- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Using Light and Electron Microscopy to Estimate Structural Variation in Thylakoid Membranes
Published: Vol 7, Iss 23, Dec 5, 2017 DOI: 10.21769/BioProtoc.2639 Views: 10035
Reviewed by: Scott A M McAdamAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The shapes of chloroplasts and the architectures of internal thylakoid membranes are altered by growth and environmental changes (Lichtenthaler et al., 1981; Kutik, 1985; Terashima and Hikosaka, 1995). These morphological alterations proceed via transitional intermediates, during which dynamic and heterogeneous thylakoid membranes are observed in cells (Nozue et al., 2017). Light microscopy is useful for the detection of morphological differences in chloroplasts. The thylakoid architecture of such morphologically variable chloroplasts is confirmed by transmission electron microscopy (TEM). The method of monitoring structural variation by light microscopy in combination with electron microscopy is described.
Keywords: Light microscopyBackground
Functional coupling of ultra-structural morphology to photosynthesis and metabolic pathways in thylakoid membranes has been suggested (Oswald et al., 2001). This is supported by the fact that thylakoid membranes are heterogeneous during leaf maturation and during the transition from the vegetative to the flowering growth phase. There is a certain time lag associated with morphological alteration (Nozue et al., 2017). Rearrangement of thylakoid membranes occurs in parallel with changes in the shapes of chloroplasts, which go from having a typically elongated lenticular appearance to a swollen appearance that is identifiable by light microscopy.
Part I. Light microscopy
Materials and Reagents
- 1.5-ml microtubes (Ina-Optika, Bio Bik, catalog number: RC-0150 )
- Mineral wool (Nippon Rockwool, Yasaihana-block50)
- Razor blades (Feather Safety Razor, catalog number: FAS-10 )
- Double-edged razor blades (Feather Safety Razor, catalog number: FA-10 )
- Cell culture dish (Violamo, AS ONE, catalog number: 2-8590-02 )
- Glass microscope slide (Matsunami Glass, catalog number: S1214 )
- Glass microscope cover slip (Trophy, Matsunami Glass, 18 m/m)
- Arabidopsis thaliana ecotype Columbia
- Hydroponic solution (OAT Agrio, OAT House Fertilizer)
- 25% glutaraldehyde (NISSHIN EM, catalog number: 3052 )
- Agar, powder (Wako Pure Chemical Industries, catalog number: 016-11875 )
- Fixing solution (see Recipes)
- 5% agar (see Recipes)
Equipment
- Extra-high-pressure mercury lamp (Nikon Instruments, model: INTENSILIGHT C-HGFI )
- Fluorescence microscope (Nikon Instruments, model: ECLIPSE 80i )
- Stainless-steel pincette (Top well) (AS ONE, catalog number: 5-1076-01 )
- Laboratory glass bottle, screw cap, 100 ml (SCHOTT, DWK Life Sciences, Duran, catalog number: 21 805 24 04 )
- High-pressure steam sterilizer (TOMY SEIKO, model: LSX-500 )
- Microwave (Nisshin EM, model: MWF-2 )
- Refrigerator (Panasonic, model: NR-B52T2-H )
- Micro-slicer (DOSAKA EM, model: DTK-Zero1 )
- Objective lens 20x (Nikon Instruments, model: Plan Fluor 20x/0.50, catalog number: MRH00201 )
- Objective lens 40x (Nikon Instruments, model: Plan Fluor 40x/0.75, catalog number: MRH00401 )
- Objective lens 100x (Nikon Instruments, model: Plan Fluor 100x/1.30 Oil, catalog number: MRH01902 )
- CCD camera (Nikon Instruments, model: DS-Ri1 )
- Condenser (Nikon Instruments, models: D-CUD , D-CUO DIC )
- Autoclave
Software
1.Imaging software (Nikon Instruments, model: NIS-Elements)
Procedure
- Growth conditions and sampling of leaves
- Arabidopsis thaliana seeds (ecotype Columbia) are germinated and cultured on mineral wool using a hydroponic solution (EC: electrical conductivity, 1.2 ds/m, pH 7.0).
- Fluorescent lighting tubes and white LEDs consisting of blue (450 nm), green (545 nm) and red (660 nm) lights are used as light sources for growth.
- The growth condition is controlled at a temperature of 22-24 °C and 50-70% humidity, and the PPFD on the growth area is adjusted to 70-80 μmol m-2 sec-1 with a 12-h light photoperiod.
- Leaves are harvested from the plants at 1-3 h of the light period after a 12-h dark period.
- Arabidopsis thaliana seeds (ecotype Columbia) are germinated and cultured on mineral wool using a hydroponic solution (EC: electrical conductivity, 1.2 ds/m, pH 7.0).
- Preparation of leaf discs
- A fresh leaf is placed on a plastic mat of semisoft material. Different sizes of leaf blocks–3 x 5 mm for light microscopy and 1-2 x 3-5 mm for electron microscopy–are cut out with a razor-sharp blade. The razor blade, in vertical contact with the leaf, is carefully drawn so as not to damage cells by excessive compression. Major veins should not be included in the leaf blocks selected (Figure 1).
Note: A slightly larger size makes light-microscopic sectioning easier, and a size of 2 mm or less makes electron-microscopic sectioning easier. - The excised leaf tissues are fixed in fixing solution (see Recipes) (a 2% glutaraldehyde-phosphate buffer solution [pH 7.0]) for a minimum of 3 h at room temperature. For convenience, the leaf tissues in fixing solution can be stored in a refrigerator overnight.
Note: To avoid tissue damage by grasping with tweezers, the leaf discs are transferred into the fixing solution by attachment to wet tweezers (Figure 1).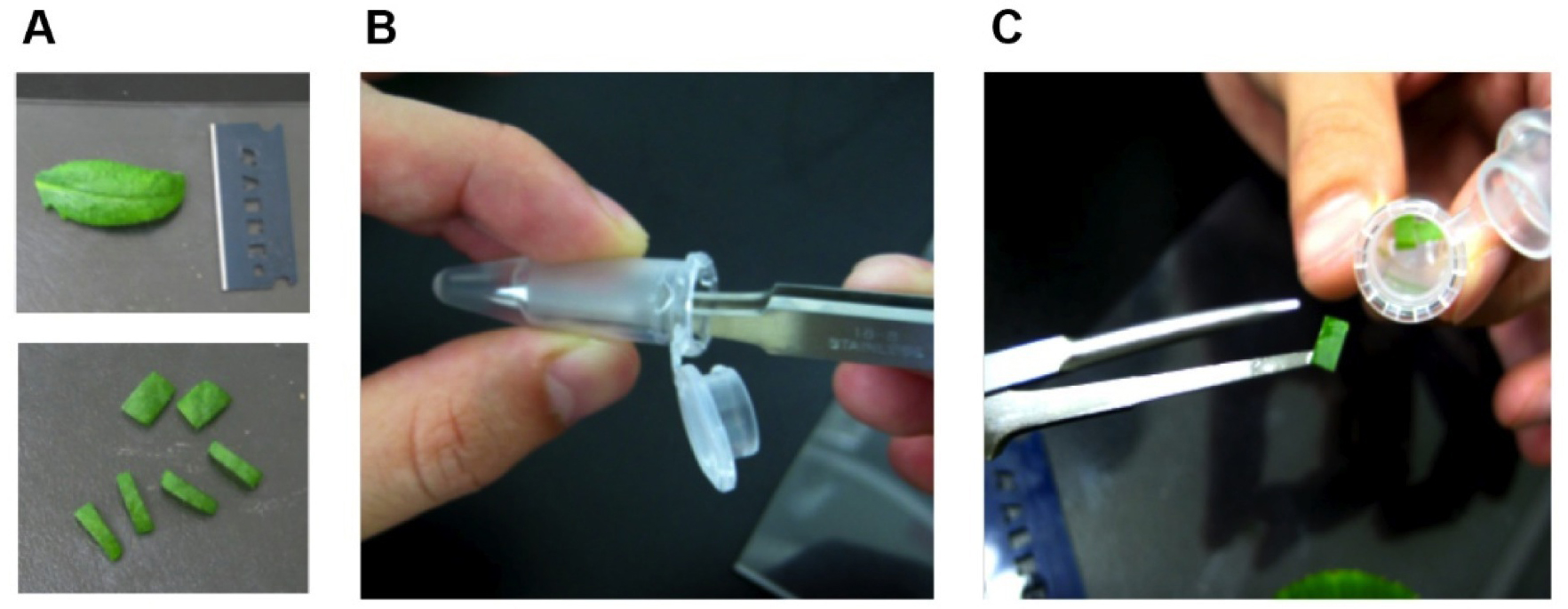
Figure 1. Preparation of Arabidopsis leaf segments for glutaraldehyde fixation. Cut out mesophyll tissues including chloroplasts (A). Wet tips of pincette (B) and carry tissue into the fixing solution (C).
- A fresh leaf is placed on a plastic mat of semisoft material. Different sizes of leaf blocks–3 x 5 mm for light microscopy and 1-2 x 3-5 mm for electron microscopy–are cut out with a razor-sharp blade. The razor blade, in vertical contact with the leaf, is carefully drawn so as not to damage cells by excessive compression. Major veins should not be included in the leaf blocks selected (Figure 1).
- Embedding and sectioning for light-microscopic observation
- The 5% solid agar (once dissolved and autoclaved according to the recipe, the agar is stored as a stock of solid agar, see Recipes) is heated until boiling in a microwave, repeating 2-3 times until all the solid agar is completely dissolved. The agar solution is allowed to cool to 50-60 °C and then poured into plastic Petri dishes approximately 50 mm high.
Note: Preparation of an agar block with similar hardness to leaf mesophyll cells is important to make tissue sections with natural, round, cellular shapes. - Tissue blocks are embedded to keep the leaf cross-sections upright.
- After cooling the agar containing leaf sections in a refrigerator at 4 °C, an agar block with the embedded leaf tissue is cut out (Figure 2).
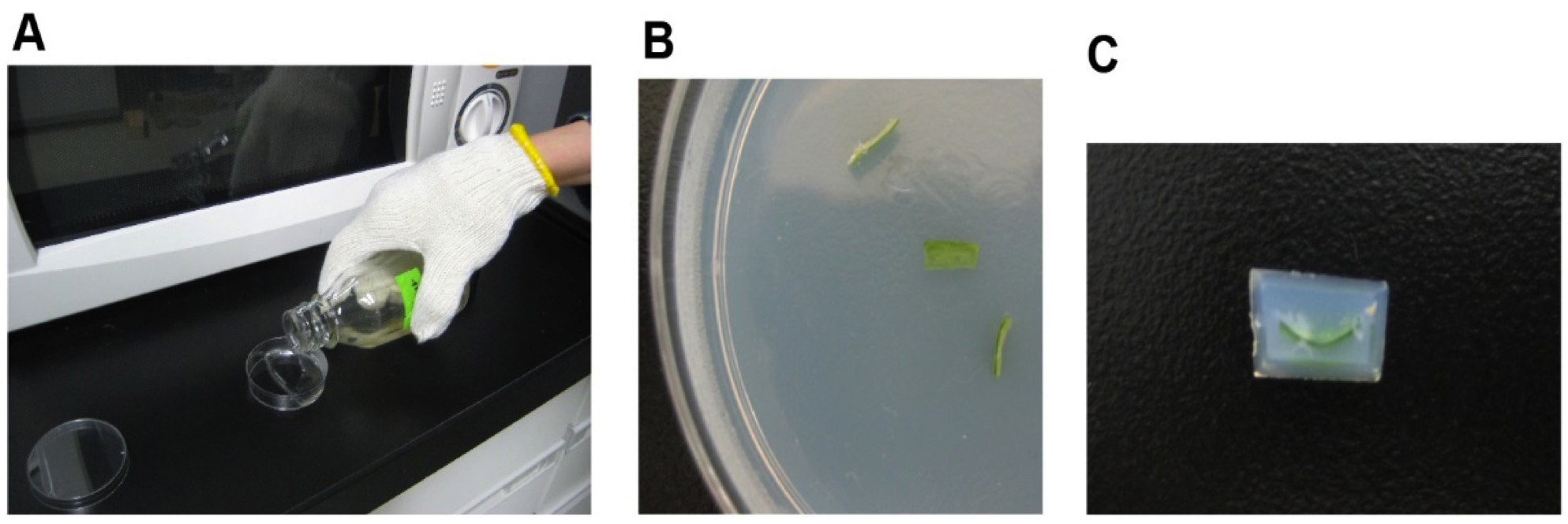
Figure 2. Preparation of agar block with embedded leaf. Pour melted agar into a Petri dish (A) and embed tissues in agar (B). Cut out agar block (C) for slicing. - The block is placed on the stage of the micro-slicer, and water is poured into the vessel. Half of a double-edged razor blade is fitted to the prescribed position to be able to precisely cut the block to a minimum thickness of 10 μm. A 100-μm-thick leaf section is scooped onto a glass slide and then subjected to light-microscopic observation (Figure 3).

Figure 3. Slicing of leaf traverse section. Place a sample block on the stage (A) of the micro-slicer (B). Scoop a leaf section (C) onto a glass slide (D).
- The 5% solid agar (once dissolved and autoclaved according to the recipe, the agar is stored as a stock of solid agar, see Recipes) is heated until boiling in a microwave, repeating 2-3 times until all the solid agar is completely dissolved. The agar solution is allowed to cool to 50-60 °C and then poured into plastic Petri dishes approximately 50 mm high.
- Microscopic observation
- A drop of water is placed on a glass microscope slide. A floating tissue slice is scooped up with the tip of a stainless-steel pincette and allowed to float on the water droplet on a glass slide. A glass microscope cover slip is placed on the sample to eliminate air bubbles (Figure 3).
- Observations are first made with the 20x objective lens, which enables whole cross-sectional observation from the surface to the underside of the leaf. The chloroplasts distributed in each cell are viewed as a whole. To verify the shape of each chloroplast, the image is magnified to 40x and 100x, focusing on a chloroplast attached to the cell membrane. Differential interference contrast (DIC) imaging makes it easier to detect the differences in membrane structures (Figure 4).

Figure 4. Light microscopic observation of Arabidopsis chloroplasts. Chloroplasts lined the inner cell membrane of a young leaf in the vegetative growth phase (A) and of a mature leaf in the floral phase (B). The Arabidopsis plants were grown under controlled conditions of a 12-h photoperiod at 22 °C. Leaves with sizes of 1 x 2 cm (A) and 2 x 5 cm (B) were subjected to microscopic observation. Scale bars represent 10 μm.
- A drop of water is placed on a glass microscope slide. A floating tissue slice is scooped up with the tip of a stainless-steel pincette and allowed to float on the water droplet on a glass slide. A glass microscope cover slip is placed on the sample to eliminate air bubbles (Figure 3).
Data analysis
Starting with an overview of chloroplasts using a wide microscopic field, a variety of different shapes were observed, which is caused by the natural placement of chloroplasts at either surface of the leaf (which reflects the different lighting conditions between the two surfaces) and the direction of observation of the chloroplasts (which reflects the non-uniformity of shape). To detect morphological differences in the outer shapes of chloroplasts, the observational analysis was carried out by focusing on chloroplasts attached to inner cell membrane like a ring (Figure 4). Chloroplasts released from membranes or attached to the membrane on the reverse side of the sample are often seen as round shapes lacking thylakoid structures.
Notes
The shapes of chloroplasts, whether lens-like or not, can be indicators of thylakoid structure. Sampling of leaves within several hours of growth in light is important in order to observe lens-like outlines of chloroplasts. Large starch granules accumulate in chloroplasts during long lighting periods due to deformation.
Recipes
- Fixing solution
10 ml of 25% glutaraldehyde
115 ml of 50 mM K-phosphate buffer, pH 7.0 - 5% agar
3.5 g of agar powder
Add 70 ml of distilled water
Dissolve agar completely by autoclaving at 105 °C for 5 min
Agitate gently to achieve a homogeneous liquid state
Cool down to 50-60 °C for use
Store at room temperature
Part II. Transmission electron microscopy
Materials and Reagents
- 1.5-ml microtubes (Ina-Optika, Bio Bik, catalog number: RC-0150 )
- Disposable Pasteur pipets (Fisher Scientific, catalog numbers: 13-678-30 , 13-678-20C )
- Filter paper (ADVANTEC No.1) (AS ONE, catalog number: 00011090 )
- Bottle for collecting waste solution
- Reagent bottle, narrow mouth, amber glass (Spectrum Chemical, Wheaton, catalog number: 989-11698 )
- Disposable plastic syringe (Terumo, catalog numbers: SS-01T , SS-20ESZ , SS-50ESZ )
- Disposable plastic beaker (Nisshin EM, catalog number: 405 )
- Silicon embedding plate (Nisshin EM, catalog number: 420-1 )
- Glass sheet for glass knife (Nisshin EM, catalog number: 5402 , 8-mm thick)
- 150 Cu Grid (Nisshin EM, Veco grids)
- Parafilm M (Pechiney Plastic Packaging, AS ONE, catalog number: 6-711-01 )
- Arabidopsis thaliana ecotype Columbia
- 99.5% ethanol (NACALAI TESQUE, catalog number: 14713-53 )
- Propylene oxide (Nisshin EM, catalog number: 311 )
- Toluidine blue (Sigma-Aldrich, catalog number: 198161 )
- Lead stain solution (Sigma-Aldrich, catalog number: 203580 )
- Glutaraldehyde, 25% aqueous ampule (Nisshin EM, catalog number: 3020-2 )
- Osmic acid solution (Nisshin EM, catalog number: 3028 )
- Potassium dihydrogen phosphate (KH2PO4) (KANTO KAGAKU, catalog number: 32379-23 )
- Dipotassium hydrogen phosphate (K2HPO4) (KANTO KAGAKU, catalog number: 32378-23 )
- Epon 812 resin embedding kit (TAAB Laboratories 500 g) (Nisshin EM, catalog number: 342-2 )
- Uranyl acetate (Merck KGaA, Darmstadt (Discontinued), Replacement: Polysciences, 400 Valley Road Warrington, PA 18976) (Polysciences, catalog number: 21447-25 )
- Molecular sieves 3A (NACALAI TESQUE, catalog number: 04176-55 )
- 2% glutaraldehyde (see Recipes)
- 2% osmium solution (see Recipes)
- 0.1 M potassium phosphate buffer (pH 7.0) (see Recipes)
- Washing buffer (see Recipes)
- 100% ethanol (see Recipes)
Equipment
- Storage bottle (sample tube bottle No.3, 10 ml) (AS one, catalog number: 9-851-05 )
- Program-controlled incubator (AS ONE, model: ICV-450P )
- Ultra-microtome (Leica, model: Leica EM UC7i )
- Tweezers (PEER-VIGOR Type 7) (Nisshin EM, catalog number: 2246 )
- Hotplate (AS ONE, model: HP-A1914B )
- Transmission electron microscope (JEOL, model: JME-1400 )
- Diamond knife (Biel Switzerland, DiATOME 2501)
- Glass knife (Leica glass Knife Strip, Nisshin EM, catalog number: 542 )
- Vacuum evaporator (JEOL, model: JEE-420T )
- CCD camera (Gatan, model: ORIUS® SC 1000, Model 832 TEM )
- Invitro shaker (Taitec, model: wave-S1 slim )
- Water purification system (Sartorius, model: Arium® 611DI )
- Automatic lab mixer (AS ONE, model: HM-10H )
- Aspirator (AS ONE, model: AS-01 )
- Rotator (Sakura, model: VEM-16 )
- Grid stick kit (Micro Star Corporation)
Procedure
Note: This part is modified from Desmond, 1965.
- Double fixation
- Pre-fixation: 4-6 pieces of 1-2 x 2-4 mm fresh leaf tissues are put into a 1.5-ml microtube filled with 0.5-1.0 ml of 2% glutaraldehyde-0.1 M phosphate buffer solution (see Recipes) for 1-2 h, and then, the tubes are placed in a vacuum desiccator for 1-2 h.
- A visual check is performed to ensure that the leaf discs sink to the bottom.
- After pre-fixation, the fixing solution is removed with a Pasteur pipet and transferred to a waste storage bottle.
- Using a new Pasteur pipet, 1-2 ml of K-phosphate washing buffer (see Recipes) is added into the microtube with the leaf discs.
- The leaves are washed for 10 min while shaking the tube gently by hand. While shaking, care is taken to prevent the leaves from getting dry. This washing is repeated 6 times.
- Post-fixation: the tissues are fixed with 2% osmium tetroxide (OsO4) (see Recipes) at 4 °C for 1 h.
- Pre-fixation: 4-6 pieces of 1-2 x 2-4 mm fresh leaf tissues are put into a 1.5-ml microtube filled with 0.5-1.0 ml of 2% glutaraldehyde-0.1 M phosphate buffer solution (see Recipes) for 1-2 h, and then, the tubes are placed in a vacuum desiccator for 1-2 h.
- Dehydration
- The tissues are dehydrated with a series of solutions containing increasing concentrations of ethanol–50%, 60%, 70%, 80%, 90% and 95%–with rotation for 15 to 30 min.
- Subsequently, the tissues are dehydrated with 100% ethanol (see Recipes) for 60 min, 3 times, with shaking.
- The tissues are dehydrated with a series of solutions containing increasing concentrations of ethanol–50%, 60%, 70%, 80%, 90% and 95%–with rotation for 15 to 30 min.
- Substitution
- The tissues are permeated twice with 100% propylene oxide for 15 min.
- To increase the concentration of epoxy resin gradually, the tissue is infiltrated into 7:3, 5:5, and 3:7 mixed solutions of propylene oxide and epoxy resin, for 60 min each time.
- Finally, the tissue is infiltrated into the pure epoxy resin for 60 min.
- The tissues are permeated twice with 100% propylene oxide for 15 min.
- Resin embedding and hardening
- The silicon embedding plate is filled with pure epoxy resin and the tissues are placed in the resin.
- The blocks of epoxy resin are hardened by heat in the program-controlled incubator.
Note: The heat program starts at 35 °C for 8 h, rises to 45 °C for 8 h, and then heats at 60 °C for 48 h.
- The silicon embedding plate is filled with pure epoxy resin and the tissues are placed in the resin.
- Preparation of specimen for light microscopy
- The epoxy resin block is attached to an ultra-microtome and cut with a glass knife to obtain 3-μm thick sections.
- Using tweezers, a section is placed on a glass slide where a drop of distilled water has been placed.
- The glass slide is dried at 60-70 °C on a hotplate.
- After drying, the tissue is stained a with 1% toluidine blue staining solution while warming for 60 sec.
- After washing with water and drying, desired portions of tissues sample are selected by optical microscopy.
- The epoxy resin block is attached to an ultra-microtome and cut with a glass knife to obtain 3-μm thick sections.
- Trimming
The sample is trimmed again under a stereoscopic microscope to an ultrathin section size of approximately 0.5 x 0.5 mm, which includes the site to be observed.
The ultrathin section size of the specimen to be observed by electron microscopy is smaller than the that of the optical microscopy specimen, and the section size depends on the blade width of the diamond knife. The minimum width size of optical microscopy specimens is 25 mm, while the maximum size of electron microscopy specimens is 3 mm. Therefore, it is necessary to select and re-trim the site for observation from the optical microscope specimen to use it for electron microscopy. - Ultrathin section preparation (Williams and Carter, 2009)
- The re-trimmed epoxy resin block is attached to the ultra-microtome, and the knife is changed from a glass knife to a diamond one.
- The parallax is readjusted because of the change in parallelism between the block and the knife.
- After adjusting the parallelism, the re-trimmed epoxy resin block is sliced with the diamond knife. The feed volume and cutting speed of the ultra-microtome are used to adjust the thickness.
- Ultrathin sections made by the diamond knife are collected on water. The thickness of the ultrathin section is set to 100 nm.
- The thin sections floating on the water in the diamond knife boat are transferred to the grid.
Extra water is wiped off with filter paper, and the grids are placed into a Petri dish.
- The re-trimmed epoxy resin block is attached to the ultra-microtome, and the knife is changed from a glass knife to a diamond one.
- Staining for electron microscopy
- The ultrathin section grid is attached to the contact tape of a stick stain holder. The stick stain holder is immersed for 10 min in a centrifuge tube filled with 1% uranyl acetate solution.
- The stick stain holder is placed in a beaker filled with distilled water.
- The grids on the stick stain holder are washed in 3 changes of water.
- After washing, the stick stain holder is wiped with filter paper.
- The stick stain holder is immersed into a lead citrate solution for 5 min.
- The stick stain holder is placed in a beaker filled with distilled water.
- After washing, the stick stain holder is wiped with filter paper.
- The ultrathin section grid is attached to the contact tape of a stick stain holder. The stick stain holder is immersed for 10 min in a centrifuge tube filled with 1% uranyl acetate solution.
- Carbon coating
- Carbon coating has the effect of preventing damage caused by electron beam irradiation to ultrathin sections during electron microscopic observation.
- The sample is placed in the carbon vacuum evaporator.
- The carbon layer thickness is set to 20 nm.
- Carbon coating has the effect of preventing damage caused by electron beam irradiation to ultrathin sections during electron microscopic observation.
Note: Procedure A-I is summarized in Figure 5.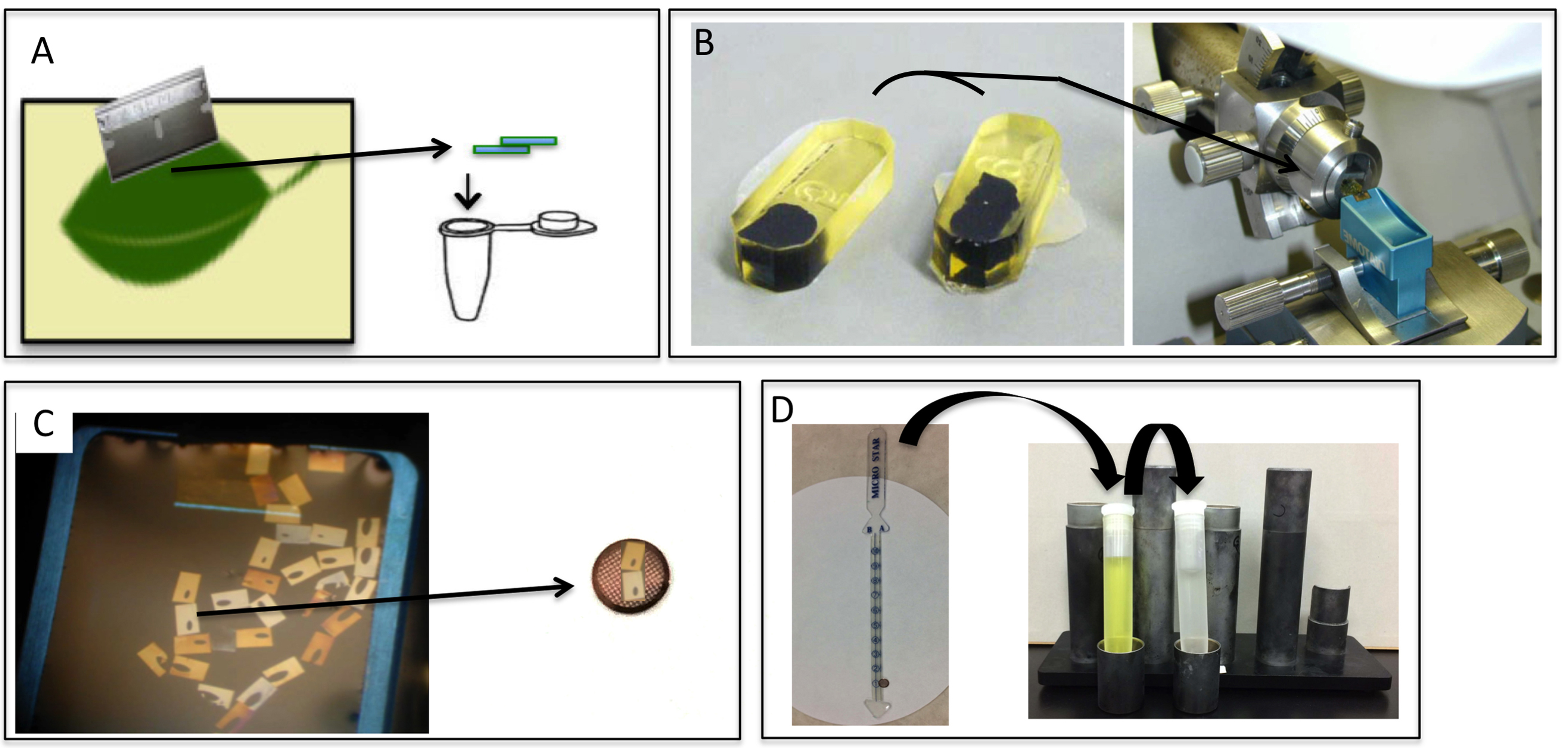
Figure 5. Ultrathin section preparation and staining. Fresh 1-2 x 2-4 mm leaf tissues are cut to a size of approximately 1 mm with a razor and placed in a 1.5-ml microtube (A), epoxy resin blocks are trimmed in preparation for ultrathin sectioning and then sectioned on an ultra-microtome (B). Ultrathin sections from the diamond knife are collected on water. The thin, sliced sections floating on the water in the diamond knife boat are picked up and transferred to the grid (C). Electron microscopic staining: The grid with the ultrathin sections grid is attached to the contact tape of a stick stain holder, which is then immersed for 10 min into a centrifuge tube filled with 1% uranyl acetate solution and for 5 min into lead citrate solution (D).
- Transmission electron microscopic observations
- For electron microscopy, low- and high-magnification observations are carried out using JEM 1400.
- Observation data are recorded and stored by the Gatan CCD camera and Digital Microscopy software (Figure 6).

Figure 6. TEM observations distinguishing the fine structures of thylakoid membranes. Stroma-grana thylakoid membrane of a young leaf (A) and isolated grana thylakoid membrane of a mature leaf (B). Scale bars represent 10 μm.
- For electron microscopy, low- and high-magnification observations are carried out using JEM 1400.
Data analysis
The TEM imaging data were in agreement with the light microscopic images. The collection of data from more than 5 repetitions of independent experiments showed similar changes in chloroplasts shape and thylakoid structure. Architectural differences in thylakoid membranes are shown as measurements of thicknesses of grana layers, lengths of stroma lamella, and angles of curvature (Nozue et al., 2017).
Recipes
- 2% glutaraldehyde
1 ml of 25% glutaraldehyde
11.5 ml of 0.1 M K-phosphate buffer - 2% osmium solution
2 ml of 4% osmic acid solution
2 ml of 0.1 M K-phosphate buffer
Note: The osmium solution is stored in a brown bottle for protection from light, at 4 °C.The stopper is sealed with Parafilm. - 0.1 M potassium phosphate buffer (pH 7.0)
A: 0.2 M KH2PO4 (27.2 g/1,000 ml H2O)
B: 0.2 M K2HPO4 (45.6 g/1,000 ml H2O)
Mix 39 ml A and 61 ml B
Adjust pH to 7.0 by adding solution A or B - Washing buffer
0.05 M K-phosphate buffer, pH 7.0, is prepared from refrigerated 0.1 M K-phosphate buffer stocks - 100% ethanol
Put an appropriate amount of molecular sieve in a 500-ml reagent bottle and then add 95% ethanol. The initially turbid ethanol solution becomes clear over time. Use the supernatant liquid - Epon 812 resin embedding kit
For 50 ml of Epon 812 resin:
23.2 ml of Epon 812
12.6 ml of DDSA (dodecenylsuccinic anhydride)
14.2 ml of MNA (Methyl nadic anhydride)
For solidification of resin, add 0.75 ml of DMP30 (2,4,6-Tris(dimethylaminomethyl) phenol)
Gently agitate the Epon resin for 30 min on a shaker
Notes:- Plastic syringes are used to measure the reagent volume and collected in a plastic centrifuge tube (50 ml).
- All Epon reagents are stored at 4 °C. The reagents warmed to room temperature (22-24 °C under temperature-controlled conditions) are used for resin preparation. Application conditions vary based on temperature, humidity and storage conditions.
- Plastic syringes are used to measure the reagent volume and collected in a plastic centrifuge tube (50 ml).
Acknowledgments
Prof. Y Kaneko provided instruction on the fundamentals of the resin embedding method for plant samples. We also thank Ms. M. Kondo for technical advice on TEM analysis. We thank Dr. K. Fukamoto for her assistance in preparing the manuscripts.
References
- Desmond, H. K. (1965). Techniques for electron Microscopy. 2nd edition. Blackwell Scientific Publications.
- Kutik, J. (1985). Photosynthesis during leaf development. In: Sestak, Z. (Ed.). Chloroplast Development. Dr W. Junk Publishers pp: 51-75.
- Lichtenthaler, H. K., Buschmann, C., Doll, M., Fietz, H. J., Bach, T., Kozel, U., Meier, D. and Rahmsdorf, U. (1981). Photosynthetic activity, chloroplast ultrastructure, and leaf characteristics of high-light and low-light plants and of sun and shade leaves. Photosynth Res 2(2): 115-141.
- Nozue, H., Oono, K., Ichikawa, Y., Tanimura, S., Shirai, K., Sonoike, K., Nozue, M. and Hayashida, N. (2017). Significance of structural variation in thylakoid membranes in maintaining functional photosystems during reproductive growth. Physiol Plant 160(1): 111-123.
- Oswald, O., Martin, T., Dominy, P. J. and Graham, I. A. (2001). Plastid redox state and sugars: interactive regulators of nuclear-encoded photosynthetic gene expression. Proc Natl Acad Sci U S A 98(4): 2047-2052.
- Terashima, I. and Hikosaka, K. (1995). Comparative ecophysiology of leaf and canopy photosynthesis. Plant Cell Environ 18: 1111-1128.
- Williams, D. B. and Carter, C. B. (2009). Transmission electron microscopy: A textbook for materials science. Springer.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Nozue, H. and Kametani, K. (2017). Using Light and Electron Microscopy to Estimate Structural Variation in Thylakoid Membranes. Bio-protocol 7(23): e2639. DOI: 10.21769/BioProtoc.2639.
Category
Plant Science > Plant cell biology > Cell structure
Cell Biology > Cell imaging > Electron microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.