- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Genotyping-free Selection of Double Allelic Gene Edited Medaka Using Two Different Fluorescent Proteins

Published: Vol 7, Iss 24, Dec 20, 2017 DOI: 10.21769/BioProtoc.2665 Views: 7970
Reviewed by: David CisnerosAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This protocol describes a simple genotyping using two different colors of fluorescent protein genes inserted at the target locus. This method makes it possible to determine the genotype of each individual simply by observing the fluorescence later than F1 generation.
Keywords: MedakaBackground
Because the conventional genotyping of genome edited animals largely depends on PCR-based genotyping, it is necessary to extract genomic DNA. In medaka, tail fin is an accessible and regenerable tissue, so that it is frequently used for genotyping to keep fish alive. To cut tail fin, fish must be fed for a few weeks to grow big enough (ca. 1 cm in body length). Feeding and taking care of many fishes are time-consuming and laborious. Therefore, reducing the number of fish to be fed in the early stage of breeding, such as embryos and early larvae, is desired to reduce the tasks. However, it is hard to take tail fin from embryos and early larvae alive because they are too small and fragile. To overcome this issue, a simple and non-invasive method for genotyping is essential. Therefore, we developed a new method that can non-invasively determine the genotype of each individual at embryonic stage simply by observing the fluorescence. We demonstrated the benefit of this method in a gene knock-in experiment targeting the growth associated protein 43 (gap43) gene expressed in the central nervous system (CNS) at 4 days post fertilization (dpf) (Murakami et al., 2017). This gene is suitable for the gene knock-in experiment, because its spatio-temporal expression pattern has been already revealed by RT-PCR and in situ hybridization in a previous study (Fujimori et al., 2008). If the expression pattern of a targeted gene in wild-type is unknown, it needs to be analyzed by RT-PCR, or any other technique, in order to confirm the correspondence between it and that of reporter gene in gene knock-in strains.
Materials and Reagents
- Plasmid pDR274 (Addgene, catalog number: 42250 )
- pBaitD-gap43-linker-EGFP (RIKEN DNA BANK, catalog number: RDB15409 )
- Ampliscribe T7-Flash Transcription Kit (Epicentre, catalog number: ASF3257 )
- RNeasy Plus Mini Kit (QIAGEN, catalog number: 74134 )
- PCR-Kit; KOD-Plus-Neo (TOYOBO, catalog number: KOD-401 )
- Sodium hydroxide (NaOH) (NACALAI TESQUE, catalog number: 31511-05 )
- Ethylenediaminetetraacetate acid (EDTA) (NACALAI TESQUE, catalog number: 14347-21 )
- Tris-HCl (pH 8.0) (NACALAI TESQUE, catalog number: 35435-11 )
- Alkaline lysis buffer (see Recipes)
- Neutralization buffer (see Recipes)
Equipment
- Forceps (DUMONT, model: 91-3869 or equivalents)
- PCR thermal cycler (NIPPON Genetics, catalog number: TC-96GHbC )
- Incubator (NKsystem, catalog number: LH-60FL3-DT )
- Fluorescent microscope (Leica, catalog number: Leica MZ8 )
Procedure
- Search target sequences for gRNAs using a gRNA-scoring algorithm (CRISPRscan; Moreno-Mateos et al., 2015), and select the one that has less off-target sites in medaka genome.
- Obtain oligonucleotides for the selected gRNA from a supplier of choice.
- Anneal the oligonucleotides and insert the annealed fragment into gRNA expression plasmid pDR274 (Addgene Plasmid #42250).
- Transcribe from the DraI-digested plasmid using the Ampliscribe T7-Flash Transcription Kit, and purify the transcript with the RNeasy Plus Mini Kit to eliminate the template DNA without DNase treatment.
- PCR-amplify the upstream and downstream regions (homology arms: ca. 500 bp) of the genomic target site using the region-specific primers with restriction enzyme sites, which help to insert into the donor plasmid described in Step 6.
- Ligate both the homology arms, the insert fragment containing a linker, a reporter gene, and a polyA signal into the backbone plasmid containing the BaitD sequence that helps to induce gene knock-in events with high efficiency in medaka (Figure 1A).
Note: The donor plasmid containing BaitD can be obtained from RIKEN DNA BANK (pBaitD-gap43-linker-EGFP #15409). The sequence of the donor plasmid is shown as Supplemental file 1.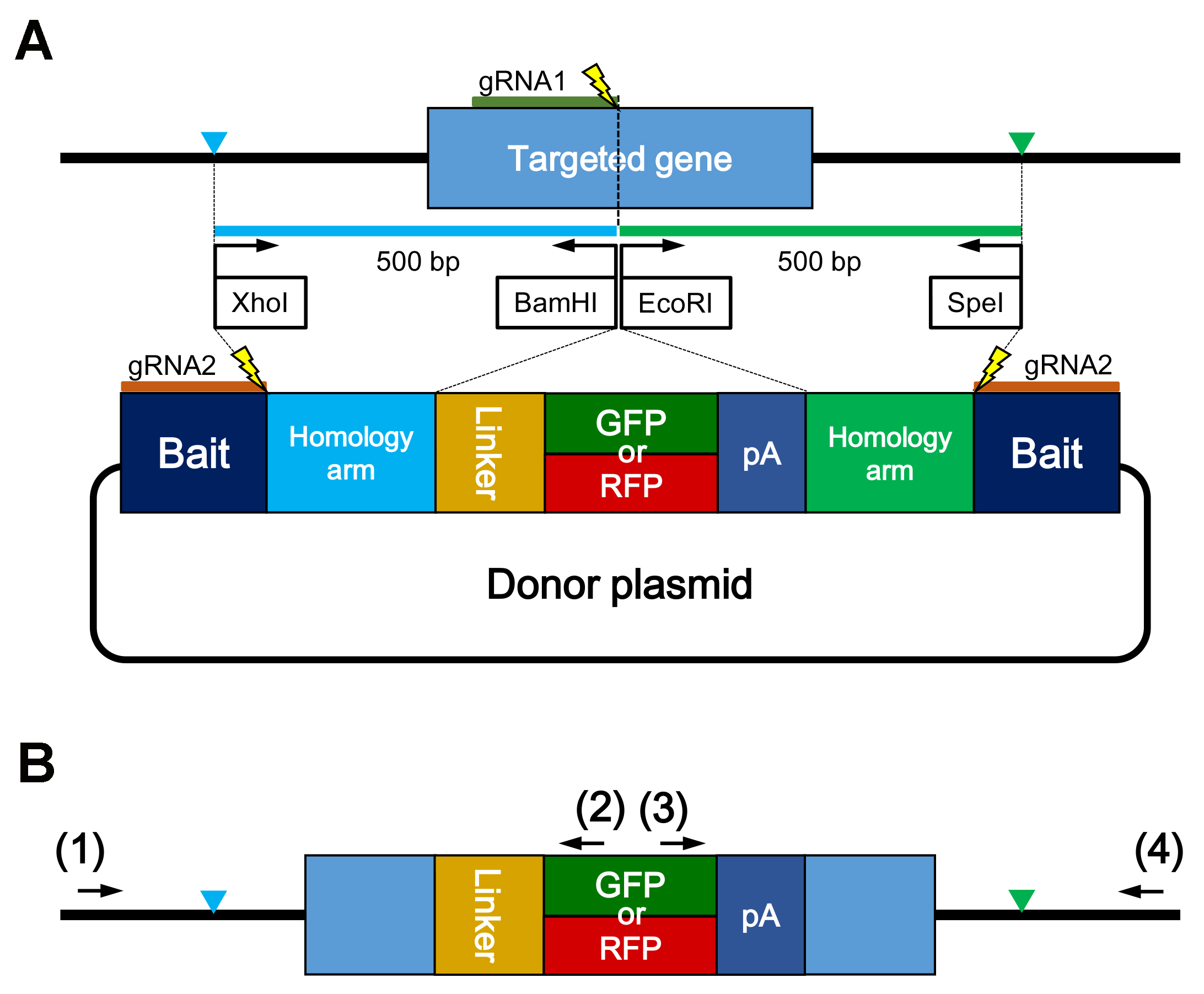
Figure 1. Schematic illustration of a gene knock-in experiment. A. Schematic design of the donor plasmid. The left (blue in (A)) and right (green in (A)) homology arms (ca. 500 bp) are amplified respectively using the region-specific primers with restriction enzyme sites (XhoI/BamHI or EcoRI/SpeI). Linker sequence stabilizes to express the reporter gene (GFP or RFP) as a fusion protein with the target gene product. Bait is a sequence to be cleaved for linearization of the donor plasmid, and enhances gene knock-in events in medaka. Thunder marks show the cleavage sites on the genome and the donor plasmid by Cas9 and gRNA1 or gRNA2, respectively. B. Schematic illustration showing precise integration of the insert gene into the genomic target site. The reporter gene (GFP or RFP) is integrated into exon and in-frame. Blue and green triangles show the junctions between the genomic target site and the fragment of donor plasmid. Primer pairs labeled (1)/(2) or (3)/(4) can be used for amplifying the upstream or downstream junction region. To confirm the precise integration into the genomic target site, primer (1) and (4) are designed on the outside of the junctions. - Inject the following solution into embryos; 100 ng/µl of Cas9 RNA, 50 ng/µl of sgRNAs for cleaving the genomic target site and the donor plasmid, and 2.5 ng/µl of each donor plasmid.
Note: Please refer to ‘Medaka-microinjection’ for the details of microinjection protocol. - Raise the embryos expressing each reporter gene to adult for about 2 months.
- Mate with wild type counterparts. (First mating)
- Extract genomic DNA from the resultant F1 embryos expressing each reporter gene, according to the following protocol:
- Put embryos in 25 µl of alkaline lysis buffer (see Recipes).
- Incubate at 95 °C for 15 min after breaking the egg envelope with forceps.
- Neutralize with 25 µl of neutralized buffer (see Recipes).
- Put embryos in 25 µl of alkaline lysis buffer (see Recipes).
- PCR-amplify the junction regions of the target site on the host genome and the introduced gene using F1 genomic DNA (Figure 1B).
Note: The PCR condition is performed as described in the protocol of KOD-Plus-Neo (Toyobo). - Perform the sequence analysis of the resultant PCR amplicons, to confirm the precise integration into the genomic target site.
- After identifying F0 founders with the desired mutation, mate with wild type counterparts again. (Second mating)
Note: First mating is for the identification of F0 founders harboring the insert gene in germ cells, while second mating is for the establishment of gene knock-in strains in F1 generation. - Collect F1 embryos expressing each reporter gene, and raise to fish with a length > 1 cm.
- Cut off tail fin from each F1 fish, and perform the genome extraction and the sequence analysis described as above.
- After confirming the precise integration into the genomic target site in each F1 fish by the sequence analysis, mate F1 fish harboring different color reporter genes with each other.
- Observe the fluorescence color of the resultant F2 embryos, to specify the individuals integrated the insert gene on the target site.
Note: The resultant F2 embryos are genotyped alive by fluorescence color: wild type without fluorescence, monoallelic mutants for integration with one fluorescence, and biallelic mutants for integration with both fluorescence.
Recipes
- Alkaline lysis buffer
25 mM NaOH
0.2 mM EDTA - Neutralization buffer
40 mM Tris-HCl (pH 8.0)
Acknowledgments
This protocol was adapted from our previous works (Murakami et al., 2017). The work was partially supported by a Grant-in-Aid for Scientific Research (A) 15H02540 (MK) and a Grant-in-Aid for JSPS fellows 13J01682 (SA). The funders had no role in the design of the study and collection, analysis, or interpretation of data or in the writing of the manuscript. The authors declare no conflicts of interest or competing interests with this manuscript.
References
- Fujimori, K. E., Kawasaki, T., Deguchi, T. and Yuba, S. (2008). Characterization of a nervous system-specific promoter for growth-associated protein 43 gene in Medaka (Oryzias latipes). Brain Res 1245: 1-15.
- Moreno-Mateos, M. A., Vejnar, C. E., Beaudoin, J. D., Fernandez, J. P., Mis, E. K., Khokha, M. K. and Giraldez, A. J. (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 12(10): 982-988.
- Murakami, Y., Ansai, S., Yonemura, A. and Kinoshita, M. (2017). An efficient system for homology-dependent targeted gene integration in medaka (Oryzias latipes). Zoological Lett 3: 10.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Murakami, Y., Ansai, S., Yonemura, A. and Kinoshita, M. (2017). Genotyping-free Selection of Double Allelic Gene Edited Medaka Using Two Different Fluorescent Proteins. Bio-protocol 7(24): e2665. DOI: 10.21769/BioProtoc.2665.
Category
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.