Viruses, catalog number: HER457 )
- Lactococcus lactis MG1363 (Félix d’Hérelle Reference Center for Bacterial Viruses, catalog number: HER1439 )
- Plasmids
- Enzymes
- Antarctic phosphatase (5,000 U per ml) (New England Biolabs, catalog number: M0289S )
- BsaI (10,000 U per ml) (New England Biolabs, catalog number: R0535S )
- Lysozyme (20,000 U per mg dry weight) (Thermo Fisher Scientific, catalog number: 89833 )
- Q5 DNA polymerase (2,000 U per ml) (New England Biolabs, catalog number: M0491S )
- T4 DNA ligase (1,000 U per ml) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15224017 )
- T4 polynucleotide kinase (10,000 U per ml) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EK0031 )
- Taq DNA polymerase (5,000 U per ml) (NBS Biologicals, catalog number: 9K-001-0034 )
- XbaI (20,000 U per ml) (New England Biolabs, catalog number: R0145S )
- Antarctic phosphatase (5,000 U per ml) (New England Biolabs, catalog number: M0289S )
- Reagents
- AccuGENETM molecular biology water (Lonza, catalog number: 51200 )
- Agarose LE (Roche Diagnostics, catalog number: 11685678001 )
- BDTM BactoTM Brain Heart Infusion (BHI) (Fisher Scientific, catalog number: DF0037-17-8 )
Manufacturer: BD, catalog number: 237500 . - Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C5080 )
- Chloramphenicol (Sigma-Aldrich, catalog number: C0378 )
- EDTA-Na2 (Sigma-Aldrich, catalog number: E5134 )
- Erythromycin (Fisher Scientific, catalog number: 10583315 )
- 95% ethanol (Commercial Alcohols, catalog number: P016EA95 )
- 100% ethanol (Commercial Alcohols, catalog number: P016EAAN )
- EZ-Vision® Three (VWR, catalog number: 97063-166 )
- Glacial acetic acid (Caledon Laboratories, catalog number: 1000-1-29 )
- Glucose monohydrate (Sigma-Aldrich, catalog number: 49159 )
- Glycerol (Merck, catalog number: GX0185-2 )
- Glycine (Merck, catalog number: 4810-OP )
- Granulated agar (Fisher Scientific, catalog number: BP1423-500 )
- Hydrochloric acid ( HCl) (Fisher Scientific, catalog number: 351285-212 )
- High DNA Mass Ladder (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10496016 )
- 1 Kb Plus DNA Ladder (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10787018 )
- Low DNA Mass Ladder (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10068013 )
- OxoidTM M17 broth (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: CM0817 )
- Magnesium sulfate heptahydrate ( MgSO4·7H2O) (Caledon Laboratories, catalog number: 4860-1-70 )
- Magnesium chloride hexahydrate (MgCl2·6H2O) (VWR, catalog number: BDH9244 )
- Sodium chloride (NaCl) (Anachemia, catalog number: 81708-380 )
- Primers (Table 1) (customized by Thermo Fisher Scientific, www.thermofisher.com)
Table 1. Primers used in this protocol
aRestriction sites for ligation and overhangs for Gibson Assembly are underlined. ‘N’ stands for any nucleotides and ‘x’ for any number of nucleotides required for the amplification of a fragment of interest. - Sodium acetate (NaAc) (Sigma-Aldrich, catalog number: S2889 )
- Sucrose (Sigma-Aldrich, catalog number: S0389 )
- Tris (Base), Ultrapure (Avantor Performance Materials, catalog number: 4109-6 )
- 0.8% and 2% agarose gel (see Recipes)
- BHI agar medium supplemented with erythromycin (see Recipes)
- BHI medium supplemented with erythromycin (see Recipes)
- 2 M CaCl2 (see Recipes)
- 10 mg/ml chloramphenicol stock solution (see Recipes)
- 10 mg/ml and 75 mg/ml erythromycin stock solution (see Recipes)
- 70% ethanol (see Recipes)
- GM17 agar medium supplemented with CaCl2 or antibiotics (see Recipes)
- GM17 medium (see Recipes)
- GM17 soft agar medium supplemented with CaCl2 (see Recipes)
- Glycine shock solution (see Recipes)
- 1 M MgCl2 (see Recipes)
- 10x Phage buffer (see Recipes)
- Recovery solution (see Recipes)
- 50x TAE buffer (see Recipes)
- Wash solution (see Recipes)
- AccuGENETM molecular biology water (Lonza, catalog number: 51200 )
- Kits
Equipment
- Autoclave
- Benchtop microcentrifuge (Eppendorf, model: 5415 D )
- Bunsen burner
- Centrifuge bottles (SS-34)
- Centrifuge Sorvall RC5C and rotor SS-34 (Thermo Fisher Scientific, catalog number: 28020 )
- Dry bath with heating block (VWR, catalog numbers: 13259-034 and 13259-130 )
- Electroporation cuvettes, 0.2 cm electrode gap (Bio-Rad Laboratories, catalog number: 1652086 )
- Gene Pulser II electroporator (Bio-Rad Laboratories, catalog number: 165-2109 )
- Incubator-shaker set to 37 °C
Note: Incubator that can be set to 30 °C (shaking not necessary) and 37 °C. - Micropipettes (Nichiryo, catalog numbers: 00-NPX2-10 , 00-NPX2-100 , 00-NPX2-1000 )
Note: Sterile filtered pipette tips to reduce contamination of the micropipettes by bioaerosols. - Microwave
- MultiDoc-ItTM Imaging System (UVP, catalog number: 97-0200-01 )
- PCR Thermal Cycler (MJ Research, model: PTC-200 )
- Power supply (Bio-Rad Laboratories, model: PowerPac 300 )
- Spectra/Por® 1 dialysis membranes, MWCO 6-8000 (VWR, catalog number: 28170-138 )
Manufacturer: Spectrum, catalog number: S632650 . - Spectrophotometer Spectronic 20D
- Tweezers
- VWR® Midi Plus 15 Horizontal Electrophoresis Systems (VWR, catalog number: 89032-296 )
Procedure
- Spacer cloning in pL2Cas9
Plasmid pL2Cas9 encodes the CRISPR-Cas9 components derived from the strain Streptococcus pyogenes SF370 as well as an erythromycin resistance gene. To avoid plasmid loss in E. coli or L. lactis, erythromycin is added to media to a final concentration of 150 µg/ml (Em 150) or 5 µg/ml (Em 5), respectively. When purchased from Addgene, pL2Cas9 is sent as a stab culture of transformed E. coli NEB5α strain. Steps A2 to A4 are adapted from Jiang et al. (2013).- Purification pL2Cas9
- Streak the bacterial strain from the stab to a BHI Em 150 plate (see Recipes).
- Incubate overnight at 37 °C.
- Pick a single colony with a sterile pipette tip using sterile tweezers and drop the tip into 500 ml of BHI Em 150 (see Recipes).
- Incubate overnight at 37 °C in a shaking incubator at 200 rpm.
- Follow QIAGEN plasmid maxi kit protocol for isolating low copy plasmid.
Note: We transfer the isopropanol precipitate in 16 x 1.5 ml micro-tubes to minimize loss of DNA during the washing step. Each DNA pellet is then redissolved in 20 µl of the provided plasmid resuspension buffer and pooled in a 1.5 ml micro-tube. - Run a sample of the purified plasmid DNA preparation on a 0.8% agarose gel (see Recipes) with the High DNA Mass Ladder to estimate DNA concentration.
Note: To visualize DNA, we use EZ-Vision® Three, a 6x loading buffer containing a fluorescent DNA dye. - Keep the purified plasmid DNA at -20 °C until needed.
- Streak the bacterial strain from the stab to a BHI Em 150 plate (see Recipes).
- Spacer design and annealing
- Search for a 5’-NGG-3’ sequence in the locus to modify (Figure 1). This signature sequence constitutes the protospacer adjacent motif (PAM) recognized by SpCas9.
- To clone the desired spacer in pL2Cas9, design and synthesize two ssDNA oligos of 35 nucleotides corresponding to both strands of the selected protospacer with BsaI restriction sites (Figure 1, Table 1).
Note: The GC content of the spacer sequences should not influence the efficiency of SpCas9 to cleave its target DNA (Tao et al., 2017). To avoid off-target activity, make sure that the protospacer is not found elsewhere in the phage or the cloning host genome next to a functional PAM. - Dilute the oligos to 50 μM in molecular biology water.
- For 5’-phosphorylation of the oligos, mix 2 μl of each diluted oligo with 10 µl of 5x T4 ligase buffer, 1 µl of T4 polynucleotide kinase (PNK) and 32 µl of distilled water in a PCR tube to a final volume of 50 µl.
Note: 5x T4 ligase buffer is supplied with T4 DNA ligase. Divide the ligase buffer into single-use aliquots as ATP in the buffer can be degraded by repeated freeze-thaw cycles. - Perform the phosphorylation for 30 min at 37 °C and heat inactivate the PNK for 20 min at 65 °C (in a PCR machine or a water bath).
- Add 2.5 µl of 1 M NaCl to the phosphorylated oligos.
- Place the tube in a PCR thermal cycler and heat the sample for 5 min at 95 °C before cooling down to 25 °C at a rate of 0.1 °C per sec.
- Keep at -20 °C for up to 2 months.
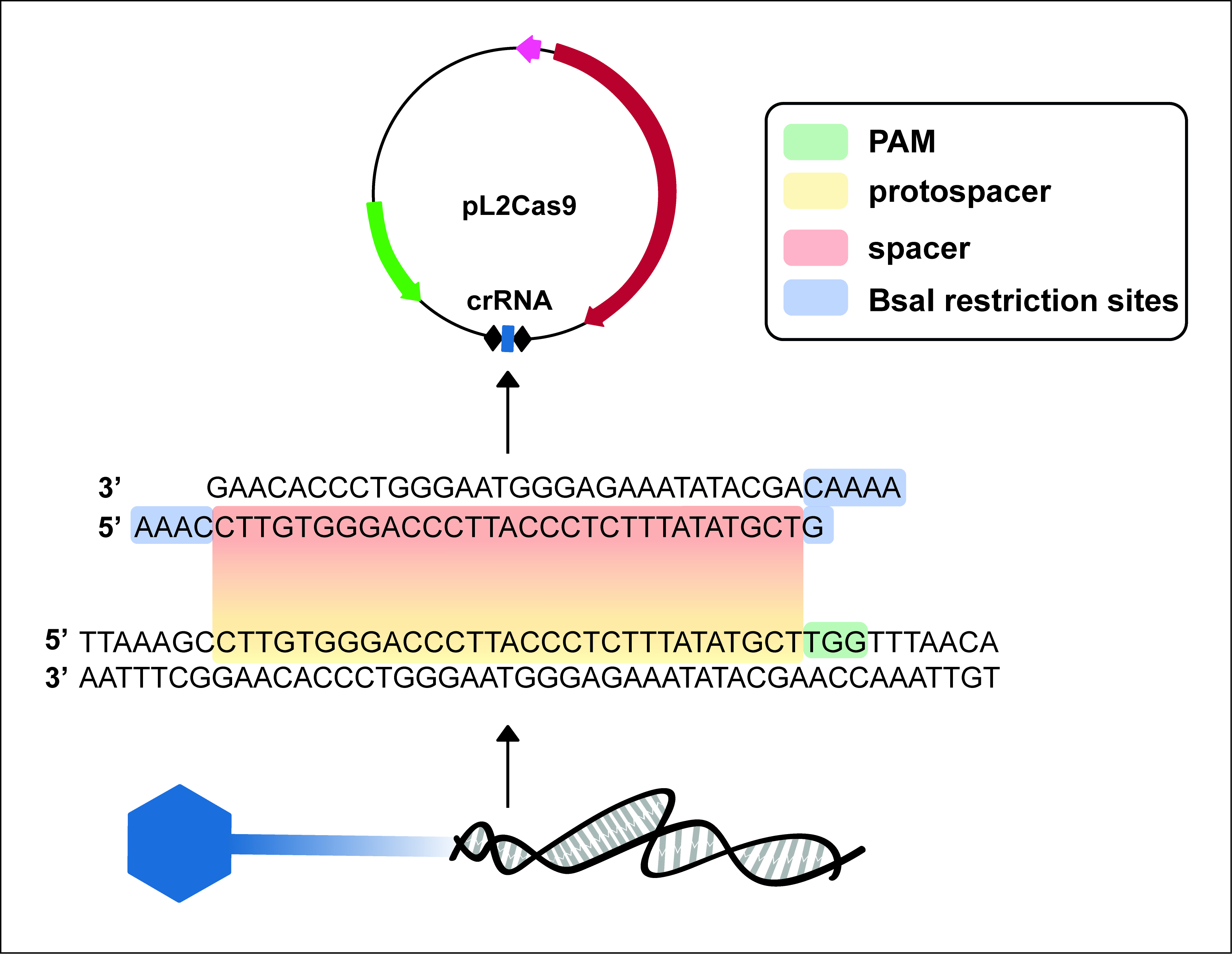
Figure 1. Graphical representation of a spacer design. In this example, a PAM is identified in the phage genome. The 30 bp sequence upstream the PAM is called the ‘protospacer’, and must be incorporated into the CRISPR array (crRNA) in the form of a ‘spacer’ to dictate target specificity. BsaI restriction sites are added to the ends of the spacer for ligation into pL2Cas9.
- Search for a 5’-NGG-3’ sequence in the locus to modify (Figure 1). This signature sequence constitutes the protospacer adjacent motif (PAM) recognized by SpCas9.
- Digestion of pL2Cas9 with BsaI
- Digest 1 µg of purified pL2Cas9 with BsaI by following the manufacturer’s instructions with an overnight incubation at 37 °C (final volume of 50 µl).
- Dephosphorylate the 5’-ends of the digested pL2Cas9 with the Antarctic phosphatase by following the manufacturer’s instructions.
- Purify the digested and dephosphorylated vector by precipitation with salts and ethanol.
Note: Purification by gel extraction removes uncut vector, but the yield is poor. - Add 1:10 volume of NaAc 3 M pH 5.2 to DNA.
- Add 2 volumes of 100% ethanol.
- Mix thoroughly and incubate on ice for 15 min.
- Centrifuge in a benchtop microcentrifuge at 16,000 x g for 15 min.
- Carefully remove the supernatant and wash the pellet with 70% ethanol (see Recipes).
- Air dry the pellet for 5-10 min.
- Resuspend the pellet in 30 µl 10 mM Tris-HCl pH 8.5.
- Run the purified vector on a 0.8% agarose gel with the annealed oligos as well as both the High DNA Mass and 1 Kb Plus DNA Ladders in separate wells to estimate concentrations.
- Store the digested pL2Cas9 at -20 °C until needed.
Note: Aliquot into smaller volumes to prevent DNA degradation that may occur during repeated freeze-thaw cycles.
- Digest 1 µg of purified pL2Cas9 with BsaI by following the manufacturer’s instructions with an overnight incubation at 37 °C (final volume of 50 µl).
- Ligation of the targeting plasmid
- Set up ligation reactions with the T4 DNA ligase following the manufacturer’s instructions. Use a molar ratio of approximately 3:1 of insert to vector.
- Prepare another ligation reaction without the insert (negative ligation control) to evaluate vector re-circularization.
- Perform the ligations overnight at 16 °C (in a PCR machine or a water bath).
- Heat inactivate the ligase at 65 °C for 10 min, put on ice, and proceed immediately to the transformation (Steps A5 and A6).
Note: Ligation products can be kept a few days at -20 °C, but transformation efficiency will decrease.
- Set up ligation reactions with the T4 DNA ligase following the manufacturer’s instructions. Use a molar ratio of approximately 3:1 of insert to vector.
- Preparation of L. lactis MG1363 competent cells
Note: If working with other phage-host, transformation of DNA constructs can be done directly into the bacterial host of interest or first into L. lactis MG1363. In the latter case, DNA constructs have to be extracted from the cloning host and then transferred into the host of interest. Cells should be transformed with one plasmid, selected with the appropriate antibiotics, made competent, transformed with the second plasmid, and selected with both antibiotics. We do not recommend double transformation.- Inoculate 10 ml of GM17 broth (see Recipes) with L. lactis MG1363 and incubate overnight at 30 °C.
- Inoculate 5 tubes containing 9.7 ml glycine shock solution (see Recipes) with 300 µl of the overnight culture from Step A5a.
Note: A 10-ml bacterial culture is sufficient to prepare 50 µl of competent cells. Typically, 5 x 50 µl competent cells are prepared for the transformation of: (1) the ligation product, (2) uncut vector (positive transformation control), (3) dephosphorylated cut vector (negative digestion control) (4) negative ligation control, and (5) no DNA (negative transformation control). If needed, additional cultures can be prepared concomitantly. - Incubate at 30 °C until the bacterial cultures reach an OD600 of 0.2.
- Transfer the cultures into sterile centrifuge bottles (SS-34) and centrifuge for 5 min at 4 °C and 12,000 x g.
Note: From here onwards, cultures and solutions should be kept on ice whenever possible. - Discard supernatant and resuspend each pellet in 1 ml of sterile cold wash solution (see Recipes).
- Transfer cells into 1.5 ml micro-tubes.
- Centrifuge the resuspensions in a benchtop microcentrifuge at 16,000 x g for 1 min.
Note: We perform this step at room temperature (RT), but a refrigerated microcentrifuge may be used to keep the cultures cold (~4 °C). - Repeat the last three steps twice.
- Resuspend each pellet of competent cells in 50 μl of sterile cold wash solution before pooling them in a 1.5 ml micro-tube and proceed immediately to Step A6.
- Inoculate 10 ml of GM17 broth (see Recipes) with L. lactis MG1363 and incubate overnight at 30 °C.
- Electroporation of L. lactis MG1363
- Dialyse assembled DNA using a membrane (Figure 2).
Note: Ligation and Gibson Assembly products should be desalted prior to electroporation.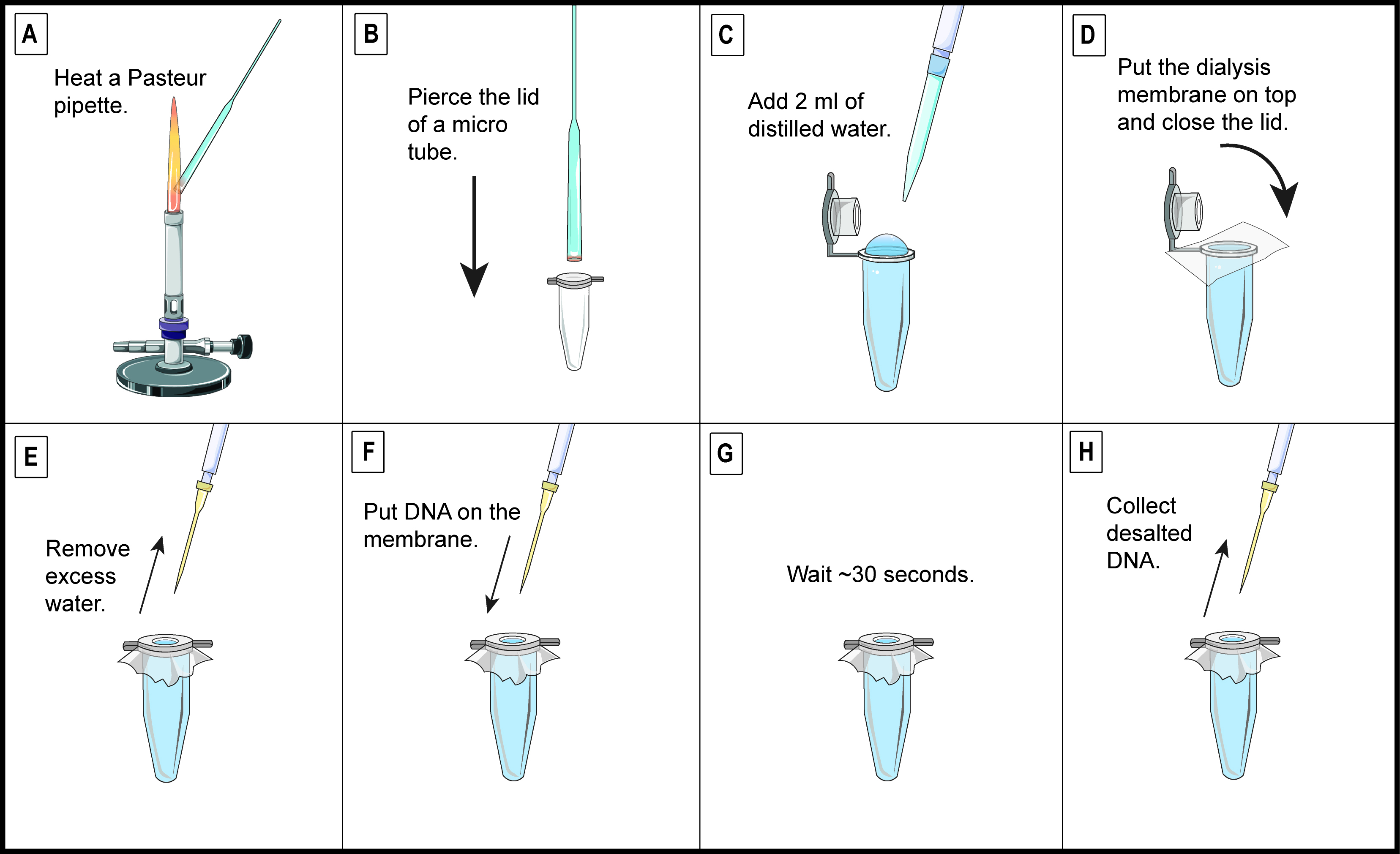
Figure 2. Quick and easy DNA dialysis. A. Heat the wide end of a Pasteur pipette with a Bunsen burner flame until the glass turns red. B. Quickly pierce the lid of a 1.5 ml micro-tube. C. Add 2 ml of distilled water in the micro-tube. Due to surface tension, a convex meniscus should be above the edge of the micro-tube. D. Place a 4-cm square of dialysis membrane (MWCO 6-8000) in distilled water. Once it is hydrated, cut the dialysis membrane tubing to the length to have two separate layers of membrane. Put a single layer of membrane onto the water and close the lid of the micro-tube. Make sure there is no air bubble between the water and the dome-shaped membrane. E. Remove excess water on top of the membrane. F. Put 5 µl of DNA sample on top of the membrane. G. Wait approximately 30 sec. H. Collect the desalted DNA. Repeat the whole process for every sample to dialyse. - Add 5 µl of DNA (or water for negative transformation control) and 45 µl of competent cells to cold electroporation cuvettes.
Note: Keep the cuvettes and the recovery solution on ice. - Set up the Gene Pulser to 25 µF, 200 Ω and 2.5 KV.
- Place a cuvette in the track and simultaneously push both red buttons until the tone.
- Quickly add 500 µl of cold recovery solution (see Recipes) to cells and leave on ice for 10 min.
- Transfer the content of the cuvettes into 1.5 ml micro-tubes.
- Repeat the last three steps for every transformation.
- Incubate for 2 h at 30 °C.
- Plate the cells on GM17 supplemented with the appropriate antibiotic (see Recipes). Use 5-10 glass beads per plate to spread the transformed cells evenly.
- Incubate for 24-48 h at 30 °C.
- Dialyse assembled DNA using a membrane (Figure 2).
- Colony PCR
- To confirm the presence of the DNA insert in the plasmid constructs, pick several single colonies with sterile pipette tips and place each of them in 50 µl molecular biology water.
Note: The number of colonies to screen will depend on the number of background colonies on the digested vector control plate. - Using the same pipette tips, streak the colonies on a GM17 plate supplemented with the appropriate antibiotic and incubate overnight at 30 °C.
Note: This plate will later serve to start liquid cultures of positive clones. - Use 5 µl of DNA template (water-bacteria suspension) per 50 µl PCR reactions with the Taq DNA Polymerase. Always follow the manufacturer’s instructions.
Note: We use the backbone-specific primers Cas9_S.pyo_F6 and crRNA_S.pyo_R (Table 1) to screen for new spacers in the CRISPR array of pL2Cas9. Amplification of the uncut vector or desired ligation products will both generate an 815 bp fragment. To screen colonies for the right repair template, we use the backbone-specific primers pNZins_F and pNZins_R (Table 1). Amplification of uncut pNZ123 will generate a 145 bp fragment, while the assembled repair template will generate a bigger fragment of variable length. - Run the PCR products on a 2% agarose gel (see Recipes) with the 1 Kb Plus DNA Ladder to determine their size.
- To confirm the sequence of the inserts, submit PCR products for Sanger sequencing.
Note: To reduce the number of PCR products sent for sequencing, positive clones can also be identified with insert-specific primers. To screen for new spacers in pL2Cas9, we use the oligo I from Step A2 (Table 1) and crRNA_S.pyo_R. While a positive clone results in the amplification of a 391 bp fragment, a negative clone results in no product. - Inoculate 10 ml of GM17 broth medium supplemented with the appropriate antibiotics with positive clones and incubate overnight at 30 °C.
- Store cells at -80 °C by adding 850 µl of overnight culture to 150 µl of sterile glycerol.
- To confirm the presence of the DNA insert in the plasmid constructs, pick several single colonies with sterile pipette tips and place each of them in 50 µl molecular biology water.
- Purification pL2Cas9
- Construction of homologous repair template
We use the broad-host-range and high-copy-number plasmid pNZ123 to construct our recombination templates. It confers chloramphenicol resistance to the bacterial cells carrying it. To avoid plasmid loss in L. lactis, chloramphenicol is supplied at a final concentration of 5 µg/ml (Cm 5). Other plasmids compatible with pL2Cas9 could be used. Most importantly, the repair template must be designed to lack the Cas9 target sequence so that recombinant phages, and the template itself, can avoid cleavage. The most effective approach is to create a deletion, which removes the whole target sequence (PAM and protospacer) (Figure 3). Otherwise, a single mutation can be introduced in the PAM and/or multiple mutations in the protospacer to prevent DNA cleavage. If the target sequence is in a coding region, mutations should be designed based on codon usage patterns.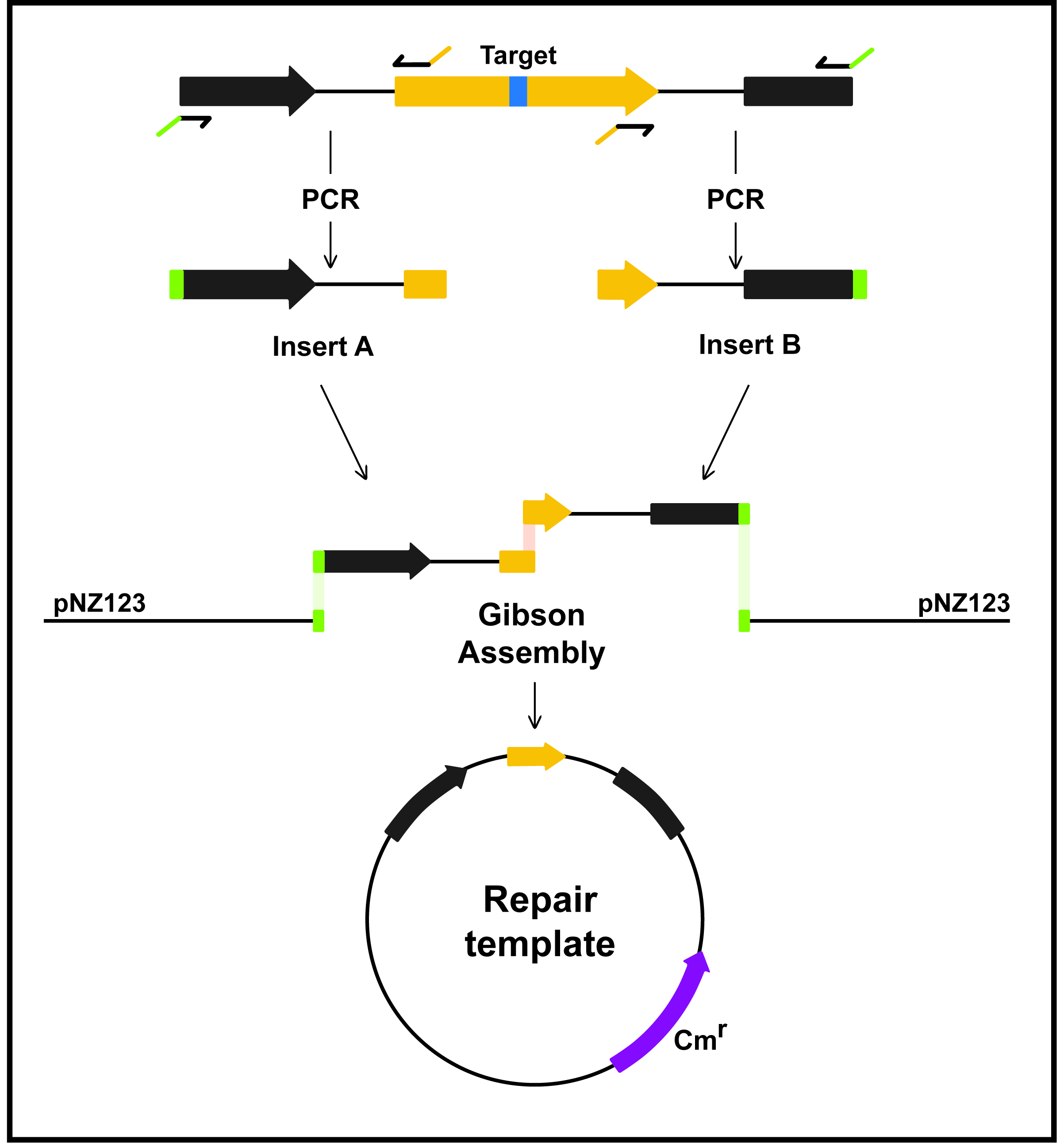
Figure 3. Construction of homologous repair template with a desired deletion. Insert A (left) contains the 5’-end of a gene of interest (yellow) and insert B (right) contains the 3’-end of the same gene. The two fragments, amplified from the phage genome, are the homologous arms of the repair template. The external primers have overlaps (green) for inserting the amplicons into linearized pNZ123 using Gibson Assembly (pNZ_insertA and pNZ_insertB, Table 1). The inner primers have complementary overhangs (yellow) for annealing together. Assembly of the two inserts removes part of the yellow gene, and the target sequence (blue box) is absent from the repair template. A similar strategy can be used to generate deletions, point mutations and insertions.- Purification pNZ123
- Streak L. lactis MG1363 (pNZ123) to a GM17 Cm 5 plate (see Recipes).
- Incubate overnight at 30 °C.
- Pick a single colony with a sterile pipette tip using sterile tweezers and drop the tip into 500 ml of GM17 Cm 5 (see Recipes).
- Incubate statically overnight at 30 °C.
- Harvest the overnight culture by centrifugation at 6,000 x g for 15 min.
- Resuspend the bacterial pellet in 20 ml buffer P1 (from QIAGEN plasmid maxi kit) supplemented with lysozyme 30 mg/ml.
- Incubate for 30 min at 37 °C.
- Follow QIAGEN plasmid maxi kit protocol.
- Run a sample of the purified plasmid DNA preparation on a 0.8% agarose gel (see Recipes) with the High DNA Mass Ladder to estimate DNA concentration.
- Keep the purified plasmid DNA at -20 °C until needed.
- Streak L. lactis MG1363 (pNZ123) to a GM17 Cm 5 plate (see Recipes).
- Linearization of pNZ123
- Digest 1 µg of purified pNZ123 with XbaI following the manufacturer’s instructions with an overnight incubation at 37 °C.
- Amplify the digested vector with primers pNZ_XbaI_F and pNZ_XbaI_R (Table 1) and the Q5 DNA polymerase following the manufacturer’s instructions. We use an annealing temperature of 58 °C and an elongation time of 75 sec.
Note: Amplification of the digested vector significantly reduces background from uncut or re-circularized vector. - Clean the resulting PCR products with the QIAquick PCR Purification kit following the manufacturer’s instructions. Elute in molecular biology water.
- Store the linearized and PCR-amplified pNZ123 at -20 °C until needed.
Note: Aliquot into smaller volumes to prevent DNA degradation that may occur during repeated freeze-thaw cycles.
- Digest 1 µg of purified pNZ123 with XbaI following the manufacturer’s instructions with an overnight incubation at 37 °C.
- Amplification of inserts (Figure 3)
- Design primers with overlaps suited for Gibson Assembly into linearized pNZ123.
Note: Even though the manufacturer recommends a 20 bp overlap between the fragments to be assembled, we found that > 30 bp overlaps significantly increased the efficiency of assembly. While 250-500 bp homologous arms are preferred, shorter arms are generally sufficient for recombination with the phage genome. - PCR amplify the inserts with the Q5 DNA polymerase using the phage genome as a template and following the manufacturer’s instructions.
- Run on a 2% agarose gel with the linearized pNZ123 and both the Low DNA Mass and 1 Kb Plus Ladders in separate wells to confirm and determine the concentration of fragments.
- Design primers with overlaps suited for Gibson Assembly into linearized pNZ123.
- Assembly and transformation of the repair template
- Assemble the fragments from Steps B1 and B2 using Gibson Assembly Master Mix according to the manufacturer’s instructions.
Note: For best results, assemble and transform the repair template on the same day. - Incubate for 1 h at 50 °C.
- Keep on ice (or -20 °C) until needed.
- Prepare competent cells of L. lactis MG1363 (Step A5).
Note: The repair template can also be electroporated into L. lactis MG1363 already harboring the targeting plasmid pL2Cas9. - Proceed immediately to the electroporation (Steps A6) of the repair template and plate the cells on GM17 supplemented with the appropriate antibiotics.
- Incubate 24-48 h at 30 °C.
Note: Selection with two antibiotics often slows down bacterial growth. - Analyze the transformants by colony PCR (Step A7).
- Assemble the fragments from Steps B1 and B2 using Gibson Assembly Master Mix according to the manufacturer’s instructions.
- Purification pNZ123
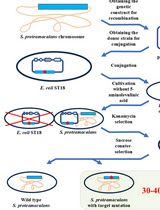
- Phage engineering (Figure 4)
We perform double layer plaque assays to obtain isolated phage plaques and purify recombinant phages. This step could easily be adapted to other phage-host pairs, provided that the bacterial host harbors the two DNA constructs obtained in Steps A and B. We do not use antibiotic selection during phage infection since the plasmids are stable in L. lactis MG1363. Plasmid stability may vary in different hosts, and antibiotic selection should be considered in some cases.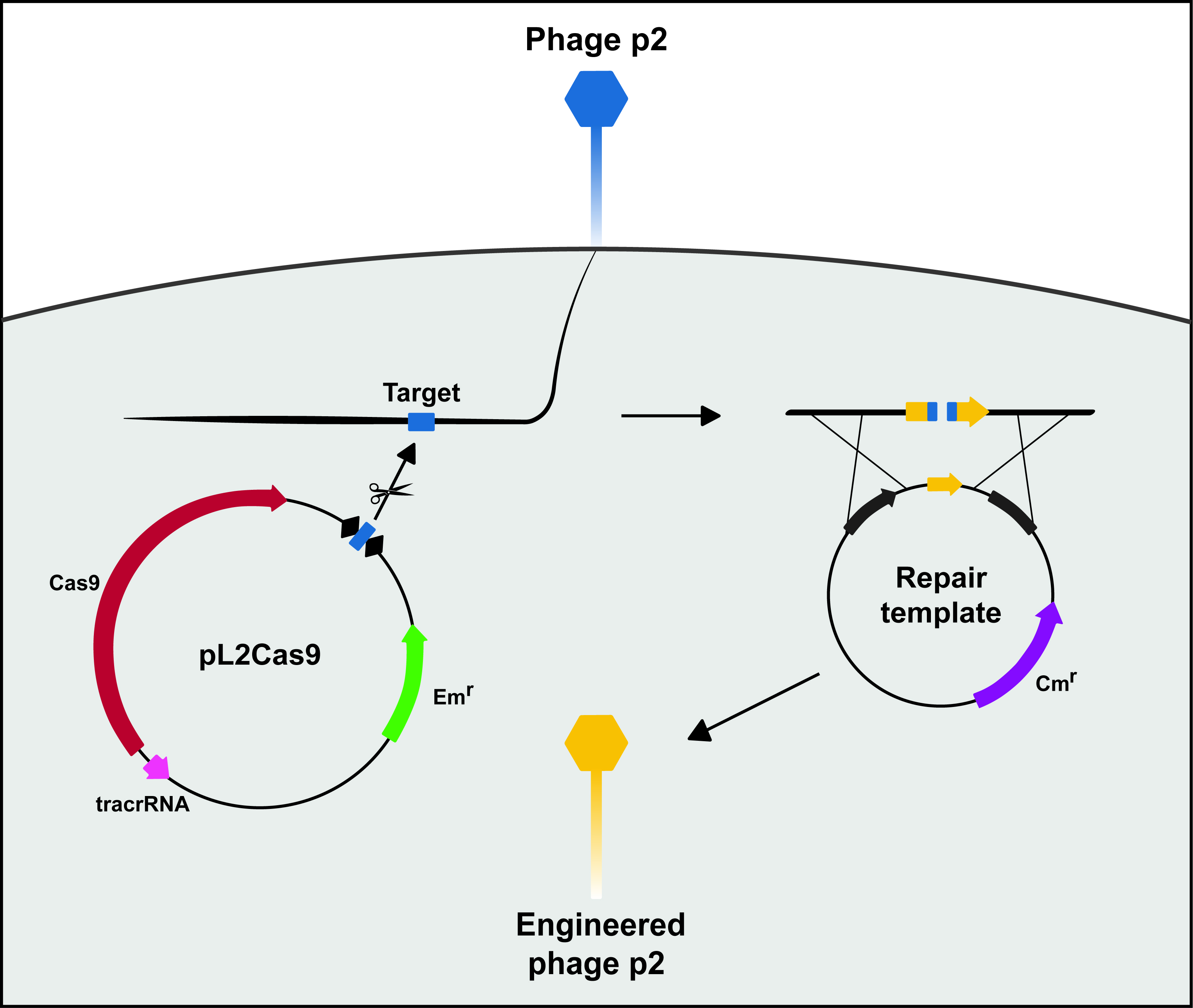
Figure 4. Targeted genome editing of phage p2 using CRISPR-Cas9. Phage p2 infects L. lactis MG1363 harboring a targeting plasmid (pL2Cas9) and a repair template. The CRISPR array is depicted as black diamonds (repeats) and a blue box (spacer). Shortly after the viral DNA enters the bacterium, the CRISPR-Cas9 complex recognizes and cleaves its target. The genomic lesion can then be repaired with a template suited for homologous recombination and harboring a desired mutation (here a deletion). Recombinant phages avoid cleavage by the CRISPR-Cas9 system as they lack the target sequence.- Phage infection
- Inoculate 10 ml of GM17 supplemented with Em 5 and Cm 5 with L. lactis MG1363 harboring the targeting plasmid and the repair template.
- Incubate overnight at 30 °C.
- Perform a ten-fold serial dilution of a phage p2 lysate. Prepare six sterile 1.5 ml micro-tubes containing 900 µl of 1x sterile phage buffer (see Recipes). Add 100 µl of undiluted phage lysate to the first micro-tube and mix by gently pipetting up and down. This is dilution 10-1. Using a new pipette tip, transfer 100 µl of the 10-1 dilution to the second micro tube (dilution 10-2), gently mix, and repeat up to dilution 10-6.
- Add 300 µl of overnight bacterial culture to 3 ml GM17 soft agar medium supplemented with CaCl2 (see Recipes) kept at 50 °C.
- Add 100 µl of undiluted phage lysate.
- Pour rapidly on top of a GM17 agar medium supplemented with CaCl2 and swirl the plate to spread the soft agar evenly.
- Repeat the last three steps for dilutions 10-2, 10-4 and 10-6.
- Incubate 24 h at 30 °C.
- Pick 3 phage plaques with a truncated sterile pipette tip and put each phage-containing agar plug into a separate sterile 1.5 ml micro-tube containing 500 µl of 1x phage buffer.
Note: Phage plaques can be kept in buffer at 4 °C for a few weeks. - Let the phages diffuse in the buffer for > 20 min at RT.
- Repeat two more rounds of infection on the same bacterial strain (Steps C1a to C1j) to purify the recombinant phages.
- Inoculate 10 ml of GM17 supplemented with Em 5 and Cm 5 with L. lactis MG1363 harboring the targeting plasmid and the repair template.
- Mutant phage analysis
- Design pairs of primers amplifying the mutated region of the phage genome and absent from the repair template.
- Use 5 µl of the phage suspension per 50 µl PCR reactions with the Taq DNA Polymerase and follow the manufacturer’s instructions. Use wild-type phage p2 as a template for positive control.
- Run the PCR products on a 2% agarose gel with the 1 Kb Plus DNA Ladder to determine their size.
Note: Gene deletions can be observed on gel by shorter PCR products obtained with recombinant phages compared to those obtained with the wild-type phage p2. - Submit PCR products for Sanger sequencing and align them with the phage genome to confirm the desired mutation in the recombinant phages.
Note: Knockout of genes that are nonessential for phage multiplication under laboratory conditions always resulted in a homogeneous population of recombinant phages. - Sequence the whole genome of the engineered phages to confirm the absence of additional mutations.
Note: To date, we have sequenced the full genome of several mutant phages and no off-target mutations were detected following CRISPR-Cas9-mediated genome engineering. Unexpected mutations could occur if genetic compensation is needed to buffer against deleterious mutations.
- Design pairs of primers amplifying the mutated region of the phage genome and absent from the repair template.
- Phage infection
Data analysis
Multiple mutants of phage p2 were generated with this methodology. We refer the readers to the original paper (Lemay et al., 2017).
Notes
- In interest of time, some of the steps from the protocol can be done concomitantly (Figure 5).
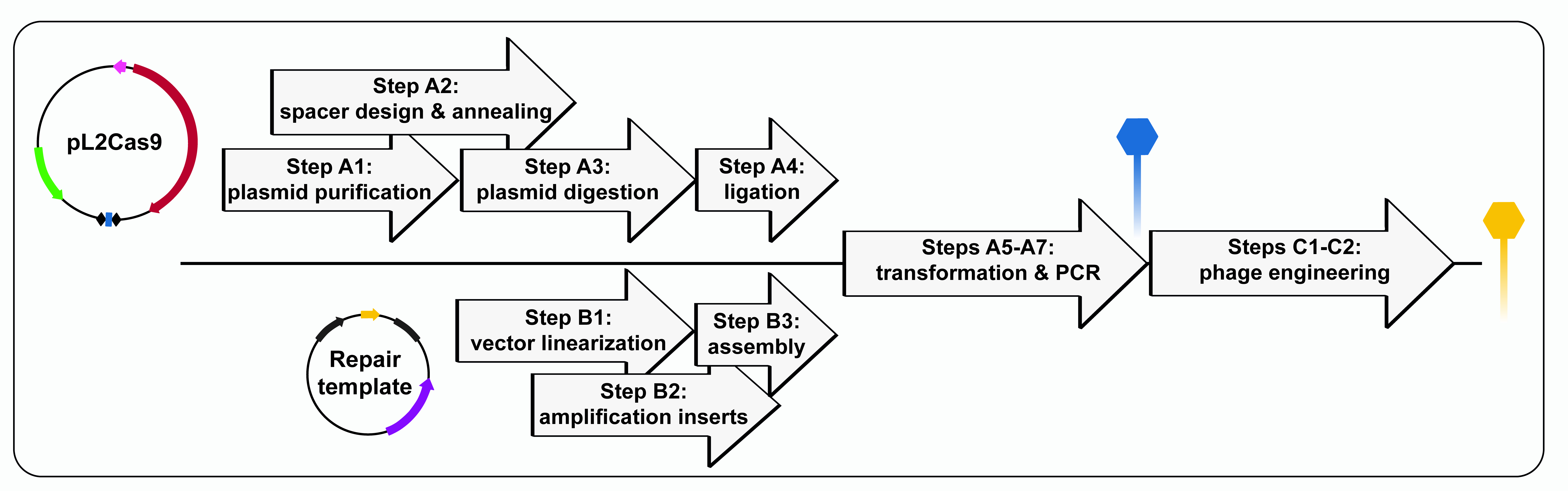
Figure 5. Flowchart illustrating the protocol to generate a mutant phage with CRISPR-Cas9 - Gene deletions are not suitable for in vivo investigation of genes essential for phage multiplication. When knockouts are not possible, non-disruptive mutations, such as point mutations or insertions, can be generated.
Recipes
- 0.8% and 2% agarose gel
- Add 0.8 g or 2 g of agarose LE to 100 ml of 1x TAE
- Heat in microwave until melted
- Cool down to ~50 °C
- Pour the appropriate volume into the electrophoresis apparatus and add the desired comb. Allow agarose to solidify, remove the comb and cover the gel with 1x TAE buffer
- Add 0.8 g or 2 g of agarose LE to 100 ml of 1x TAE
- BHI agar medium supplemented with erythromycin
Note: Important to use BHI as E. coli is more resistant to erythromycin in lysogeny broth (LB).- Dissolve 3.7 g of BHI and 1 g of agar in 75 ml of distilled water
- Complete to 100 ml with distilled water
- Sterilize by autoclaving and cool down to 50 °C
- Add 200 µl of 75 mg/ml erythromycin (150 µg/ml)
- Pour approximately 20 ml of medium per plate
- Store at 4 °C in the dark
- Dissolve 3.7 g of BHI and 1 g of agar in 75 ml of distilled water
- BHI medium supplemented with erythromycin
- Dissolve 18.5 g of BHI in 500 ml of distilled water
- Sterilize by autoclaving and cool down
- Store at room temperature (RT)
- When ready to grow the bacterial culture, add 1 ml of 75 mg/ml erythromycin (150 µg/ml)
- Dissolve 18.5 g of BHI in 500 ml of distilled water
- 2 M CaCl2
Note: CaCl2 is required for phage p2 infection.- Dissolve 29.4 g of CaCl2·2H2O in 100 ml of distilled water
- Sterilize by autoclaving and cool down
- Aliquot in smaller volumes to limit contamination of the solution
- Store at RT
- Dissolve 29.4 g of CaCl2·2H2O in 100 ml of distilled water
- 10 mg/ml chloramphenicol stock solution
Note: Chloramphenicol is required for selection of bacteria harboring pNZ123 and derivatives.- Dissolve 0.1 g chloramphenicol in 10 ml 95% ethanol
- Store at -20 °C
- Dissolve 0.1 g chloramphenicol in 10 ml 95% ethanol
- 10 mg/ml and 75 mg/ml erythromycin stock solution
Note: Erythromycin is required for selection of bacteria harboring pL2Cas9 and its derivatives.- Dissolve 0.1 g or 0.75 g erythromycin in 10 ml 95% ethanol
- Store at -20 °C
- Dissolve 0.1 g or 0.75 g erythromycin in 10 ml 95% ethanol
- 70% ethanol
- Mix 2.8 L 100 % ethanol with 1.2 L of distilled water
- Store at RT
- Mix 2.8 L 100 % ethanol with 1.2 L of distilled water
- GM17 agar medium supplemented with CaCl2 or antibiotics
- Dissolve 37.25 g of M17, 5 g glucose monohydrate and 10 g agar in 850 ml distilled water
- Complete to 1 L with distilled water
- Sterilize by autoclaving and cool down to 50 °C
- Add 5 ml of sterile 2 M CaCl2 (final concentration of 10 mM) or 0.5 ml of the appropriate 10 mg/ml antibiotic stock (final concentration of 5 µg/ml)
- Pour approximately 20 ml of medium per plate
- Store at RT or at 4 °C in the dark if supplemented with antibiotics
- Dissolve 37.25 g of M17, 5 g glucose monohydrate and 10 g agar in 850 ml distilled water
- GM17 medium
- Dissolve 37.25 g of M17 and 5 g glucose monohydrate in 1 L of distilled water
- Sterilize by autoclaving and cool down
- Store at RT
- Dissolve 37.25 g of M17 and 5 g glucose monohydrate in 1 L of distilled water
- GM17 soft agar medium supplemented with CaCl2
- Dissolve 3.73 g M17, 0.5 g glucose monohydrate and 0.75 g agar in 85 ml distilled water
- Complete to 100 ml with distilled water
- Sterilize by autoclaving and cool down to 50 °C
- Add 0.5 ml of sterile 2 M CaCl2 (final concentration of 10 mM)
- Pour 3 ml of medium in as many 13 x 100 mm sterile glass tubes as needed
- Keep the aliquots at 50 °C in a dry bath with heating block until ready to use
- Store remaining of the media at RT and melt in the microwave whenever needed
- Dissolve 3.73 g M17, 0.5 g glucose monohydrate and 0.75 g agar in 85 ml distilled water
- Glycine shock solution
- Dissolve 1 g of glycine and 17.1 g of sucrose in GM17 broth medium for a final volume of 100 ml
- Filter sterilize through 0.2 µm sterile PES syringe filter
- Store the solution at RT
- Dissolve 1 g of glycine and 17.1 g of sucrose in GM17 broth medium for a final volume of 100 ml
- 1 M MgCl2
- Dissolve 203.3 g of MgCl2·6H2O in 1 L of distilled water
- Sterilize by autoclaving
- Store at RT
- Dissolve 203.3 g of MgCl2·6H2O in 1 L of distilled water
- 10x Phage buffer
- Dissolve 58 g of NaCl and 20 g of MgSO4·7H2O in 500 ml of 1 M Tris-HCl pH 7.5
- Complete to 1 L with distilled water
- Sterilize by autoclaving
- Store at RT
- For 1x phage buffer, dilute 100 ml of 10x phage buffer with 900 ml distilled water, sterilize it by autoclaving and aliquot in smaller volumes to prevent contamination
- Dissolve 58 g of NaCl and 20 g of MgSO4·7H2O in 500 ml of 1 M Tris-HCl pH 7.5
- Recovery solution
- For a final volume of 100 ml, dissolve 17.1 g of sucrose in GM17 broth medium supplemented with 2 ml of 1 M MgCl2 and 100 µl of 2 M CaCl2
- Filter sterilize through a 0.2 µm sterile PES syringe filter
- Store the solution at 4 °C
- For a final volume of 100 ml, dissolve 17.1 g of sucrose in GM17 broth medium supplemented with 2 ml of 1 M MgCl2 and 100 µl of 2 M CaCl2
- 50x TAE buffer
- Mix 242 g Tris Base, 57.1 ml glacial acetic acid, 100 ml 0.5 M EDTA pH 8.0
- Complete to 1 L with distilled water
- Store at RT
- For 1x TAE buffer, dilute 20 ml of TAE 50x with 980 ml of distilled water
- Mix 242 g Tris Base, 57.1 ml glacial acetic acid, 100 ml 0.5 M EDTA pH 8.0
- Wash solution
- Mix 100 ml of glycerol and 171 g of sucrose (0.5 M) in 800 ml of distilled water
- Complete to 1 L with distilled water
- Sterilize by autoclaving
- Store the solution at 4 °C
- Mix 100 ml of glycerol and 171 g of sucrose (0.5 M) in 800 ml of distilled water
Acknowledgments
This protocol details the methodology used in the original paper (Lemay et al., 2017). We thank Witold Kot and Hanne Hendrix for insightful discussions. M.-L.L. is supported by a scholarship from the Natural Sciences and Engineering Research Council of Canada (NSERC). S.M. acknowledges funding from the NSERC Discovery program. S.M. holds a Tier 1 Canada Research Chair in Bacteriophages. The authors declare no competing financial interest.
References
- Bari, S. M. N., Walker, F. C., Cater, K., Aslan, B. and Hatoum-Aslan, A. (2017). Strategies for editing virulent staphylococcal phages using CRISPR-Cas10. ACS Synth Biol 6(12): 2316-2325.
- Box, A. M., McGuffie, M. J., O'Hara, B. J. and Seed, K. D. (2015). Functional analysis of bacteriophage immunity through a type I-E CRISPR-Cas system in vibrio cholerae and its application in bacteriophage genome engineering. J Bacteriol 198(3): 578-590.
- Breitbart, M. and Rohwer, F. (2005). Here a virus, there a virus, everywhere the same virus? Trends Microbiol 13:278-284.
- Deveau, H., Labrie, S. J., Chopin, M. C. and Moineau, S. (2006). Biodiversity and classification of lactococcal phages. Appl Environ Microbiol 72(6): 4338-4346.
- De Vos, W. M. (1987). Gene cloning and expression in lactic streptococci. FEMS Microbiol Lett 46: 281-295.
- Hurwitz, B. L., U'Ren, J. M. and Youens-Clark, K. (2016). Computational prospecting the great viral unknown. FEMS Microbiol Lett 363(10).
- Jiang, W., Bikard, D., Cox, D., Zhang, F. and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31(3): 233-239.
- Kiro, R., Shitrit, D. and Qimron, U. (2014). Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol 11(1): 42-44.
- Lemay, M. L., Tremblay, D. M. and Moineau, S. (2017). Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS Synth Biol 6(7): 1351-1358.
- Mahony, J., Murphy, J. and van Sinderen, D. (2012). Lactococcal 936-type phages and dairy fermentation problems: from detection to evolution and prevention. Front Microbiol 3: 335.
- Manor, M. and Qimron, U. (2017). Selection of genetically modified bacteriophages using the CRISPR-Cas system. Bio Protoc 7(15) e2431.
- Martel, B. and Moineau, S. (2014). CRISPR-Cas: an efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res 42(14): 9504-9513.
- Paez-Espino, D., Eloe-Fadrosh, E. A., Pavlopoulos, G. A., Thomas, A. D., Huntemann, M., Mikhailova, N., Rubin, E., Ivanova, N. N. and Kyrpides, N. C. (2016). Uncovering Earth’s virome. Nature 536: 425-430.
- Pires, D. P., Cleto, S., Sillankorva, S., Azeredo, J. and Lu, T. K. (2016). Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol Rev 80(3): 523-543.
- Suttle, C. A. (2005). Viruses in the sea. Nature 437(7057): 356-361.
- Tao, P., Wu, X., Tang, W. C., Zhu, J. and Rao, V. (2017). Engineering of bacteriophage T4 genome using CRISPR-Cas9. ACS Synth Biol 6(10): 1952-1961.
Article Information
Copyright
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lemay, M., Renaud, A., Rousseau, G. M. and Moineau, S. (2018). Targeted Genome Editing of Virulent Phages Using CRISPR-Cas9. Bio-protocol 8(1): e2674. DOI: 10.21769/BioProtoc.2674.
Category
Microbiology > Microbial genetics > DNA
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.