- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Using CRISPR-ERA Webserver for sgRNA Design

Published: Vol 7, Iss 17, Sep 5, 2017 DOI: 10.21769/BioProtoc.2522 Views: 13050
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2015

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The CRISPR-Cas9 system is emerging as a powerful technology for gene editing (modifying the genome sequence) and gene regulation (without modifying the genome sequence). Designing sgRNAs for specific genes or regions of interest is indispensable to CRISPR-based applications. CRISPR-ERA (http://crispr-era.stanford.edu/) is one of the state-of-the-art designer webserver tools, which has been developed both for gene editing and gene regulation sgRNA design. This protocol discusses how to design sgRNA sequences and genome-wide sgRNA library using CRISPR-ERA.
Keywords: sgRNA designBackground
Genome engineering is essential to the study of biology, which attracted several new technological breakthroughs (Doudna and Charpentier, 2014). CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR associated protein 9) technology has proven to have great efficiency and generalizability both in gene editing and gene regulation (Qi et al., 2013; La Russa and Qi, 2015). CRISPR-Cas9 system consists of Cas9 endonuclease and a target-identifying CRISPR RNA duplex (crRNA and trans-activating crRNA (tracrRNA)) that can be simplified into a single guide RNA (sgRNA). sgRNA sequence can match and target with an 18- to 25-bp DNA sequence, with a required DNA motif termed the protospacer-adjacent motif (PAM) adjacent to the binding site. The most commonly used type of Cas9 is derived from Streptococcus pyogenes, and the PAM sequence is NGG (N represents any nucleotide), while NAG works sporadically with lower efficiency.
In CRISPR-Cas9 system, sgRNA with a general 20 bp custom designed sequence determines target specificity and efficiency. Designing sgRNA is an indispensible part of CRISPR related projects. Of the published tools that enable automated sgRNA design, CRISPR-ERA can provide sgRNA searching approaches for both gene editing and gene regulation applications, and provide additional genome-wide sgRNA library building protocol (Liu et al., 2015). Currently, CRISPR-ERA supports sgRNA design for nine organisms with different kinds of manipulations. It provides a user-friendly webserver to enable sgRNA searching in preassembled databases. The preassembled genome-wide sgRNA databases are built by seeking all targetable sites with patterns of N20NGG. To evaluate the efficiency and specificity of each sgRNA, CRISPR-ERA utilizes criteria summarized from published data, and then computes an efficacy score (E-score) and a specificity score (S-score). Criteria will have a slight change within different kinds of manipulation and organisms.
Equipment
- Personal computer for CRISPR-ERA website searching
- High performance computing cluster for building genome-wide sgRNA library. Taken genome version hg19 as an example, the minimum storage space is 500 G
Software
- CRISPR-ERA (http://crispr-era.stanford.edu/)
- USCS genome browser (Kent et al., 2002; http://genome.ucsc.edu/)
- Bowtie2 (Langmead et al., 2012; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
- NCBI (https://www.ncbi.nlm.nih.gov/)
- Perl scripts (Programming language, https://www.perl.org/)
- Shell scripts (Programming language, Command Line Interface shell, https://www.linux.org/)
Procedure
- Using CRISPR-ERA webserver for sgRNA searching
- CRISPR-ERA webserver input (Figure 1)
- Choose the type of objective gene manipulation: gene editing using nuclease, gene editing using nickase, gene repression, or gene activation.
- Choose the host organism: Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans, Danio rerio, Rattus norvegicus, Mus musculus, or Homo sapiens. Different type of choice in step A1a presents different optional organisms.
- Choose the input format: official gene name, gene location (target region for gene editing or transcriptional start sites (TSS) location for gene regulation), or gene sequence in FASTA format (using a textbox or uploading files).
Note: By clicking ‘?’ in choice box of every step, users could find detailed instruction.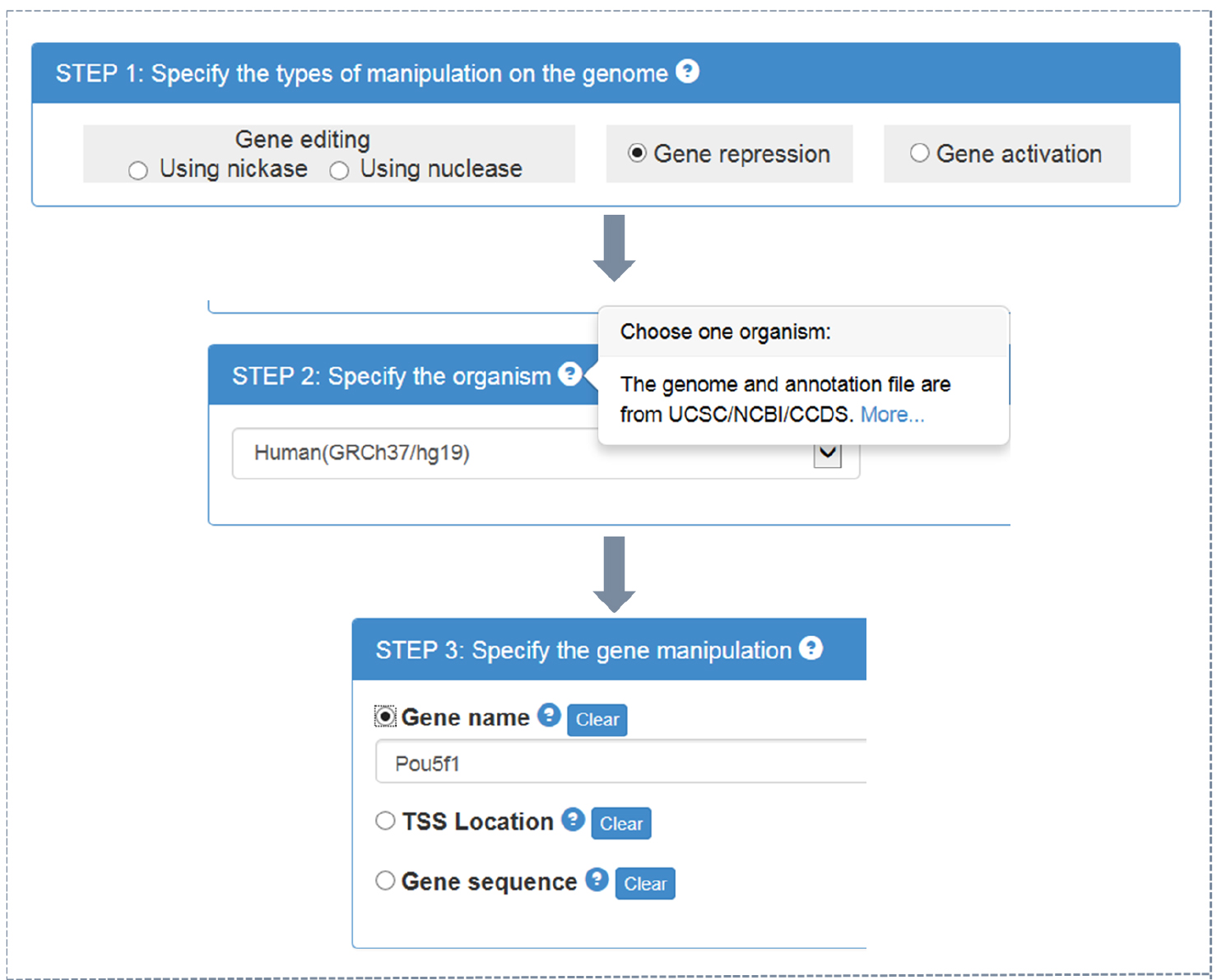
Figure 1. CRISPR-ERA input process - Choose the type of objective gene manipulation: gene editing using nuclease, gene editing using nickase, gene repression, or gene activation.
- CRISPR-ERA webserver output (Figure 2)
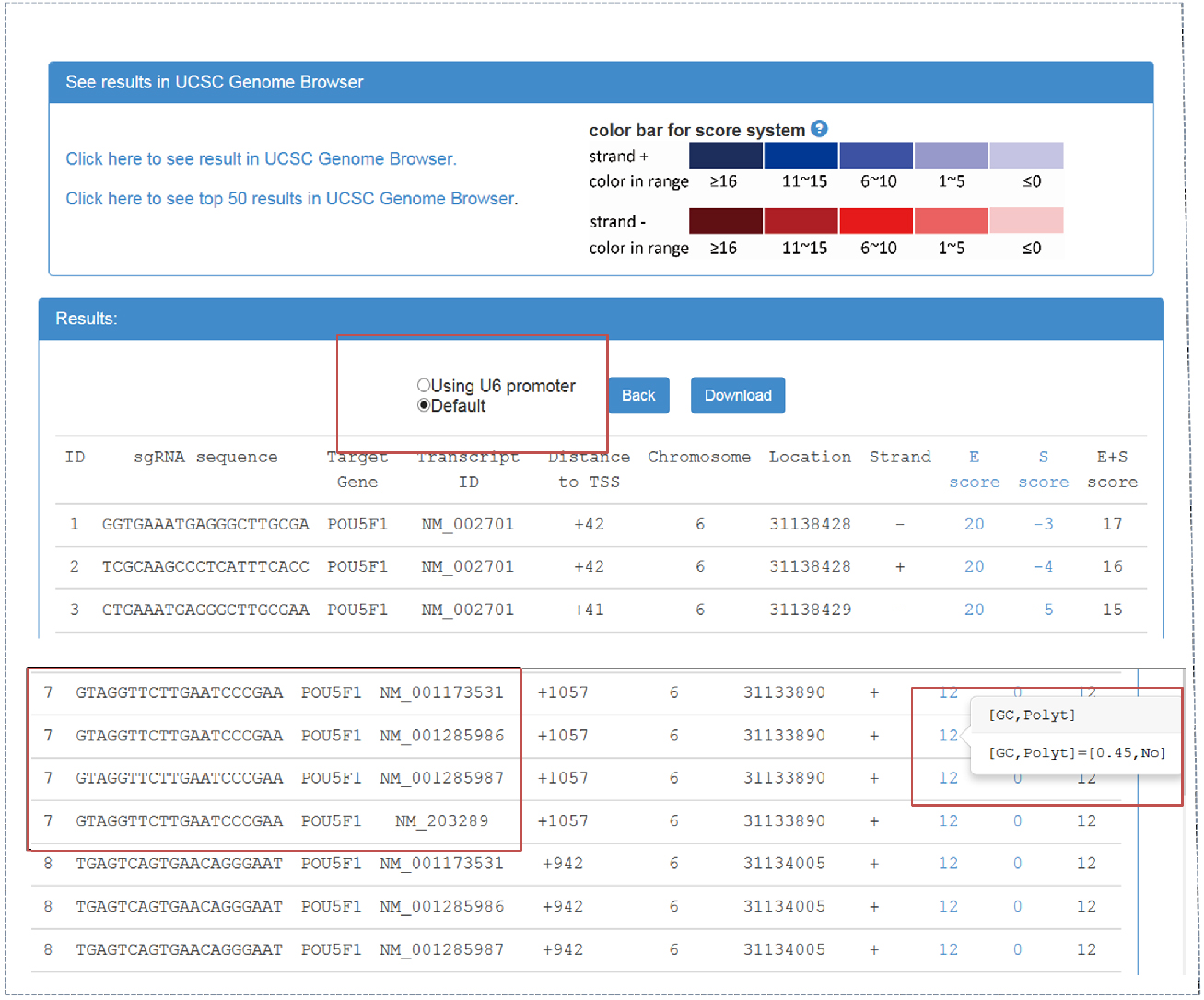
Figure 2. CRISPR-ERA output webpage
Output webpage contains two parts, ‘See results in UCSC Genome Browser’ and ‘Results’. - By clicking ‘click here to see result in UCSC Genome Browser’, CRISPR-ERA can show all the sequences on UCSC Genome Browser. sgRNA is identified by ‘ID’. The sum of E-score and S-score is represented by color shades referred to the color bar.
- Result table contains sgRNA sequences and their properties, such as target gene, transcript ID, distance to TSS, location, strand, etc. The sgRNAs starting with ‘G’ can be screened out, which could be applied in a CRISPR system using U6 promoter. When targetable region belongs to more than one transcript, the result table will show the information of all the transcripts, as shown in Figure 2.
- E-score and S-score columns contain the features that affect sgRNA efficiency and specificity. E-score and S-score are computed based on the criteria summarized from published data. E-score could represent the sgRNA efficacy, which contains GC content, poly-T presence and other sequence features. S-score shows the specificity of sgRNA sequence which is based on genome-wide off-target information. All sgRNA sequences can be downloaded.
- CRISPR-ERA webserver input (Figure 1)
- Genome-wide sgRNA library building pipeline
- Download genome sequence files in FASTA format and genome annotation files in RefFlat or GFF format, from UCSC genome browser or NCBI website. With genome version hg19 as an example, genome sequence and annotation files can be downloaded in http://hgdownload.soe.ucsc.edu/downloads.html.
- The Perl scripts can be received after the material transfer form is submitted, which allow 20 bp sgRNA searching with a default PAM (NGG) sequence and pattern (N20NGG). During the searching step, locations and strand information of all potential sgRNA target sites will be recorded.
Run Perl program:
perl find_all_sgRNA_z_f_c_y.pl hg19_dna.fa out_sgRNA.txt out_sgRNA_fasta.txt out_sgRNA_gc_t.txt out_nag_fasta.txt out_no_sgRNA.txt
# out_sgRNA: all potential sgRNA sequences
# out_sgRNA_fasta.txt: all potential sgRNA sequences with FASTA format for bowtie next step (with PAM sequence NGG)
#out_sgRNA_gc_t.txt: all sgRNA sequences with GC content and Poly T information
#out_nag_fasta.txt: all potential sgRNA sequences with FASTA format for bowtie next step (different with out_sgRNA_fasta.txt, PAM sequence here is NAG)
# out_no_sgRNA.txt: Number of sgRNA sequences in each chromosome.
Note: Help information of Perl can be found by commands ‘perl –h’, ‘perldoc perl’, or in http://learn.perl.org/. - Run Bowtie to find all possible off-target sequences (both PAM = NGG, PAM = NAG are considered) containing up to 3-bp mismatches for each sgRNA.
bowtie -v 2 -k 100 ./hg19 -f out_sgRNA_fasta.txt sgRNA_bowtie_fasta.txt
bowtie -v 2 -k 100 ./hg19 -f out_nag_fasta.txt sgRNA_nag_bowtie_fasta.txt
Note: Parameter setting of Bowtie can be found in http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml. - Compute the E-score and S-score by analyzing the sgRNA sequence features. E-score is computed by GC content and poly-T presence (mammalian only), and S-score is computed based on off-target information derived in step B3. Criteria can be customized, and differ in different organisms and gene manipulations (Figure 3).

Figure 3. An example of E-score and S-score computation. Sequence: GGTGAATGAGGGCTTGCGA. - Extract gene TSS location and coding region in genome annotation files. For gene editing, sgRNA target region is coding region. For gene repression, sgRNA targets a region from upstream -1.5 kbp to downstream 1.5 kbp from TSS, while the target region is -1.5 kbp upstream from TSS for gene activation. By hash searching the eligible sgRNA of these regions in the genome-wide sgRNA library derived in step B4, details of sgRNA for all genes are derived. Then update the E-score and S-score scores according to the additional target location information. Figure 3 is an E-score and S-score computation example of one sgRNA for Pou5f1 repression. The sgRNA database for different gene manipulations formed after the information above integrated.
- Download genome sequence files in FASTA format and genome annotation files in RefFlat or GFF format, from UCSC genome browser or NCBI website. With genome version hg19 as an example, genome sequence and annotation files can be downloaded in http://hgdownload.soe.ucsc.edu/downloads.html.
Data analysis
After finding the objective sgRNA sequences, the essential step is to evaluate the efficiency and specificity of each sgRNA sequence. In this protocol, we provide a general method to compute the E-score and S-score when building genome-wide sgRNA libraries. For sgRNA database for specific gene manipulations, other criteria should be included except the criteria for genome-wide sgRNA libraries, such as exon locations for gene editing and the distance to TSS for gene regulation. For example, efficiency reduces with a longer distance relative to TSS for gene regulation. The more detailed description of E-score and S-score could be found on the ‘Help’ webpage of CRISPR-ERA webserver (http://crispr.stanford.edu/help.jsp).
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 31371341), Tsinghua University Initiative Scientific Research Program (No. 20141081175), and the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University.
References
- Doudna, J. A. and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213): 1258096.
- Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M. and Haussler, D. (2002). The human genome browser at UCSC. Genome Res 12(6): 996-1006.
- Langmead, B., Trapnell, C., Pop, M. and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10(3): R25.
- La Russa, M. F. and Qi, L. S. (2015). The new state of the art: Cas9 for gene activation and repression. Mol Cell Biol 35(22): 3800-3809.
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X. and Qi, L. S. (2015). CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics 31(22): 3676-3678.
- Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P. and Lim, W. A. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5): 1173-1183.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Liu, H., Wang, X. and Qi, L. S. (2017). Using CRISPR-ERA Webserver for sgRNA Design. Bio-protocol 7(17): e2522. DOI: 10.21769/BioProtoc.2522.
Category
Molecular Biology > DNA > DNA cloning
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.