- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Macrophage Survival Assay Using High Content Microscopy
Published: Vol 7, Iss 16, Aug 20, 2017 DOI: 10.21769/BioProtoc.2509 Views: 7889
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Macrophages maintain tissue homoeostasis by regulating inflammation and tissue repair mechanisms. Thus, the fate of macrophages has an impact on the state of the tissue. The aim of this protocol is to quantify macrophage survival using high content microscopy and image processing software. Here, we describe a high-content image based protocol to assess the effect of diverse stimuli in combination with pharmacological treatments on macrophage survival in a quantitative, unbiased and high-throughput manner.
Keywords: MacrophageBackground
Macrophages are phagocytic innate immune cells and are the main drivers of inflammation in tissue (Medzhitov, 2008). These cells are associated with cancer together with autoimmune, autoinflammatory, infectious, neurodegenerative and metabolic diseases (Ginhoux and Jung, 2014). In this context, the role of macrophages in inflammation is well-studied, however, the impact of macrophage survival in non-infectious and infectious diseases is largely unknown. Our study showed that the activation of certain pathogen-associated receptors (PRRs) can induce macrophage survival (Eren et al., 2016). We described a molecular mechanism that demonstrated how an obligate intracellular pathogen exploits PRR-induced cell survival (Eren et al., 2016). Thus, further studies are necessary to understand the role of macrophage survival in different disease settings.
Materials and Reagents
- Sterile 1.5 ml tubes (Corning, Axygen®, catalog number: MCT-175-C )
- 25 G-needle
- 50 ml syringe (B. Braun Medical, catalog number: 4617509F-02 )
- Polypropylene conical 50 ml centrifuge tube (TPP Techno Plastic Products, catalog number: 91050 )
- 40 µM cell strainer (Corning, Falcon®, catalog number: 431750 )
- 90 mm Petri dish (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 101RTC )
- 96-well clear bottom cell-culture grade black imaging plates (Corning, Falcon®, catalog number: 353219 )
- Sterile reagent reservoir (VWR, catalog number: 89094-664 )
- 10 ml serological pipette (SARSTEDT, catalog number: 86.1254.001 )
- 25 ml serological pipette (SARSTEDT, catalog number: 86.1685.001 )
- 10 μl filtered barrier tip (Biotix, Neptune®, catalog number: BT10XL )
- 200 μl filtered tip low retention (CLEARLINE, catalog number: 713117 )
- 0.22 μm syringe-filter (Carl Roth, catalog number: P668.1 )
- Adhesive plate seal
- 6-to-9 week old specific-pathogen free C57BL/6 mice
- ddH2O
- Ethanol
- Macrophage colony stimulating factor (M-CSF) (ImmunoTools, catalog number: 12343115 )
- EDTA 0.5 M pH 8.0 solution (as described Reference 1)
- Pharmacological inhibitor
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10270106 )
- HEPES buffer (BioConcept, catalog number: 5-31F00-H )
- Penicillin-streptomycin (P/S) (BioConcept, catalog number: 4-01F00-H )
- Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific, GibcoTM, catalog number: 31966021 )
- Sodium hydroxide (NaOH)
- Hydrochloric acid (HCl)
- Paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: 76240 )
Note: This product has been discontinued. - Cell-culture grade Ca/Mg-free Dulbecco’s PBS (DPBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14040091 )
- Saponin (Sigma-Aldrich, catalog number: 84510 )
- DAPI (Thermo Fisher Scientific, InvitrogenTM, catalog number: D1306 )
- Phalloidin (Thermo Fisher Scientific, InvitrogenTM, catalog number: A12379 )
- Complete DMEM cell medium (see Recipes)
- 4% PFA solution (see Recipes)
- 5% Saponin solution (see Recipes)
- Staining solution (see Recipes)
Equipment
- 10-11 cm long stainless steel dissecting scissors
- 10-11 cm long stainless steel dissecting straight forceps
- Refrigerator centrifuge with 50 ml tube adapter
- Cell culture incubator
- Pipette controller
- Pipettes
- Cell counting chamber
- Finnpipette® 50-300 μl 12 channel multi-pipette
- Chemical hood
- Luminal flow hood
- Plate washer (BioTek Instruments, model: EL406 )
- High content microscope (such as Molecular Devices, model: ImageXpress Micro XL )
- 40x Plan Apo λ 0.95 NA objective (Nikon, catalog number: MRD00405 )
- pH meter
Procedure
- Preparation of murine bone-marrow derived macrophages
- Euthanize mice using carbon-dioxide. Keep scissors and forceps in 70% ethanol during the procedure. Make a small incision on the central dorsum. Peel the skin from the top of each hind leg and down over the foot. Cut off the hind legs at the hip bone with scissors. Carefully cut off the foot and hip bones without compromising the integrity of the tibia and femur. Separate the tibia and femur by severing at the joint.
- Remove excess muscle from the tibia and femur. Hold bones with forceps and spray with 70% ethanol.
- Place bones in sterile tubes containing ice-cold DMEM with P/S. Keep tubes on ice.
- Using forceps, soak the bones in 70% ethanol for a few seconds. Wait for the excess ethanol to drip off before cutting each extremity of the bone with scissors to expose the bone marrow cavity.
- Flush the bone marrow out, using a 25 G-needle attached to a 50 ml syringe containing 25 ml of ice-cold complete DMEM medium (see Recipes), into a sterile 50-ml canonical centrifuge tube. Filter the sample through a 40 μm cell strainer into a fresh 50-ml tube on ice.
Note: All liquid steps must be performed using sterile and pyrogen-free material under a laminar flow hood following good cell culture practice to avoid the activation of macrophages. - Centrifuge cells at 450 x g for 8 min at 4 °C.
- Discard the supernatant by inverting the tube in one single move. Resuspend cells in ice-cold complete DMEM containing 50 ng/ml M-CSF.
- Euthanize mice using carbon-dioxide. Keep scissors and forceps in 70% ethanol during the procedure. Make a small incision on the central dorsum. Peel the skin from the top of each hind leg and down over the foot. Cut off the hind legs at the hip bone with scissors. Carefully cut off the foot and hip bones without compromising the integrity of the tibia and femur. Separate the tibia and femur by severing at the joint.
- Culturing macrophages
- Count cells and adjust cell number to ~7 x 106/ml in complete DMEM containing M-CSF. Add ~7 x 105 cells per Petri dish in a final volume of 10 ml complete DMEM containing M-CSF. Incubate Petri dishes in a 37 °C, 5% CO2 incubator.
- On day 3, add 5 ml of fresh pre-warmed (37 °C) complete DMEM containing 50 ng/ml M-CSF to each plate.
- Count cells and adjust cell number to ~7 x 106/ml in complete DMEM containing M-CSF. Add ~7 x 105 cells per Petri dish in a final volume of 10 ml complete DMEM containing M-CSF. Incubate Petri dishes in a 37 °C, 5% CO2 incubator.
- Harvest macrophages
- On day 6, discard the culture supernatants. Immediately add 10 ml of ice-cold cell-culture grade sterile 1x PBS without calcium and magnesium, pH 7.4 containing 5 mM EDTA. Incubate Petri dishes ~3 min on ice.
- Hold the Petri dish at a 30 °C angle. Flush the Petri dish with its contents using a pipette controller and a sterile pipette. Transfer the detached cells to a 50 ml sterile tube containing one-sixth of the transferred volume of complete DMEM.
- Centrifuge the tube at 200 x g for 8 min at 4 °C.
- Count cells using a cell counting chamber or an automated cell counter. Adjust the cell concentration to 1.25 x 106 cells/ml.
- On day 6, discard the culture supernatants. Immediately add 10 ml of ice-cold cell-culture grade sterile 1x PBS without calcium and magnesium, pH 7.4 containing 5 mM EDTA. Incubate Petri dishes ~3 min on ice.
- Plating macrophages
Pipette 100 μl of cells into 96-well clear bottom black plates using a multi-channel pipette or an automated cell dispenser under a laminar flow hood (Figure 1). Incubate cells overnight in an incubator (37 °C, 5% CO2) to ensure attachment of cells.
Note: Cells should be dispensed on the edge of wells to have homogeneous distribution. Shaking or swirling of the plates must be avoided to prevent the accumulation of cells in certain areas of the wells (Figure 2).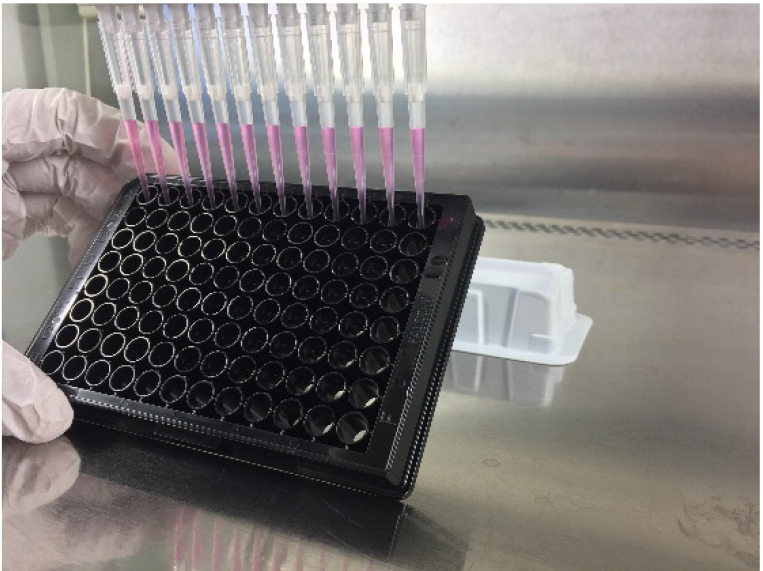
Figure 1. Aspiration and dispension of liquids should be performed on the edge of the plate. Hold the plate at an approximately 30° angle to the surface and hold the multipipette at a 120° angle to the plate when aspirating or dispensing liquid into the wells to avoid cell detachment.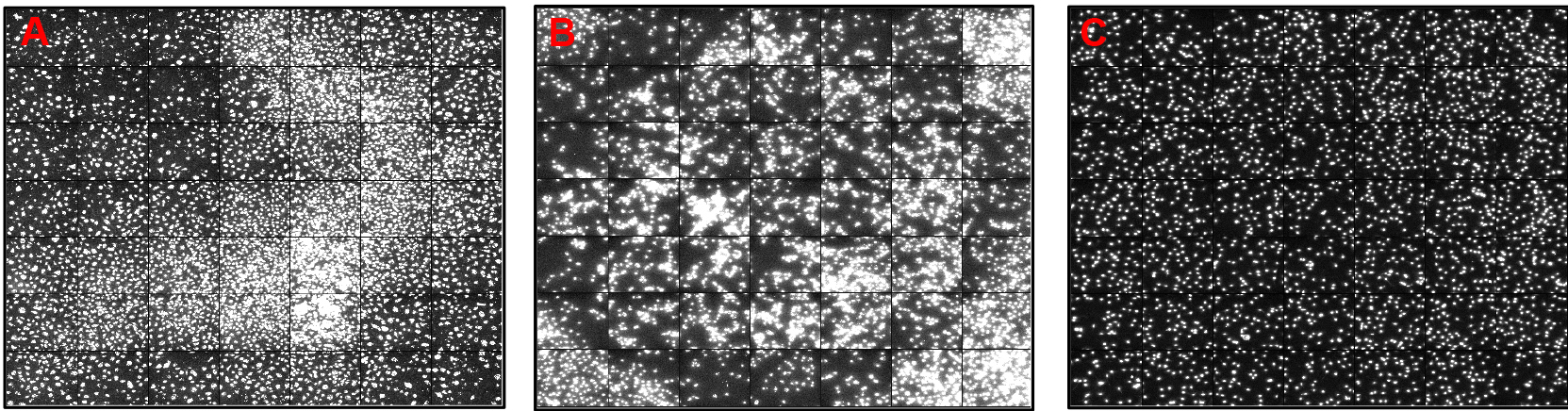
Figure 2. An example of 7 x 7 tile images from wells. A and B. Show a heterogeneous cell distribution that can be caused by cell plating, plate shaking/swirling, and/or aspiration/dispension. C. Shows a homogenous cell distribution. Images display DAPI-stained fluorescent nuclei of macrophages. - Treatment of macrophages
- Cells can be pre-treated or treated with a pharmacological inhibitor at an optimized concentration and for an optimized duration. In the case of pre-treatment, wash the wells with pre-warmed 37 °C complete DMEM, and add 100 μl of complete DMEM.
Notes:- Aspiration and dispension steps of media must be performed on the edge of wells to avoid detaching the cells (Figure 2).
- Following this protocol, perform a two-fold serial-dilution of a selective pharmacological inhibitor to determine non-toxic concentrations for primary macrophages. Test the potency of the pharmacological inhibitor at determined concentrations in the time course of the experiment using appropriate biochemical and/or cellular experimental approaches. We can recommend to use 1 μM staurosporine (negative control) to induce macrophage death and 10-50 ng/ml M-CSF (positive control) to induce macrophage survival and proliferation.
- Aspiration and dispension steps of media must be performed on the edge of wells to avoid detaching the cells (Figure 2).
- Treat the cells with stimuli of interest in 100 μl complete DMEM. Incubate cells at 37 °C, 5% CO2.
Note: The duration of incubation needs to be optimized for each individual experimental set-up. It should be noted that there is a basal reduction in macrophage number after 48 h of incubation in cell culture media.
- Cells can be pre-treated or treated with a pharmacological inhibitor at an optimized concentration and for an optimized duration. In the case of pre-treatment, wash the wells with pre-warmed 37 °C complete DMEM, and add 100 μl of complete DMEM.
- Fixation, staining, and washing steps
- Aspirate the cell supernatant using a multi-channel pipette or an automated plate washer.
- Dispense 50 μl of 4% PFA (see Recipes) in 1x PBS, pH 7.4 and incubate the plates at room temperature for 10 min.
- Wash the wells twice with 200 μl 1x PBS using a multi-channel pipette or an automated plate washer.
- Aspirate the supernatant, and dispense 50 μl staining solution. Incubate the plate with the staining solution (see Recipes) in a dark place for 10 min.
- Wash the wells twice with 200 μl 1x PBS using a multi-channel pipette or an automated plate washer.
- Seal plates with an adhesive plate seal either manually or using an automated plate sealer.
- Aspirate the cell supernatant using a multi-channel pipette or an automated plate washer.
- Image acquisition
- Clean the bottom of each 96-well plate with 70% ethanol.
- Acquire 49 images per well that consist of 7 x 7 images, 200 μm apart from each other, using a high content microscope equipped with a 40x objective.
- Clean the bottom of each 96-well plate with 70% ethanol.
- Counting cells with nuclear and F-actin staining
- Cell scoring object function of Metaexpress was used to segment and define all nuclei (DAPI channel). As a positive marker, cytoplasm staining (phalloidin channel) was used (Figure 3A). Approximate minimum and maximum width were set at 5 μm and 30 μm for ‘all nuclei’ and ‘positive marker’ parameters (Figure 3B).
Note: Image analysis can be performed using ImageJ, Icy or Cell-profiler that are freeware image-analysis programs. - A logical operation that merges nuclei and cytoplasmic objects was performed to define a cell as an object in the algorithm (Figure 3B).
- Cells that had cytoplasm bordering the image were removed from the image to count cells only in the field of view (Figure 3B).
- The cell count was taken as output data from the image analysis pipeline.
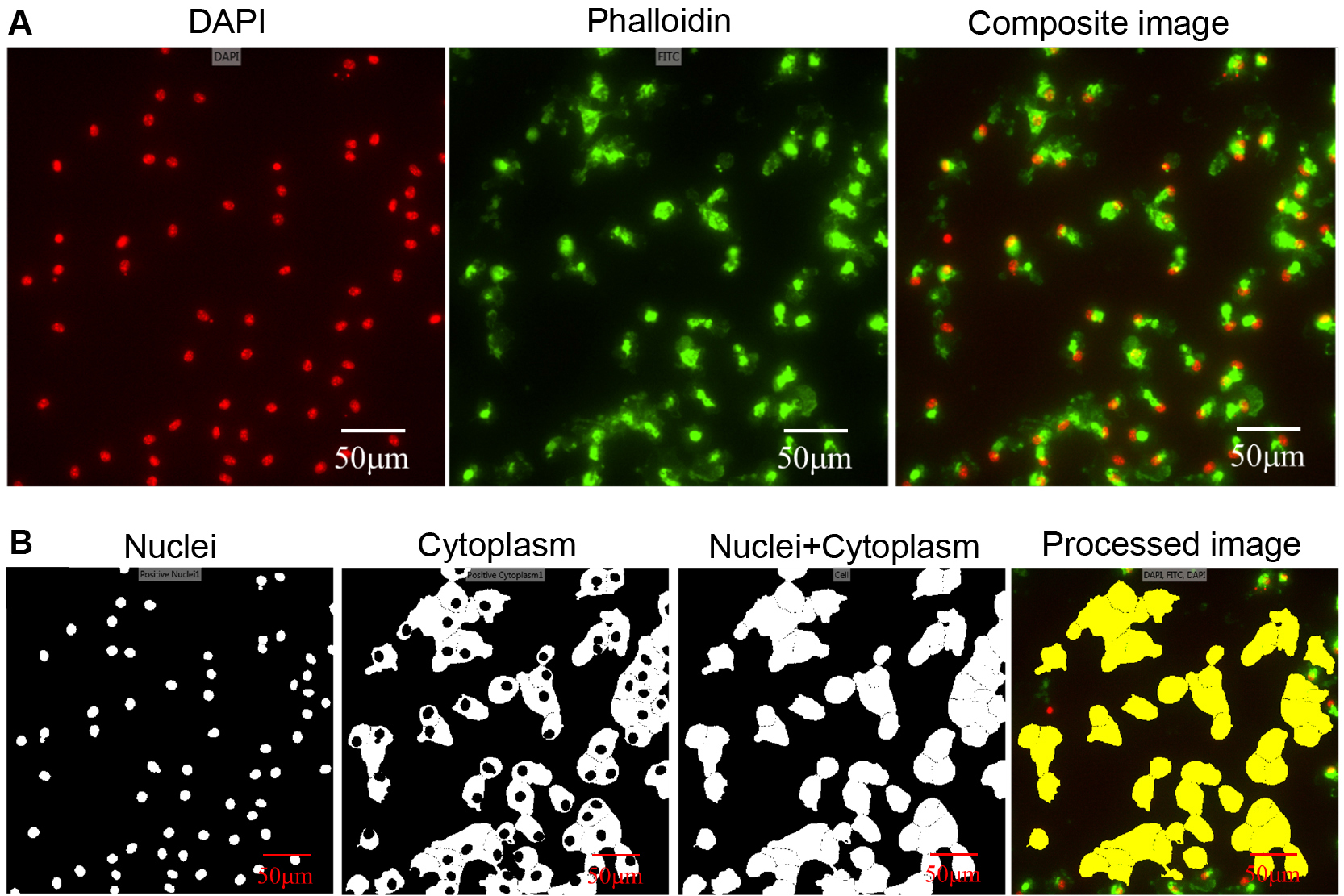
Figure 3. The image processing and object segmentation pipeline. A. Unprocessed DAPI, phalloidin, and composite image; B. Object segmentation of nuclei and cytoplasm is shown. A cell is defined by merging nuclei and cytoplasm binary images. Bordering cells were removed in the final processed image that was used to quantify cell number.
- Cell scoring object function of Metaexpress was used to segment and define all nuclei (DAPI channel). As a positive marker, cytoplasm staining (phalloidin channel) was used (Figure 3A). Approximate minimum and maximum width were set at 5 μm and 30 μm for ‘all nuclei’ and ‘positive marker’ parameters (Figure 3B).
Data analysis
Each independent experiment should be performed with three technical or biological replicates. In order to pool the independent experiments, the data can be normalized to the cell count in untreated condition or to the cell count in vehicle-treated condition if a pharmacological inhibitor was used. A bar graph and the appropriate statistical test can be used to represent the data.
Recipes
- Complete DMEM cell medium
50 ml FBS
5 ml HEPES
5 ml P/S
500 ml DMEM
Filter FBS, HEPES and P/S into DMEM using a 0.22 μm filter and a 50 ml syringe - 4% PFA solution
45 ml ddH2O
1 N NaOH
1 N HCl
5 ml 10x PBS
2 g PFA
Heat ddH2O to 60 °C. Add 2 g PFA into the tube while it is stirring under a chemical hood. Add a few drops of 1 N NaOH. Wait until the solution becomes clear. Add 10x PBS and adjust the pH to 7.4 using a pH meter and 1 N HCl. Filter the solution using a 0.22 μm and a 50 ml syringe. Aliquot and store at -20 °C - 5% Saponin solution
50 ml 1x DPBS
2.5 g Saponin
Dissolve Saponin by stirring. Filter the solution using a 0.22 μm and a 50 ml syringe. Aliquot and store at -20 °C - Staining solution
5 μg/ml DAPI (nucleic acid stain) (stock concentration 5 mg/ml)
1 U/ml Phalloidin (F-actin stain) (stock concentration 200 U/ml)
0.05% (w/v) Saponin (stock concentration 5%, w/v)
1x DPBS, pH 7.4
Acknowledgments
We thank Dimitri Monreau and Cansel Ustunel Eren for assistance with the high-content microscopy and Slavica Masina for critical reading of the manuscript. We thank the NCCR Geneva Access platform for providing the equipment for the high-content microscopic experiments. This work was funded by grants from the Swiss National fund for research (FNRS 310030-153204 and IZRJZ3_164176, N.F.), the Institute for Arthritis Research (iAR), and the COST action (CM1307 SEFRI: C14.0070, N.F.). This protocol was adapted from the protocol described in Eren et al. (2016).
References
- EDTA. (2006). Cold Spring Harbor Protoc pdb.rec8030.
- Eren, R. O., Reverte, M., Rossi, M., Hartley, M. A., Castiglioni, P., Prevel, F., Martin, R., Desponds, C., Lye, L. F., Drexler, S. K., Reith, W., Beverley, S. M., Ronet, C. and Fasel, N. (2016). Mammalian innate immune response to a Leishmania-resident RNA virus increases macrophage survival to promote parasite persistence. Cell Host Microbe 20(3): 318-328.
- Ginhoux, F. and Jung, S. (2014). Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14(6): 392-404.
- Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature 454(7203): 428-435.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Eren, R. and Fasel, N. (2017). Macrophage Survival Assay Using High Content Microscopy. Bio-protocol 7(16): e2509. DOI: 10.21769/BioProtoc.2509.
Category
Immunology > Immune cell imaging > High content microscopy
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.