- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Brief Protocol for EDGE Bioinformatics: Analyzing Microbial and Metagenomic NGS Data
(*contributed equally to this work) Published: Vol 7, Iss 23, Dec 5, 2017 DOI: 10.21769/BioProtoc.2622 Views: 11537
Reviewed by: Modesto Redrejo-RodriguezChao JiangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Next-generation sequencing (NGS) offers unparalleled resolution for untargeted organism detection and characterization. However, the majority of NGS analysis programs require users to be proficient in programming and command-line interfaces. EDGE bioinformatics was developed to offer scientists with little to no bioinformatics expertise a point-and-click platform for analyzing sequencing data in a rapid and reproducible manner. EDGE (Empowering the Development of Genomics Expertise) v1.0 released in January 2017, is an intuitive web-based bioinformatics platform engineered for the analysis of microbial and metagenomic NGS-based data (Li et al., 2017). The EDGE bioinformatics suite combines vetted publicly available tools, and tracks settings to ensure reliable and reproducible analysis workflows. To execute the EDGE workflow, only raw sequencing reads and a project ID are necessary. Users can access in-house data, or run analyses on samples deposited in Sequence Read Archive. Default settings offer a robust first-glance and are often sufficient for novice users. All analyses are modular; users can easily turn workflows on/off, and modify parameters to cater to project needs. Results are compiled and available for download in a PDF-formatted report containing publication quality figures. We caution that interpreting results still requires in-depth scientific understanding, however report visuals are often informative, even to novice users.
Keywords: GenomicsBackground
EDGE bioinformatics was developed to help biologists rapidly process next-generation sequencing (NGS) data even if they have little to no bioinformatics expertise. EDGE is a highly integrated and interactive web-based platform that is capable of running many of the standard analyses that biologists require for viral, bacterial/archaeal, and metagenomic samples. EDGE provides an intuitive web-based interface for user input, allows users to visualize and interact with selected results, and generates a final detailed PDF report. Results in the form of tables, text files, graphic files, and PDFs, together with the raw output files of executed programs, can all be downloaded. A user management system allows tracking of an individual’s EDGE runs, along with the ability to share, post publicly, delete, or archive their results. Users can explore ongoing data processing within a user-friendly, intuitive web-based environment and interactive results are presented on a sample-by-sample basis. While EDGE was intentionally designed to be as simple as possible for the user, there is still no single ‘tool’ or algorithm that fits all use cases in the bioinformatics field. Our intent is to provide a detailed panoramic view of the user’s sample from various analytical standpoints. The initial release of EDGE in January 2017 provides six analytical workflows: pre-processing (data QC and host removal), assembly and annotation, reference-based analysis, taxonomy classification, phylogenetic analysis, and PCR analysis (validation and design). The latest release (version 1.5) includes several new features: identification of antimicrobial resistance and virulence genes, 16S/18S/fungal ITS analysis using QIIME, metadata collection/storage, and comparative analysis of taxonomic classification of multiple metagenomic samples. EDGE Bioinformatics is an ongoing effort to provide best of breed bioinformatics tools for NGS data analysis. Updates to current modules are continuous and more modules are under development.
Equipment
- Installing EDGE on a local server
- Hardware requirements: For high-throughput users that desire to process several large (50-500 million reads) samples at once, computers with 256 GB memory and 64 computing CPUs with 8 TB of local storage are highly recommended. The current computational hardware for one of the demonstration servers (https://bioedge.lanl.gov) is a Dell, PowerEdge R720 with 4 x Intel(R) Xeon(R) CPU E5-2630 v2 @ 2.60GHz = 24 cores (48 threads) and 512 GB RAM
- System requirements: EDGE bioinformatics has been tested on a Linux server with Ubuntu 14.04 or CentOS 7 operating systems, and requires 64 bit Linux environments. EDGE will not natively run on Mac OS X but, if enough computational resources are available for analyses, a Dockerized version of EDGE could be installed on a Mac in a Linux environment
- Essential libraries and dependencies required prior to EDGE software install are detailed in step-by-step command line instructions at https://edge.readthedocs.io/en/v1.5/system_requirement.html
- To simplify installation, a Docker image or a VM in OVF is also available. Information and links are located at http://edge.readthedocs.io/en/v1.5/installation.html#edge-docker-image and http://edge.readthedocs.io/en/v1.5/installation.html#edge-vmware-ovf-image
- Useful Resources: A web-based video tutorial series describing how to set-up and run each EDGE module can be found at http://tutorial.getedge.org. Written documentation for software install and how to run EDGE is available at https://edge.readthedocs.io/en/v1.5/
- Hardware requirements: For high-throughput users that desire to process several large (50-500 million reads) samples at once, computers with 256 GB memory and 64 computing CPUs with 8 TB of local storage are highly recommended. The current computational hardware for one of the demonstration servers (https://bioedge.lanl.gov) is a Dell, PowerEdge R720 with 4 x Intel(R) Xeon(R) CPU E5-2630 v2 @ 2.60GHz = 24 cores (48 threads) and 512 GB RAM
- Demo versions of EDGE
Los Alamos National Laboratory (LANL) and the Naval Medical Research Center (NMRC) host and/or support outward facing demo versions of EDGE bioinformatics for prospective users. Any computer with internet access can use the demo web-based EDGE bioinformatics platforms. To run analyses on provided test data, or samples deposited in the Sequence Read Archive (SRA), visit https://bioedge.lanl.gov. To upload your own data (maximum file size is 5 Gbp) and run EDGE, visit http://hobo-nickel.getedge.org
Software
- EDGE source code is open-source and can be located at https://github.com/LANL-Bioinformatics/EDGE/tree/v1.5
- FaQCs software (Lo and Chain, 2014)
- PhaME (Ahmed et al., 2015) software
Procedure
Note: The procedure described herein assumes a user has obtained access to a demo version of EDGE bioinformatics, or has installed EDGE locally. Users must be logged-in to an EDGE account to upload data, run analyses, or view past submissions. If you wish to install EDGE, step-by-step command line instructions are detailed https://edge.readthedocs.io/en/v1.5/installation.html. Feel free to contact any member of the development team directly, or visit our google group at edge-users@googlegroups.com. The procedure is outlined in the following sections: Accessing EDGE, Upload Data Files, Run EDGE Input Sample, Run EDGE Choose Processes, Run EDGE Job Submission, and Navigating Projects. A web-based video tutorial series describing how to set-up and run each EDGE module can be found at http://tutorial.getedge.org.
- Accessing EDGE
Note: To run EDGE on any platform (i.e., in-house or demo versions), users are required to create an account and log-in. User permissions can be set to manage access levels to data and analyses.- User Accounts & Log-in
- Open a web browser and access EDGE (see demo versions above, or access local version). Web addresses (URLs) will depend on the internal network configuration for locally installed versions.
- At the EDGE interface, click on the silhouette in the top right-hand corner. For new users, submit information for a new account. For returning users, sign-in using EDGE credentials.
- Open a web browser and access EDGE (see demo versions above, or access local version). Web addresses (URLs) will depend on the internal network configuration for locally installed versions.
- User Accounts & Log-in
- Upload data files
Click on ‘Upload Files’ tab. Drag and drop files for upload, or click the ‘+ Add Files’ button. Select ‘Start Upload’ to complete. Note that the maximum file size for upload is 5 GB. This is configurable on a local installation of EDGE. Allowed file types include FASTQ, FASTA, GenBank, and test (txt, config, ini), and can be in gzip format. These files can be located in the MyUploads directory. - Run EDGE: Input sample (see Figure 1)
Note: EDGE parameter configurations are optimized for Illumina data. To run EDGE, sequence data files are needed in FASTQ format for a single sample.
Click ‘Run EDGE’ to set-up and submit jobs to the EDGE bioinformatics pipeline. The first section, ‘Input Raw Reads’, requires users to provide a project name, and sequencing data at a bare minimum. EDGE accepts raw FASTQ files (single or paired-end), or Sequence Read Archive (SRA) accession numbers. Details for each setting are provided herein.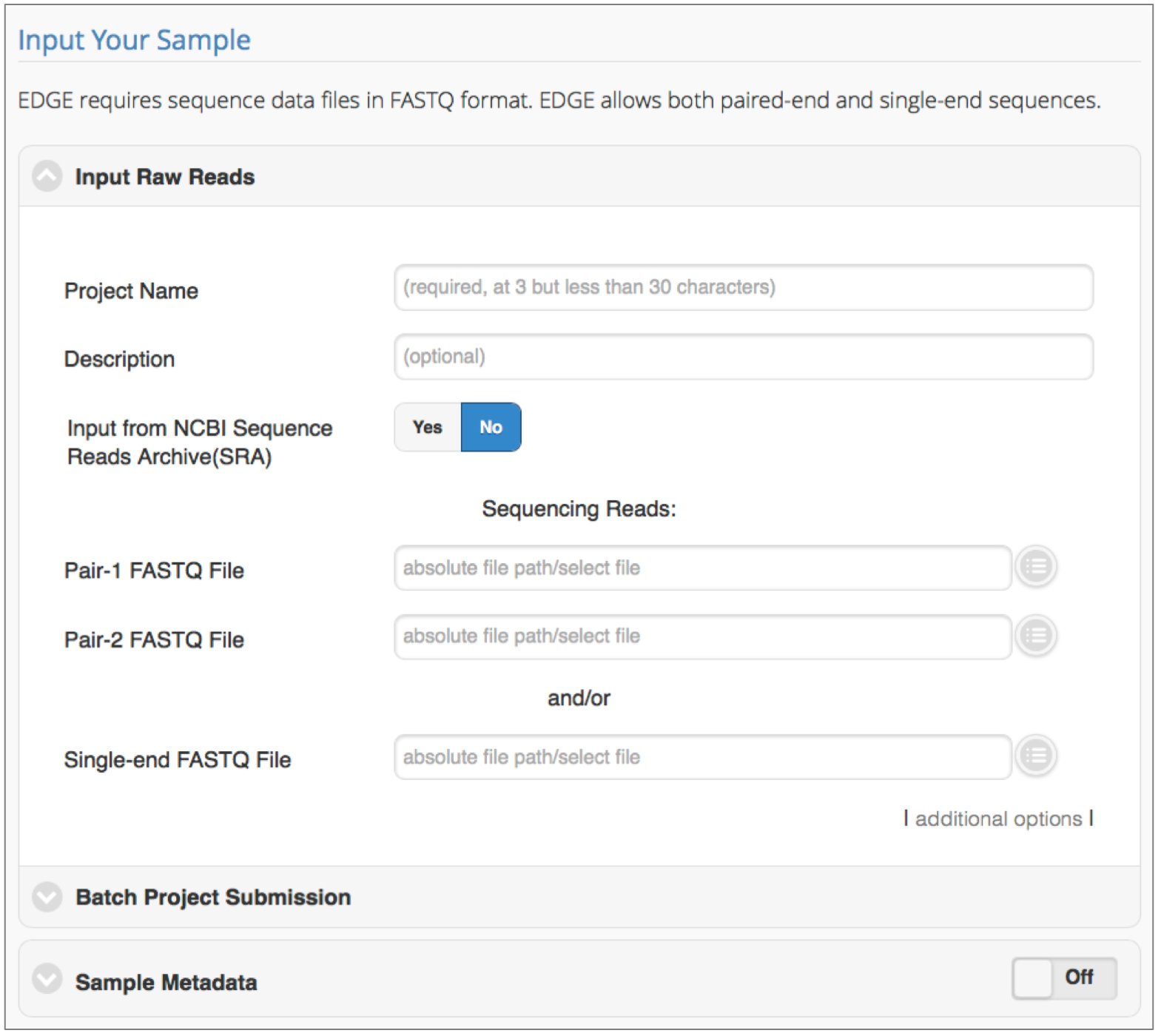
Figure 1. Initiating a run in EDGE. Required input includes a unique project name and the location of sequencing reads to be processed/analyzed. Batch submission is possible and metadata collection is encouraged.- Input Raw Reads
- Project Name
Required field. Enter project name. Note that the same project name can be reused, hence unique identifiers are encouraged. There is a 3-character minimum and 30-character limit for the project name. Avoid using spaces, but dashes and underscores are acceptable. - Description
Optional field. Entry space to describe project/sample in more detail. - Input from Sequencing Read Archive (SRA)
Clickable Yes/No toggle that controls options for accessing location of input sequencing reads. The default setting is No; this requires users to use single- or paired-end FASTQ files from the EDGE Input Directory or Upload File directory. (See step C1d for instructions on how to proceed when set to No). When this toggle is set to Yes, reads are obtained from SRA accession numbers. Internet access is required to use the Yes option. Supported SRA accession formats include: studies (SRP*/ERP*/DRP*), experiments (SRX*/ERX*/DRX*), samples (SRS*/ERS*/DRS*), runs (SRR*/ERR*/DRR*), or submissions (SRA*/ERA*/DRA*). - Sequencing Reads
Required field. Default settings require users to indicate the direct path for data (see step C1c for SRA input). EDGE accepts sequence data files in FASTQ format; compressed files (.gz) are also acceptable. Both paired-end and single-end sequences are permissible. Absolute file paths are required to run EDGE. Users can click the round button to the right of the file path text box to access data from the Upload File directory, or other directory structures configured internally (i.e., directly linked to an in-house Illumina sequencer). - Additional Options
In most cases, the additional options can be ignored. If a user wants to add more input read files, or increase the CPUs for a job, this field provides those options. Clicking on this field will expose the following parameters:- Add paired-end input: add absolute path for additional paired-end input read files.
- Add single-end input: add absolute path for additional single-end input read files.
- Use # of CPUs: Specify the number of CPUs to be used; default and minimum value is ¼ of total number of CPUs on the server.
- Config file: a configuration file is generated automatically for every EDGE run. In the event that a job was interrupted and unfinished, submitting the config file will re-run the job and ensure that the submission runs exactly the same, with the same options.
- Add paired-end input: add absolute path for additional paired-end input read files.
- Project Name
- Batch Project Submission
Batch submission allows a user to run multiple samples using the same configuration, rather than submitting jobs one-by-one. Batch submission is off by default. If this module is turned on, the ‘Input Sequence’ module will be turned off. To implement batch submission, an Excel file with project name, inputs, and project descriptions must be submitted; a sample is available for download.
Batch Excel File: If turned on, user must provide absolute path to Batch Excel File. - Sample Metadata
EDGE supports the input and storage of metadata associated with the genomic or metagenomic sample being analyzed. This currently includes sample type (human, animal, or environmental), isolation source, sample collection date, sample collection location, sequencing platform and sequencing date.
- Input Raw Reads
- Run EDGE: Choose processes/analyses (see Figure 2)
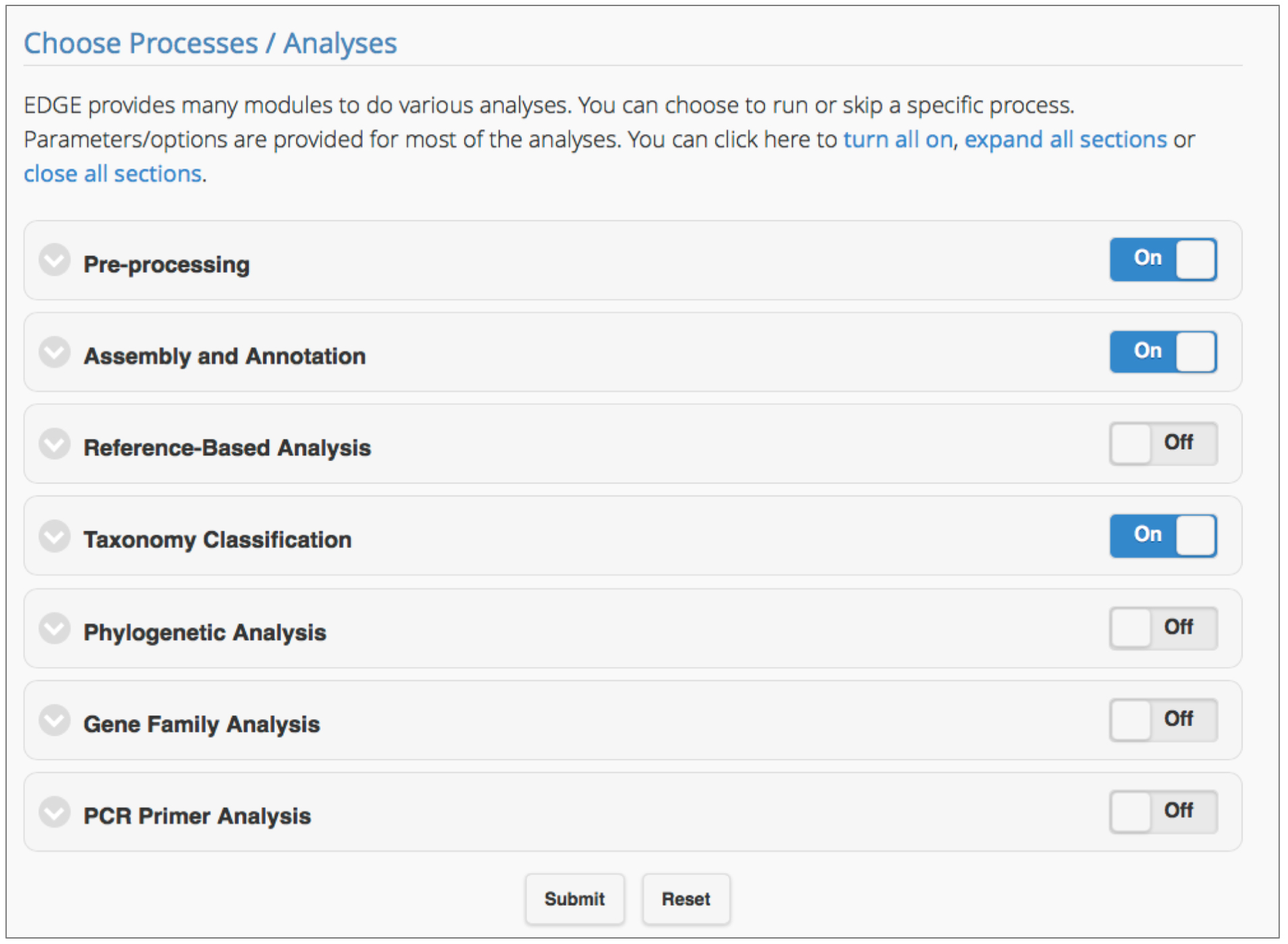
Figure 2. Selecting the EDGE modules for analysis. Users can include any module in a workflow by simply clicking the toggle ‘On’ for the module. Clicking on the arrow to the left of the module title shows subsections of the module and parameters which can be adjusted.
EDGE v1.5 has seven modules: Pre-processing, Assembly and Annotation, Reference-based Analysis, Taxonomy Classification, Phylogenetic Analysis, Gene Family Analysis, and PCR Primer Tools. After input files have been selected and a project name has been assigned, click ‘Submit’ to run the EDGE suite using default parameters. The following analyses are automatically turned on: Pre-processing Quality Trim and Filter, Assembly and Annotation, Taxonomy Classification. Users have full control over which modules to run, and can modify parameter values according to project needs. Each module, and its key parameters are outlined below.
Note: Modules can be expanded or collapsed by clicking the module header. To turn modules on or off, use the toggle button within each header. Expand each module to see/edit settings. Module parameters and default settings are listed in Table 1. Modules/processes which are set ON by default are shown in Table 1 in green.
Table 1. EDGE modules and default settings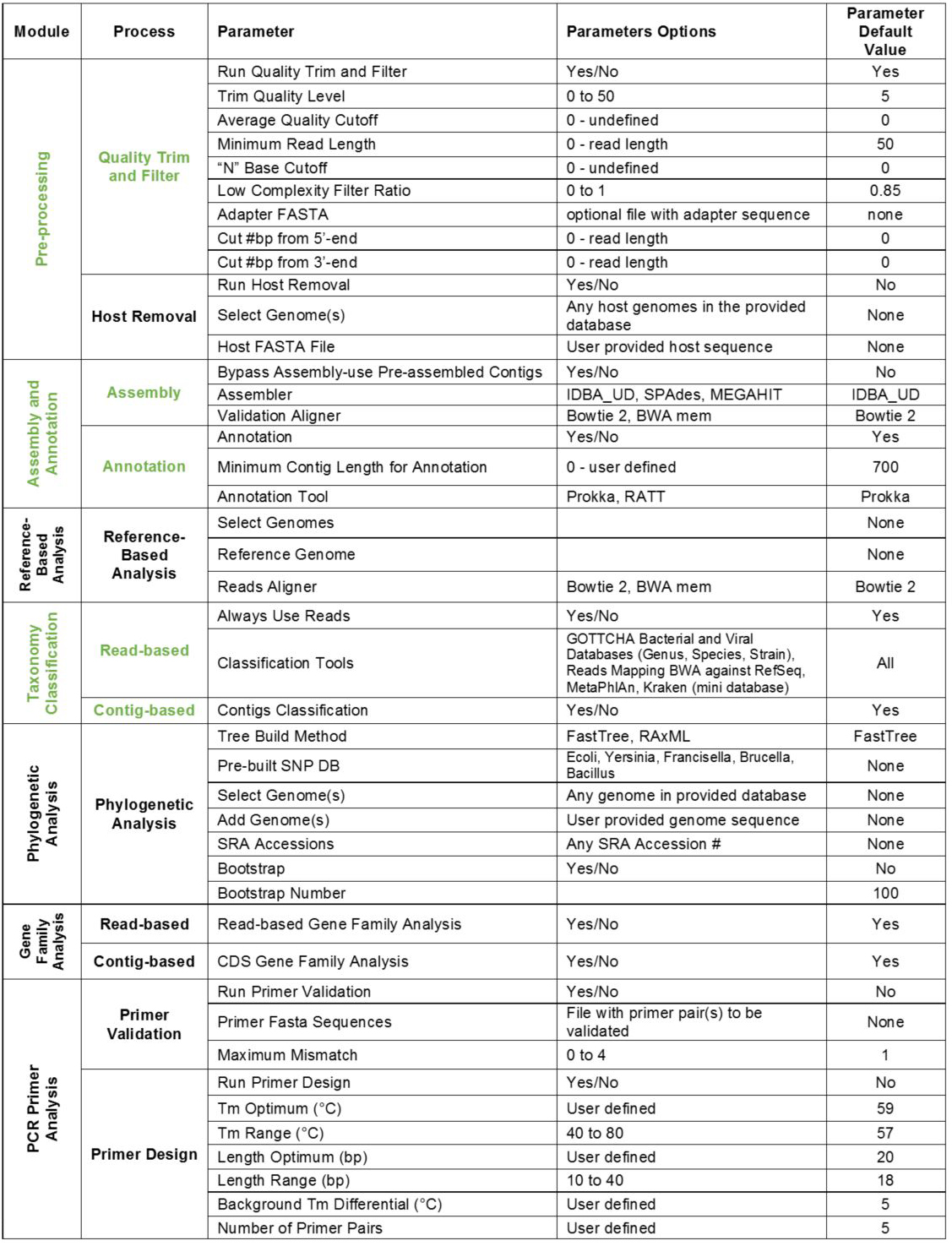
- Pre-processing (Default is ON)
Note: Pre-processing contains two components: Quality Trim and Filter, and Host Removal. By default, Quality Trim and Filter is turned ON, and Host Removal is turned OFF. This module is not required for downstream analyses, but highly recommended when processing raw reads.- Quality Trim and Filter
FaQCs software (Lo and Chain, 2014) is used to rapidly analyze reads for quality, then trim or filter those with poor quality. Pre-set parameter values are appropriate to filter unwanted reads in most cases. Exceptions can arise when specialized adapter sequences have been added. In this case, a user can supply FASTA files or specify the number of base pairs to trim from each end of the reads. After this step, only high-quality reads are passed to downstream analyses. Each parameter is described below; default settings can be found in Table 1.- Run Quality Trim and Filter: Yes/No command to execute pipeline
- Trim Quality Level: minimum quality threshold based on Phred scores
- Average Quality Cutoff: filter based on average quality score of entire read
- Minimum Read Length: filter reads based on length
- ‘N’ Base Cutoff: discard reads with more than this number of continuous ‘N’ bases
- Low Complexity Filter Ratio: indicate maximum fraction of mono-/di-nucleotide sequence permissible
- Adapter FASTA: adapters can be removed from sequences; user must provide FASTA file containing adapter sequences
- Cut #bp from 5’-end: define a set number of base pairs to remove from the 5’ end of each read
- Cut #bp from 3’-end: define a set number of base pairs to remove from the 3’ end of each read
- Run Quality Trim and Filter: Yes/No command to execute pipeline
- Host Removal
While called ‘Host Removal’, this module is used to subtract unwanted reads that align to any selected reference. For example, unwanted reads derived from hosts and/or from positive controls, such as PhiX, can be filtered out at this step. Whether to employ this step and if so, which genomes to select for use in host removal, depend on each sample’s origin and therefore is left to the user’s discretion. Reads are mapped to reference genome(s) using BWA, and removed based on the similarity threshold parameter. At the EDGE interface, this module must be turned on to run. Parameter descriptions and uses:- Run Host Removal: Yes/No command to execute pipeline
- Select Genome(s): Click on dropdown menu to select common hosts or search for additional hosts from RefSeq by typing in the search text box. Choose relevant host genomes by clicking; blue checkmarks will indicate selected hosts. The number of hosts selected is unlimited. Click on the X in the top left corner of the selection menu to save results.
Note: This RefSeq database refers to all complete bacterial and archaeal genomes plus complete viral genomes and near neighbors. - Host FASTA File: a user also has the option to upload a specific host sequence (i.e., sequenced in-house, or host is not present in the EDGE database) for removal. To do so, provide the direct path to the FASTA formatted file containing the host sequence.
- Similarity (%): Minimum percent similarity threshold is used in host removal when calculated by [Reads aligned bases]/[Reads length] x 100. 90% similarity is the default and lowest recommended setting.
- Run Host Removal: Yes/No command to execute pipeline
- Quality Trim and Filter
- Assembly and Annotation (Default is ON)
Note: Assembly and Annotation pipelines are turned on by default in EDGE. In order to annotate a genome or perform any downstream contig-based analysis, assembly must be completed. There is an option to upload pre-assembled contigs in the form of a FASTA file, then bypass the assembly module. If assembly fails, downstream modules requiring contig files will be bypassed.- Assembly
Three different de novo assembler options are provided: IDBA_UD (Peng et al., 2012), SPAdes (Bankevich et al., 2012), and MEGAHIT (Li et al., 2015). Optimal selection of an assembler can depend on sample type (e.g., isolate vs. metagenome), data size, and time available for analysis. Pre-set parameters for each assembler are robust and perform well in the majority of cases. Users can set the minimum cut-off value for final contigs. As default, contigs smaller than 200 bp are filtered out. If using sequence reads longer than 200 bp (for instance 2 x 300 bp), this threshold should be adjusted to the read length. Read-alignment validation is used to ensure confidence in assembly.- Bypass Assembly and use Pre-assembled Contigs: Yes/No command to execute
- Assembler: Three de novo assemblers are built into EDGE. IDBA_UD (default) performs efficiently using either isolates and metagenomic samples, however it is not ideal for large genomes. There are multiple preset configurations in SPAdes (tailored for single cells, metagenomes, plasmids, or RNA-Seq) and SPAdes performs well on isolates and metagenomes, but can be very computationally intensive for any large dataset. SPAdes can additionally take in long read data (PacBio or Nanopore) with the Illumina short read data and produces a hybrid assembly; this option increases the computational resources required. MEGAHIT is a fast, robust solution for large and complex metagenomic samples.
- Validation Aligner: Bowie 2 (default) or BWA mem can be selected to map reads back to assembled contigs for validation.
- Bypass Assembly and use Pre-assembled Contigs: Yes/No command to execute
- Annotation
Successful assembly is a prerequisite for annotation. EDGE offers users two annotation tools: PROKKA (Seemann, 2014) and RATT (Otto et al., 2011). PROKKA is appropriate for most cases; it has been designed for rapid annotation of prokaryotic genomes. Alternatively, users can use RATT to transfer annotation from an annotated reference genome to an unannotated sample.- Annotation: Yes/No command to execute pipeline
- Minimum Contig Length for Annotation: User defined length of contig to include in annotation
- Annotation Tool: Two annotation tools are provided. If PROKKA is selected, the user must also choose the genome type to annotate under Specify the Kingdom (Archaea, Bacteria, Mitochondria, Viruses, Others). RATT, on the other hand, will transfer the annotation from a reference genome to the sample of interest. The reference genome must be a close relative to the sample. If RATT is selected, a user must provide the GenBank formatted reference/source annotation file.
- Annotation: Yes/No command to execute pipeline
- Assembly
- Reference-based Analysis
Reference-based analysis is a useful tool for investigating samples of known composition, for instance, studying a pure bacterial culture. This module maps reads and contigs to references selected by the user to obtain coverage information, and identify uncovered regions where sample reads or assembled contigs, do not align to the reference. The output of this module provides information on variants, such as single nucleotide polymorphisms (SNPs), and uncovered regions (potential insertion/deletions) that do not align to the reference. Variants are identified using SAMtools (Li et al., 2009). The in-depth results can be interactively explored using the genome browser, JBrowse (Skinner et al., 2009).
Note: Reference-based analysis is off by default. Users can turn this module on using the toggle button.- Select Genome(s): Pre-built reference list. Click on dropdown menu and search for microbial species of interest. It is important to choose closely related organisms. Click on desired references and blue checkmarks will indicate selected genomes. The number of hosts selected is unlimited. Click on the X in the top left corner of the selection menu to save results.
- Reference Genome: If a reference organism is not in the pre-constructed list, users can upload an appropriate FASTA or GenBank file for your experiment.
- Reads Aligner: Users can choose Bowtie 2 or BWA-MEM as the read mapper. The two algorithms will yield very similar results and can be set based on user preference.
- Select Genome(s): Pre-built reference list. Click on dropdown menu and search for microbial species of interest. It is important to choose closely related organisms. Click on desired references and blue checkmarks will indicate selected genomes. The number of hosts selected is unlimited. Click on the X in the top left corner of the selection menu to save results.
- Taxonomy classification (Default is ON)
The EDGE Taxonomy module will perform sequence classification and determine sample composition. This module is useful for identifying organisms in complex samples. Similarly, taxonomy classification is useful for analyzing purified cultures to detect contamination coming from lab reagents or mishandling of samples.
Note: By default, taxonomic classification is turned on for both reads and contigs. EDGE implements several different databases and algorithms for taxonomy assignments. By default, all of the tools are turned on to take advantage of their strengths and provide users with cross-validated assessments. The tools vary in sensitivity and classification.- Read-based taxonomy classification
- Always Use All Reads: Yes/No command to indicate what reads will be used in taxonomy. Yes (default) indicates that all reads that pass pre-processing will be used. If the user has provided a reference for Reference-based analysis, and selects No, then results will include only reads that are different from the reference.
- Classification Tools: Drop down menu with checkbox selection for different profiling tools. Default settings implement all databases, which is recommended in order to take advantage of all tools which vary significantly in terms of sensitivity and specificity. GOTTCHA will provide the most specific results (a very low false positive rate), while BWA and Kraken will provide the most sensitive results (a very low false negative rate).
1) GOTTCHA Bacterial Databases (Genus, Species, Strain) (specific) (Freitas et al., 2015), version 20150825.
2) GOTTCHA Viral Databases (Genus, Species, Strain) (specific) (Freitas et al., 2015), version 20150825.
3) Read Mapping using BWA against RefSeq (see Note above for description) (sensitive) (Chen et al., 2010)
4) MetaPhlAn, searches clade-specific marker genes (specific) (Segata et al., 2012)
5) Kraken mini, exact alignment of k-mers (sensitive) (Wood and Salzberg, 2014)
- Always Use All Reads: Yes/No command to indicate what reads will be used in taxonomy. Yes (default) indicates that all reads that pass pre-processing will be used. If the user has provided a reference for Reference-based analysis, and selects No, then results will include only reads that are different from the reference.
- Contig-based Taxonomy Classification
Contigs classification: Yes/No command to execute. Yes (default) indicates contigs will be mapped against NCBI databases for taxonomy and functional annotations.
- Read-based taxonomy classification
- Phylogenetic Analysis
EDGE will construct phylogenetic trees for reads and contigs. PhaME (Ahmed et al., 2015) software is implemented to align core conserved sections of genomes, perform whole-genome SNP discovery, and build a phylogenetic tree. Users can choose from pre-computed pathogen databases or build their own by selecting genomes provided in EDGE databases, or uploading their own.
Note: Phylogenetic analysis will not automatically run. Users must turn the toggle switch on and address required parameters for this module. Due to the nature of the tool, users should choose closely related strains or species only; this will ensure that the user’s target genome falls within the final tree build. Additionally, although this module has been successfully applied to metagenomes, the phylogeny tool was engineered with isolate genome projects in mind. If a user selects genomes with little to no similarity, the tool will exclude them from the analysis.- Tree Build Method: Users have two options for generating phylogenetic tree. FastTree is fast and selected as default (Price et al., 2010). RAxML is more accurate, but more time consuming (Stamatakis, 2014).
- Pre-built SNP DB: EDGE supports 5 pre-computed pathogen databases for SNP phylogeny analysis (Escherichia coli, Yersinia, Francisella, Brucella, Bacillus).
- Select Genome(s): RefSeq genomes (see Note above for description) are available in this dropdown menu. Open menu, search, and click on desired genomes to select.
- Add Genome(s): User can provide FASTA entries for genomes to be built into tree. A maximum of 20 reference genomes can be used and including an outgroup is recommended.
- SRA Accessions: SRA entries are allowed for specifying references to be used in phylogenetic analysis.
- Bootstrap: Yes/No command to execute bootstrap in analysis.
- Bootstrap Number: Can be modified if user indicates Yes for Bootstrap method.
- Tree Build Method: Users have two options for generating phylogenetic tree. FastTree is fast and selected as default (Price et al., 2010). RAxML is more accurate, but more time consuming (Stamatakis, 2014).
- Gene Family Analysis
Note: The Gene Family Analysis module searches reads and annotated coding sequences (CDS) for specific gene families (currently, antibiotic resistance and virulence gene families). There are two components to this module: read-based and contig-based profiling. To perform either analysis, users must first turn this module on using the toggle switch, then ensure each sub-analysis is set to Yes. Contig-based analysis requires successful assembly.- Read-based Gene Family Analysis
The read-based analysis uses the ShortBRED (Kaminski et al., 2015) algorithm to search for antimicrobial resistance genes in the Antibiotic Resistance Genes Database, ARDB, (Lui and Pop, 2009) and Resfams (Gibson et al., 2014) databases. Similarly, ShortBRED will search for virulence genes using a version of the Virulence Factor Database, VFDB, (Chen et al., 2005) curated by an EDGE developer.
Read-based Gene Family Analysis: Yes/No command to execute analysis. - Contig-based (CDS) Gene Family Analysis
Similarly, virulence genes are called using ShortBRED and VFDB. However, for antibiotic resistance gene finding, the Comprehensive Antibiotic Resistance Database (CARD) program RGI (Jia et al., 2017) is used for CDS-based analysis performed on contigs.
Contig-based (CDS) Gene Family Analysis: Yes/No command to execute analysis.
- Read-based Gene Family Analysis
- PCR Primer Analysis
Note: The PCR Primer Analysis module consists of two processes: validation of existing primers and design of new primers. PCR Primer Analysis will not automatically run. Users must turn the toggle switch on, and turn on each independent component of this module.- Primer Validation
In the validation pipeline, users upload a file containing existing primer sequences. EDGE maps these primers to the sample’s assembly using BWA to determine if the amplicon is generated. Users can define the number of mismatches allowed in this validation.- Run Primer Validation: Yes/No command to execute analysis.
- Primer FASTA Sequences: Provide absolute path to input file with existing primers to be validated. Must contain an even number of forward and reverse primers saved in FASTA format.
- Maximum Mismatch: Indicate the maximum number of mismatches allowed per primer sequence. Click on the number to select (i.e., 0, 1, 2, 3, or 4).
- Run Primer Validation: Yes/No command to execute analysis.
- Primer Design
To design primers for newly assembled contigs, EDGE identifies unique regions of the sample using BWA then generates primer sets using Primer3 (Untergasser et al., 2012). Primers are designed, and compared to RefSeq to ensure the selected regions in the sample genome are indeed unique. Users indicate desired primer reaction parameters for melting temperature, amplicon sizing, and the number of primer pairs.- Run Primer Design: Yes/No command to execute analysis.
- Primer Settings: All reaction parameters can be modified. Parameters for primer design include: Tm Optimum (°C), Tm Range (°C), Length Optimum (bp), Length Range (bp), Background Tm, Differential (°C).
- The number of Primer Pairs: Desired amount of new primer pairs to be designed.
- Run Primer Design: Yes/No command to execute analysis.
- Primer Validation
- Pre-processing (Default is ON)
- Run EDGE: Job submission
After input files, project name, and desired process options have been defined (Procedure A-Procedure D), a job is ready for submission. To submit a job, click on the ‘Submit’ button at the bottom of the page. Red and/or green indicators will immediately appear to indicate successful job submission. If errors occur, the message (in red) is clickable and will return users to the section needing attention where the data entry box(es) with errors are highlighted with a yellow glow. - Navigate Projects
Upon successful submission, users can monitor job status by clicking ‘Projects’ in the navigation bar on the left-hand side of the user interface. This provides a list of projects submitted by the user. Job status of each project in the list is indicated by a color-coded system: grey = not yet begun, red = error, orange = in progress (running), green = completed. Errors normally only occur if there are issues in retrieving or reading the input data files. If a job is in progress, clicking on the project in the left navigation bar will open the Project page where results are posted in real time.
The Project page has links to other pages and to the project list on the left (which can be hidden with a link in the upper left corner). Project information and results are shown in the center section. The information on this page is static and allows users to access portions of the run that are already complete, however the page needs to be refreshed for any updates to the project. A small square icon in the upper right corner of the screen opens a sidebar on the right that displays active monitoring of job progress and server usage. Options to interrupt, delete, rerun or share the project and view the live log are available in this sidebar. Figure 3 shows a screen shot of a project page from https://bioedge.lanl.gov/ and originally published by Oxford University Press in Nucleic Acids Research (Li et al., 2017).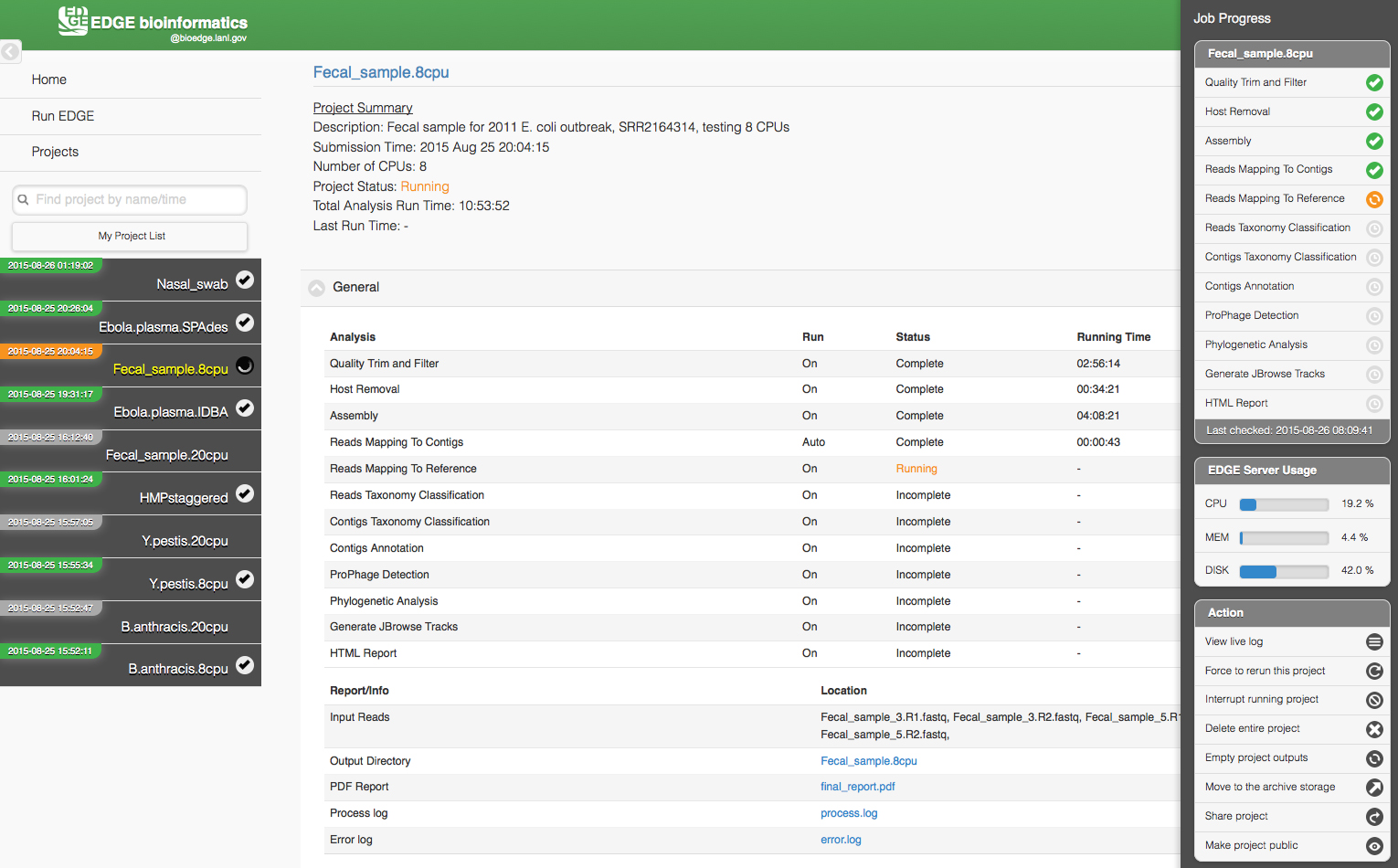
Figure 3. The EDGE Project page displays an analysis in progress
Data analysis
In addition to the several examples provided in our original publication (Li et al., 2017) which can be viewed at https://bioedge.lanl.gov, two additional use cases were run to demonstrate much of the functionality within EDGE; the full analyses for these runs can be viewed at http://hobo-nickel.getedge.org. The first use case is a reduced dataset for E. coli (10x coverage) which will run quickly to test the modules and is entitled EDGE_tutorial_FASTQ_3. The second use case is a metagenomic sample from an E. coli outbreak in 2011 with the data downloaded from the Sequence Read Archive (SRA) and is entitled EDGE_tutorial_SRA_2. Table 2 details precise EDGE parameter settings for these two runs.
Table 2. Use Case Parameter Settings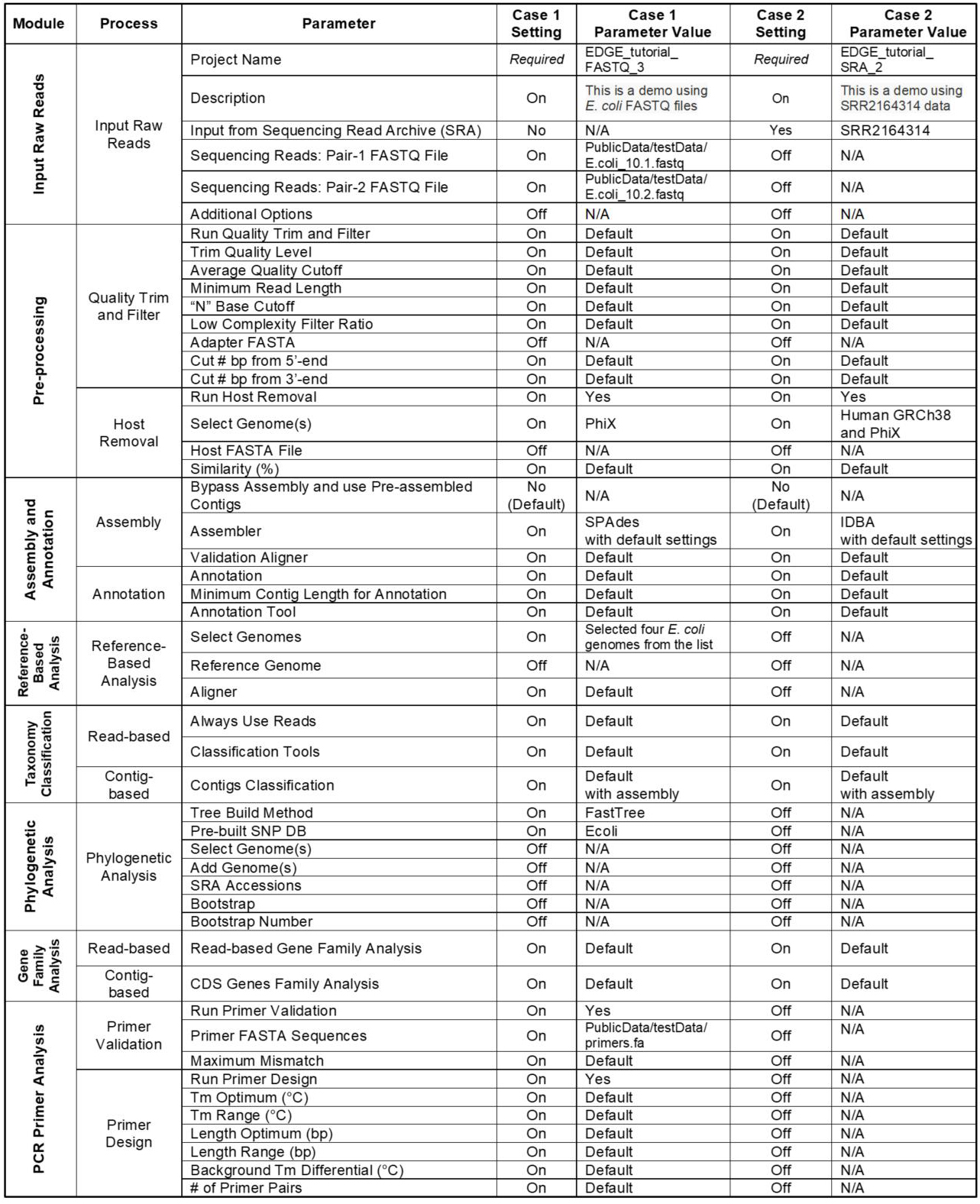
The Project page includes the statistics of the run (each module and time to completion) and links to the output directory with all the results, summary log files, and a PDF summary of results (see Figure 3). Each module produces summarized results with both text and figures along with some interactive graphics within the context of the Project page. Figure 4 shows some examples of graphical output from the two use cases described in Table 2. 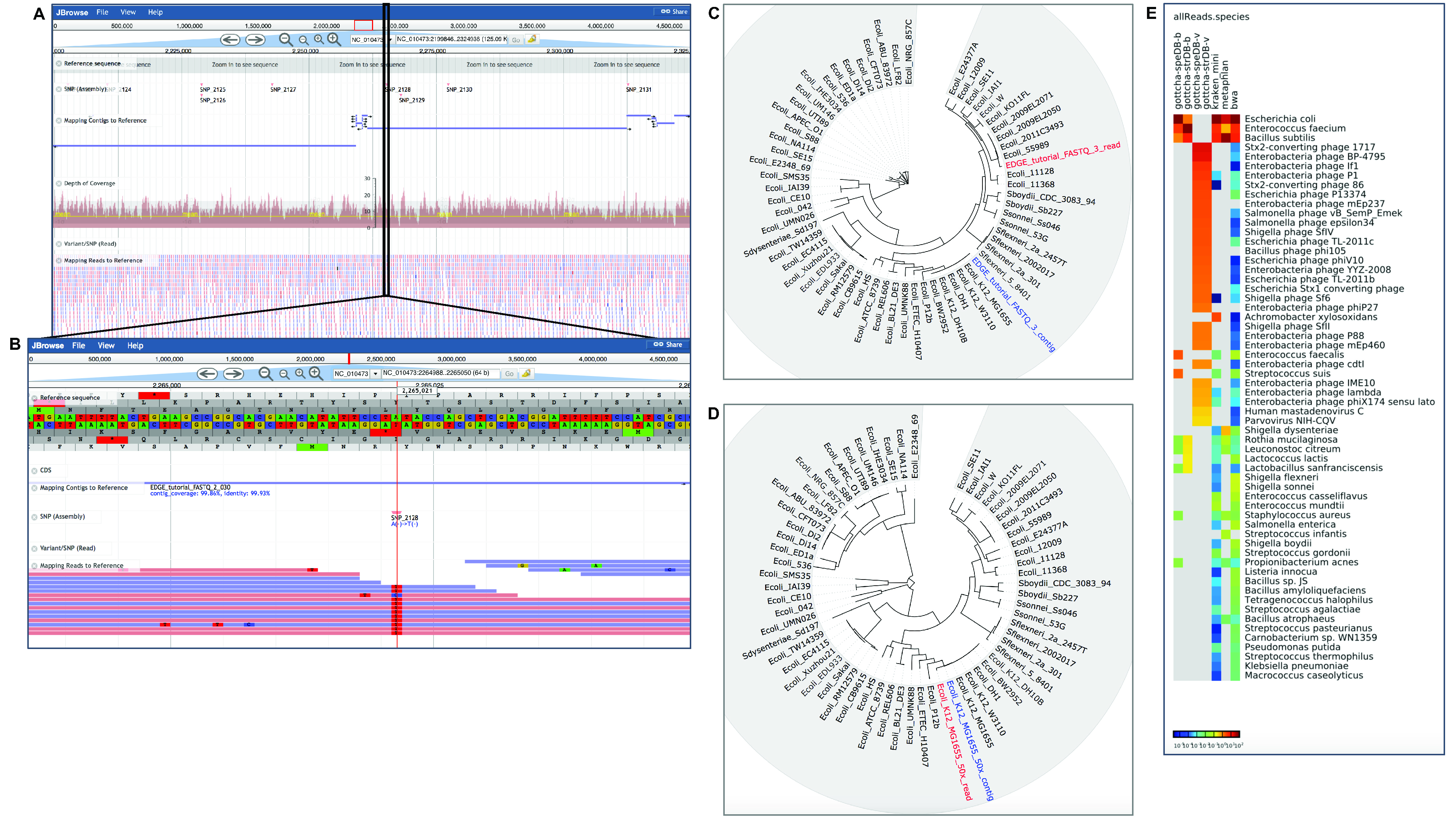
Figure 4. Examples of graphical output in EDGE for Use Cases 1 and 2. EDGE provides many results as visual graphics within the Project page. Use Case 1 is an isolate E. coli dataset with approximately 10x coverage. This reduced coverage data set was created to test EDGE installation. A. Shows the interactive view of reads and contigs mapped to a reference in the genome browser. B. Shows a closer inspection of a region with an SNP/variant highlighted in the same reference-based analysis. *C. Shows both reads and assembled contigs placed into a phylogenetic tree based on whole genome SNP analysis. D. Shows results for a larger dataset (50x) for the same genome; reads and contigs are placed directly adjacent to one another and to E. coli K12 MG1655 with the higher coverage dataset. Use Case 2 is a clinical fecal sample from an E. coli outbreak in 2011. E. shows results from multiple tools for taxonomy classification in a heatmap.
*Note: At this low depth of coverage the reads and contigs are not placed immediately adjacent to one another in the tree, but the contigs are placed adjacent to the correct reference genome, E. coli K12 MG1655.
Each module also provides links within the integrated visualization for that module for primary desired output from the analyses (e.g., assembled contigs, reads mapped to a reference, SNPS/variants, abundance tables). Figure 5 shows an example of tabular output with links for downloading output files by the project owner or links to external sources of information (e.g., NCBI, ARDB). This is a screen shot of a portion of project page from https://bioedge.lanl.gov/.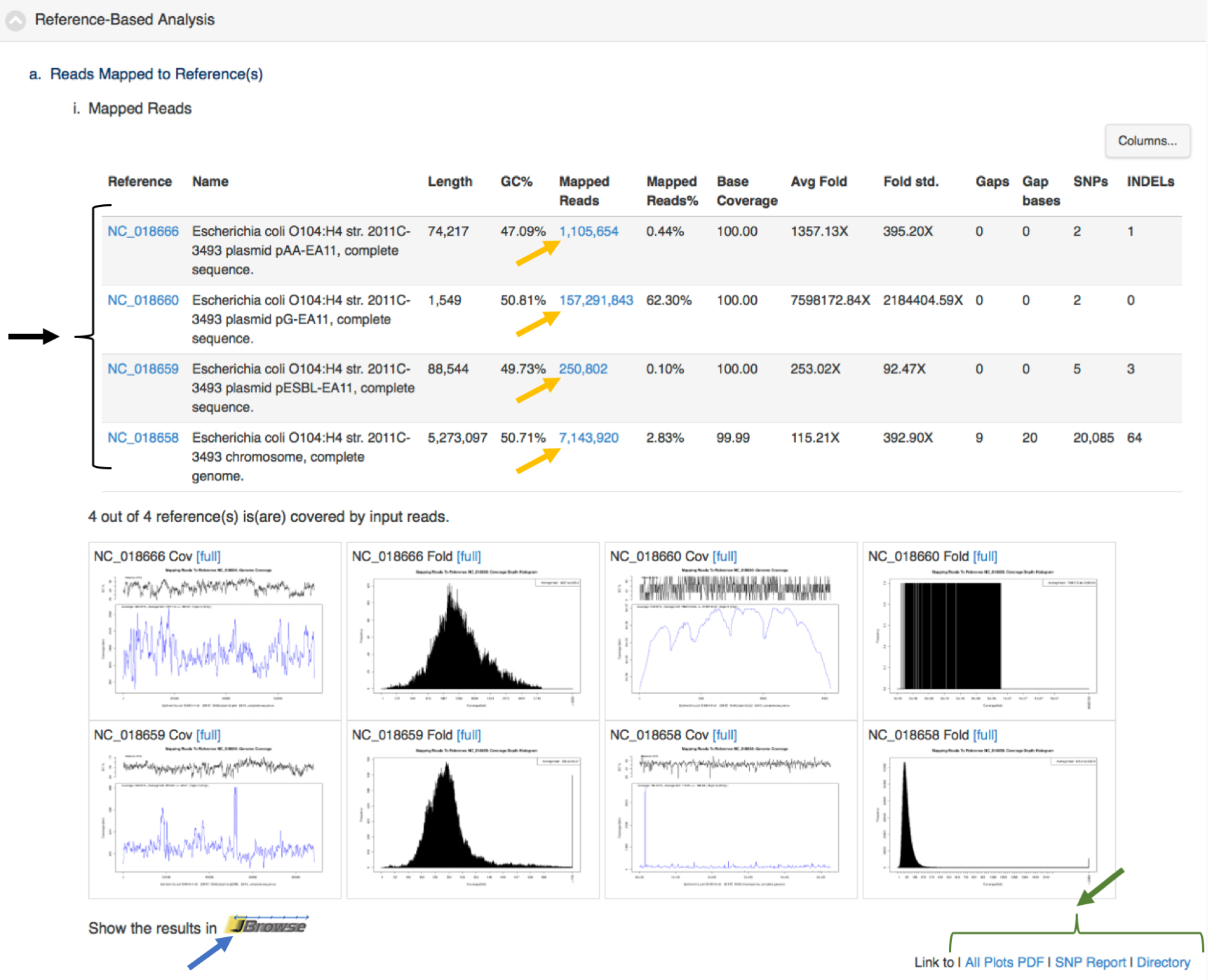
Figure 5. Results of reference-based analysis showing links with the tabular output of data mapped to an E. coli genome (one chromosome and three plasmids). This clinical sample came from an E. coli outbreak in 2011. This is the same sample from Use Case 2. The data was mapped back to a reference genome from the same outbreak. The black arrow and bracket on the left highlight links to NCBI for more information about each of the replicons. The yellow arrows indicate links to download output files of reads mapped to each of the replicons. The blue arrow is a link to an interactive view of the reads and contigs mapped to the reference E. coli. replicons. The green arrow and bracket in the lower right highlight links to graphics, tables and the full output of the reference-based analysis module. Similar links are available for all the modules.
Notes
The tools included in EDGE have been selected for robustness, speed, and accuracy. Kmer-based assemblers may display some variation from run to run due to inherent non-deterministic properties of the assemblers, but all other results are fully reproducible. While EDGE provides even novice NGS users with the ability to easily perform complex analyses, we encourage users to understand the tools and algorithms and to have some insight into how results should be interpreted.
Acknowledgments
This work is funded by the Defense Threat Reduction Agency. The views expressed in this manuscript are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, the Department of Defense, the National Institutes of Health, the Department of Health and Human Services, nor the U.S. Government. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
References
- Ahmed, S. A., Lo, C. C., Li, P. E., Davenport, K. W. and Chain, P. S. G. (2015). From raw reads to trees: Whole genome SNP phylogenetics across the tree of life. bioRxiv.
- Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., Lesin, V. M., Nikolenko, S. I., Pham, S., Prjibelski, A. D., Pyshkin, A. V., Sirotkin, A. V., Vyahhi, N., Tesler, G., Alekseyev, M. A. and Pevzner, P. A. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5): 455-477.
- Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., Jin, Q. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 33: D325-8.
- Chen, P. E., Cook, C., Stewart, A. C., Nagarajan, N., Sommer, D. D., Pop, M., Thomason, B., Thomason, M. P., Lentz, S., Nolan, N., Sozhamannan, S., Sulakvelidze, A., Mateczun, A., Du, L., Zwick, M. E. and Read, T. D. (2010). Genomic characterization of the Yersinia genus. Genome Biol 11(1): R1.
- Freitas, T. A., Li, P. E., Scholz, M. B. and Chain, P. S. (2015). Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res 43(10): e69.
- Gibson, M. K., Forsberg, K. J., Dantas, G. (2015). Improved annotation of antibiotic resistance functions reveals microbial resistomes cluster by ecology. ISME J 9(1): 207-16.
- Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., Lago, B. A., Dave, B. M., Pereira, S., Sharma, A. N., Doshi, S., Courtot, M., Lo, R., Williams, L. E., Frye, J. G., Elsayegh, T., Sardar, D., Westman, E. L., Pawlowski, A. C., Johnson, T. A., Brinkman, F. S., Wright, G. D. and McArthur, A. G. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45(D1): D566-D573.
- Kaminski, J., Gibson, M. K., Franzosa, E. A., Segata, N., Dantas, G. and Huttenhower, C. (2015). High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput Biol 11(12): e1004557.
- Li, D., Liu, C. M., Luo, R., Sadakane, K. and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31(10): 1674-1676.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and 1000 Genome Project Data Processing Subgroup. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-9.
- Li, P. E., Lo, C. C., Anderson, J. J., Davenport, K. W., Bishop-Lilly, K. A., Xu, Y., Ahmed, S., Feng, S., Mokashi, V. P. and Chain, P. S. (2017). Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Res 45(1): 67-80.
- Lo, C. C. and Chain, P. S. (2014). Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinformatics 15: 366.
- Lui, B. and Pop, M. (2009). ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res 37(Database issue): D443-7.
- Otto, T. D., Dillon, G. P., Degrave, W. S. and Berriman, M. (2011). RATT: Rapid annotation transfer tool. Nucleic Acids Res 39(9): e57.
- Peng, Y., Leung, H. C., Yiu, S. M. and Chin, F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28(11): 1420-1428.
- Price, M. N., Dehal, P. S. and Arkin, A. P. (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5(3): e9490.
- Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14): 2068-2069.
- Segata, N., Waldron, L., Ballarini, A., Narasimhan, V., Jousson, O. and Huttenhower, C. (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9(8): 811-814.
- Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J. and Holmes, I. H. (2009). JBrowse: a next-generation genome browser. Genome Res 19: 1630-1638.
- Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9): 1312-1313.
- Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M. and Rozen, S. G. (2012). Primer3--new capabilities and interfaces. Nucleic Acids Res 40(15): e115.
- Wood, D. E. and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15(3): R46.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Philipson, C., Davenport, K., Voegtly, L., Lo, C., Li, P., Xu, Y., Shakya, M., Cer, R. Z., Bishop-Lilly, K. A., Hamilton, T. and Chain, P. S. G. (2017). Brief Protocol for EDGE Bioinformatics: Analyzing Microbial and Metagenomic NGS Data. Bio-protocol 7(23): e2622. DOI: 10.21769/BioProtoc.2622.
Category
Microbiology > Community analysis > Metagenomics
Systems Biology > Genomics > Sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.