- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A High-throughput Assay for mRNA Silencing in Primary Cortical Neurons in vitro with Oligonucleotide Therapeutics
Published: Vol 7, Iss 16, Aug 20, 2017 DOI: 10.21769/BioProtoc.2501 Views: 11354
Reviewed by: Longping Victor TseEhsan KheradpezhouhSébastien Gillotin
Original research article
The authors used this protocol in:

Oct 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Primary neurons represent an ideal cellular system for the identification of therapeutic oligonucleotides for the treatment of neurodegenerative diseases. However, due to the sensitive nature of primary cells, the transfection of small interfering RNAs (siRNA) using classical methods is laborious and often shows low efficiency. Recent progress in oligonucleotide chemistry has enabled the development of stabilized and hydrophobically modified small interfering RNAs (hsiRNAs). This new class of oligonucleotide therapeutics shows extremely efficient self-delivery properties and supports potent and durable effects in vitro and in vivo. We have developed a high-throughput in vitro assay to identify and test hsiRNAs in primary neuronal cultures. To simply, rapidly, and accurately quantify the mRNA silencing of hundreds of hsiRNAs, we use the QuantiGene 2.0 quantitative gene expression assay. This high-throughput, 96-well plate-based assay can quantify mRNA levels directly from sample lysate. Here, we describe a method to prepare short-term cultures of mouse primary cortical neurons in a 96-well plate format for high-throughput testing of oligonucleotide therapeutics. This method supports the testing of hsiRNA libraries and the identification of potential therapeutics within just two weeks. We detail methodologies of our high throughput assay workflow from primary neuron preparation to data analysis. This method can help identify oligonucleotide therapeutics for treatment of various neurological diseases.
Keywords: Primary cortical neuronsBackground
Oligonucleotide therapeutics represent a new class of drug that can target any genetically defined disorder, by silencing the expression of mutant proteins. Specifically, siRNAs are double stranded oligonucleotides that are loaded into the RNA induced silencing complex (RISC) and can silence mRNA before it is translated. However, unmodified siRNAs are unstable and cannot enter cells without the help of cationic lipid formulation, which can be toxic to primary cells such as neurons. In this protocol, we use self-delivering, hydrophobically modified siRNAs (hsiRNAs) for mRNA silencing. Recent progress in the chemistry of oligonucleotides has enabled the design of these stabilized hsiRNAs, which promote cellular internalization, efficient entry into RISC, and potent knockdown of target genes (Byrne et al., 2013; Alterman et al., 2015; Ly et al., 2017). These compounds contain 2’-O-methyl and 2’-fluoro modifications on all sugars and phosphorothioate backbone modifications; the oligonucleotides are often conjugated to a hydrophobic moiety, such as cholesterol, to support membrane binding and cellular internalization without toxicity. This new class of compounds offers researchers a straightforward method for silencing various genes in the context of biologically relevant, and hard to transfect, primary cortical neurons (Alterman et al., 2015).
Today, in the early stage of drug discovery, most high-throughput tests are performed in cell-based assays for lead oligonucleotide identification and validation. Cell-based assays improve and accelerate drug screening, providing more relevant in vivo biological information than biochemical assays and reducing the need for animal testing. High-throughput assays performed in primary neurons have emerged as a powerful tool to discover new therapies for the treatment of neurodegenerative disorders, such as Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) or Alzheimer’s disease (AD) (Sharma et al., 2012). Cell based assays using primary neurons provide a more natural (relevant) environment for studying neurodegenerative disorders than clonal neuronal lines. Transfecting oligonucleotide-based therapeutics into primary neurons generally relies on approaches such as electroporation (Mertz et al., 2002; Gresch et al., 2004; Zhang et al., 2016), viral transduction (Naldini et al., 1996; Hughes et al., 2002; Janas et al., 2006) or lipid-mediated transfection (Ohki et al., 2001; Dalby et al., 2004; Zhang et al., 2016). However, these methods can be laborious, show low efficiency, and induce cellular toxicity. Thus, the self-delivering properties of hsiRNAs represent an effective method to identify new leads in vitro in complex cellular models, such as primary neurons. We have recently demonstrated that hsiRNAs efficiently bind neuronal-cell membranes within seconds after treatment, enter cells, and induce potent gene silencing, both in vitro, in primary neurons, and in vivo, in mouse brain, all without the use of transfection reagents (Alterman et al., 2015; Ly et al., 2017).
Our laboratory has established a rapid high-throughput platform to identify and validate hsiRNA leads in primary neurons in vitro in 96-well format. To quantify the amount of target mRNA upon hsiRNA treatment, we use the QuantiGene 2.0 branched DNA (bDNA) assay, a high-throughput, 96-well plate-based mRNA quantification assay. This technique is designed to directly quantify the target mRNA from sample lysate without the need to purify the RNA, thus minimizing sample manipulation (Kern et al., 1996; Coles et al., 2015). This assay enables accurate and precise detection of even low abundance mRNAs, minimizing experimental variability and error (Collins et al., 1997; Canales et al., 2006). The combination of both self-delivering hsiRNAs and high-throughput quantification of mRNA accelerates the identification of efficacious oligonucleotides in primary neurons.
Here, we describe the workflow of this high throughput assay for performing large-scale screening and dose response validation of hsiRNAs in primary cortical neurons. We detail our methods for primary cortical neuron preparation in 96-well plate format (Figure 2), neuronal treatment with hsiRNA, assay plate management, mRNA quantification, and data analysis. This platform can be used for the screening of oligonucleotide therapeutics in primary neurons for the potential treatment of neurodegenerative diseases.
Materials and Reagents
- Poly-L-lysine pre-coated tissue culture treated 96-well plate (Corning, catalog number: 356516 )
- Deep 96-well sterile polypropylene plate (Corning, Axygen®, catalog number: 391-04-062 )
- Tips (from 0.2 μl to 1 ml) (VWR)
- Serological pipettes, individually wrapped (from 5 ml to 50 ml) (Costar)
- Fire-polished Pasteur pipet with cotton plug (made in-house)
- Tissue culture treated 10 cm dish (Corning, catalog number: 430167 )
- 50 ml conical centrifuge tubes (Corning, Falcon®, catalog number: 352097 )
- 15 ml conical centrifuge tubes (Corning, Falcon®, catalog number: 352098 )
- 1.7 ml microcentrifuge tubes (Genesee Scientific, catalog number: 22-282 )
- 1 ml sterile syringe (BD, catalog number: 309659 )
- Adhesive plate seals (VWR, catalog number: 60941-126 )
- Pregnant wild-type mice (THE JACKSON LABORATORY)
- Cholesterol-conjugated hsiRNAs (designed and produced in-house)
- Ice-cold block (Koolit® Refrigerants) (Cold Chain Technologies, catalog number: 305F )
- Poly-L-lysine (Sigma-Aldrich, catalog number: P4707 )
- Phosphate-buffered saline (PBS) (Mediatech, catalog number: 21-031-CV )
- 200-Proof ethanol (Decon Labs, catalog number: 2701 )
- DMEM cell culture medium (Mediatech, catalog number: 10-013-CV )
- Hibernate E (BrainBits, catalog number: HE )
- DNase I (Worthington, catalog number: 54M15168 )
- Papain (Worthington, catalog number: 54N15251 )
- Trypan blue stain solution (Thermo Fisher Scientific, GibcoTM, catalog number: 15250061 )
- QuantiGene 2.0 Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: QS0011 )
- QuantiGene 2.0 Probe sets (varies by gene)
- Lysis mixture (Thermo Fisher Scientific, AffymetrixTM, catalog number: 13228 )
- Proteinase K (Thermo Fisher Scientific, AffymetrixTM, catalog number: QS0103 )
- Neurobasal medium NbActiv4 (BrainBits, catalog number: Nb4-500 )
- Fetal bovine serum (FBS) (Mediatech, catalog number: 35-010-CV )
- NeuralQTM medium (Sigma-Aldrich, catalog number: N3100 ) supplemented with GS21TM supplement (50x) (Sigma-Aldrich, catalog number: G0800 ), 0.5 mM dipeptide Ala-Gln (Sigma-Aldrich, catalog number: A8185 )
- 5’UTP (Sigma-Aldrich, catalog number: U6625 )
- 5’FdU (Sigma-Aldrich, catalog number: F3503 )
- Papain/DNase solution (see Recipes)
- Plating medium (see Recipes)
- Feeding medium (see Recipes)
Equipment
- Micropipettes from 0.5 μl to 1 ml (Labnet International, model: BioPetteTM Plus )
- Multichannel (8 or 12 channels) micropipettes from 10 μl to 300 μl (Eppendorf, model: Research® plus )
- Tissue culture incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: HeracellTM 150i )
- Biological safety cabinet connected to vacuum (Thermo Fisher Scientific, Thermo ScientificTM, model: 1300 Series Class II, Type A2 )
- Bunsen burner
- Germinator 500 (Braintree Scientific, model: Germinator 500, catalog number: GER 5287-120V )
- Water bath at 37 °C (Fisher Scientific, model: Model 2332 )
- 4 °C fridge
- Microscissors (Fine Science Tools, catalog numbers: 14060-10 and 14002-12 )
- Set of two forceps (Fine Science Tools, catalog number: 11251-30 )
- Dissection microscope (Motic, model: SMZ168 Series )
- Pipet-aid (Drummond Scientific, model: Portable Pipet-Aid® XP )
- Tissue culture phase-contrast inverted microscope (Motic, model: AE2000 )
- Hemocytometer (0.1000-0.0025 mm2) (Neubauer)
- µPlate carrier (Beckman Coulter, model: SX4750 )
- Autoplate washer (BioTek Instruments, model: ELx405 )
- Refrigerated swing rotor benchtop centrifuge (Beckman Coulter, model: Allegra X-15R )
- Allegra X-15R rotor (Beckman Coulter, model: SX4750 )
- Plate reader spectrophotometer (Tecan Trading, model: Infinite M1000 Pro )
Software
- Microsoft Office Excel (Microsoft Office)
- GraphPad Prism 7 software (GraphPad Software)
Procedure
- Poly-L-lysine coating of tissue culture treated 96-well plates
- To coat the plates, transfer 100 μl of 0.01% poly-L-lysine to each well using a multichannel micropipette.
- Incubate overnight in the tissue culture incubator at 37 °C.
- The next day, before the dissection, aspirate the coating solution using a vacuum-connected Pasteur pipet (or a 200 μl multichannel micropipette) and rinse three times with 100 μl PBS.
- Remove the final wash and let the plate dry in the biological safety cabinet.
Note: It is also possible to purchase and use 96-well plates pre-coated by the manufacturer, to store at room temperature. We did not observe any difference between data from cells plated in 96-well plates pre-coated in our laboratory or by the manufacturer.
- To coat the plates, transfer 100 μl of 0.01% poly-L-lysine to each well using a multichannel micropipette.
- Preparation of cotton-plugged, fire-polished Pasteur pipet
- Using a Bunsen burner, polish the cotton plugged Pasteur pipet extremity by holding the pipet oriented with the capillary tube in the flame for 2-3 sec, rotating rapidly.
- Check the opening of the capillary tube, ensuring that the edge is round and the hole is not too small. If the hole is too small, discard the pipet, as it will sheer neurites during the trituration (Figure 1).
- Autoclave the Pasteur pipets to sterilize before use.
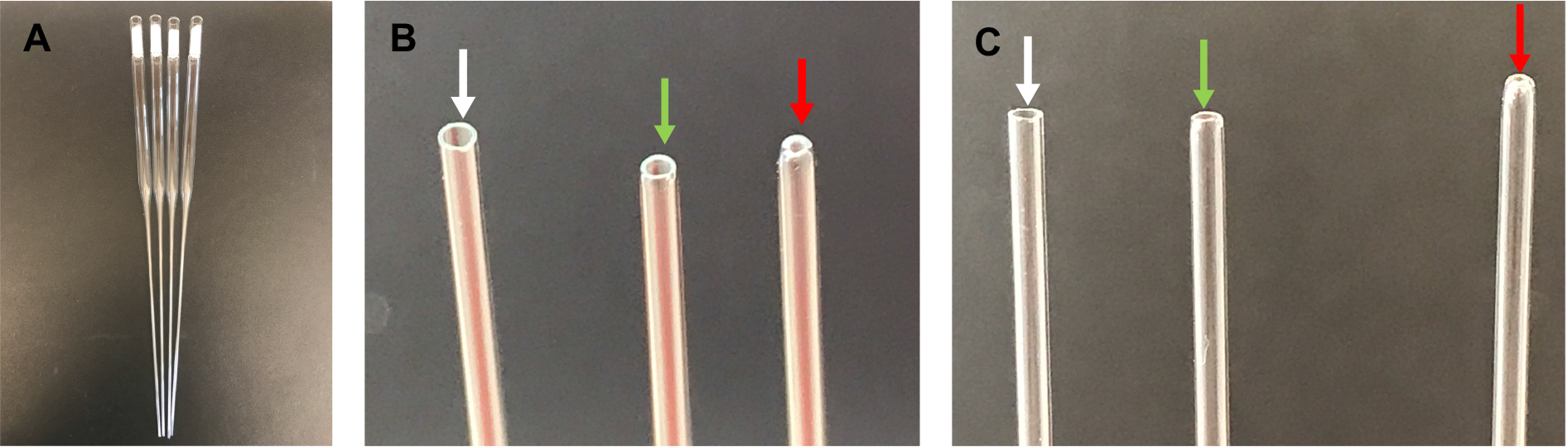
Figure 1. Fire-polished pipettes. A. Cotton-plugged, fire-polished pipettes; B. From left to right: unpolished tip (edges are too sharp–pointed with white arrow), correctly polished tip (edges are smooth–pointed with green arrow), over-polished tip (opening is too small–pointed with red arrow); C. Same as in B.
- Using a Bunsen burner, polish the cotton plugged Pasteur pipet extremity by holding the pipet oriented with the capillary tube in the flame for 2-3 sec, rotating rapidly.
- Isolation of mouse primary cortical neurons (Figure 2)
Note: To avoid contamination, sterilize instruments for use during the preparation. Between each step, tools can be kept in 70% ethanol or sterilized by heat in a Germinator.
Primary cortical neurons are isolated from E15.5 mouse embryos of pregnant mice. Before starting, prepare plating medium required during the procedure and, pre-warm at 37 °C by incubation in a water bath.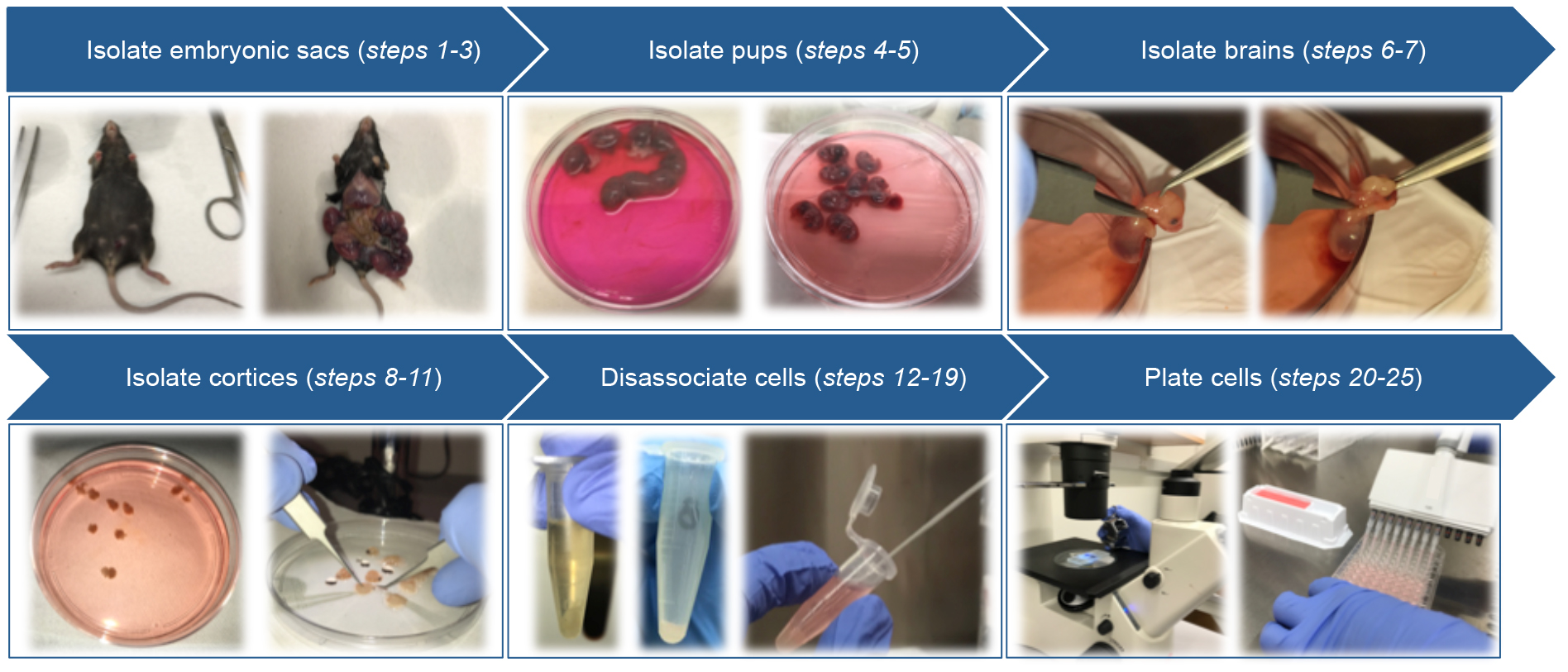
Figure 2. Schematic of primary cortical neuron isolation- Euthanize pregnant mice with CO2 followed by cervical dislocation.
Note: This is one example of proper procedure, each investigator should follow his or her approved IACUC protocol. - Lay the mouse on her back and clean the abdominal surface with 70% ethanol to sterilize the area.
- Using microscissors, make a vertical incision in the abdominal muscles to open the abdominal cavity of the mouse. Using forceps, carefully remove the embryonic sacs and rinse them in cell culture medium (DMEM, ice cold, no additives).
- Place the embryonic sacs in a 10 cm dish containing 10 ml of ice-cold DMEM.
- Using microscissors and forceps, carefully open the embryonic sacs, remove the embryos and transfer to a 10 cm dish containing 10 ml of ice-cold DMEM.
- Place the forceps near the back of the embryos skull and pull forward toward the nose.
- Place the forceps on either side of the head, beneath the brain, and carefully lift the brain out of the skull (Video 1).
 Video 1. Removal of the brain from the skull
Video 1. Removal of the brain from the skull - Transfer the brains to a 10 cm dish containing 10 ml of ice-cold Hibernate E.
- Transfer brains to single droplets of Hybernate E in 10 cm dishes under the dissecting microscope for easy access.
- Under a dissecting microscope, use forceps to carefully peel away the meninges from the upper surface of each brain hemisphere. The cortex will fall away from the brain after the meninges are removed (Figure 3B).
- Using the forceps, carefully separate the cortices from the brain.
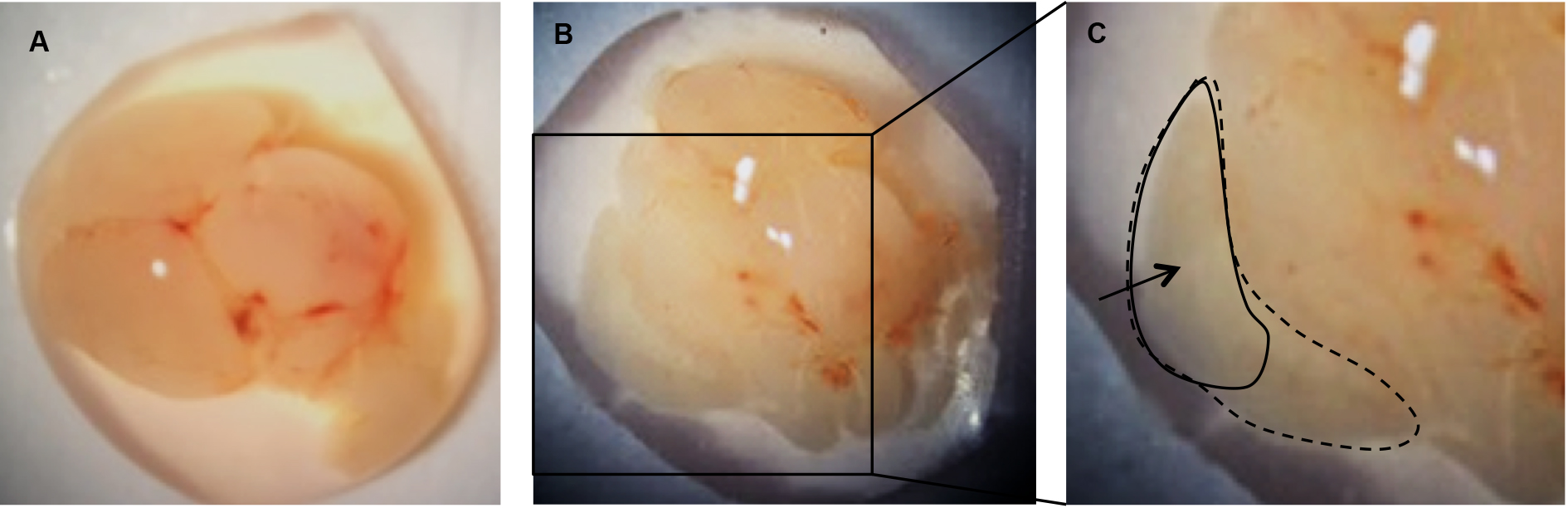
Figure 3. Remove the meninges to release the cortices. A. Individual brain removed from skull; B. Cortex from one hemisphere following removal of the meninges; C. Cortex from one hemisphere (dotted line). Area of cortex to be transferred to papain/DNase (solid line). - Transfer the upper, outer portion of the cortices (Figure 3C) into a 1.5 ml microcentrifuge tube containing 1 ml of pre-warmed papain/DNase solution and close the tube while collecting the remainder of the cortices to keep away as much contamination as possible.
- Incubate the tube open, in a sterile 10 cm dish for 30 min at 37 °C to dissolve the tissue. This allows the tissue to breathe while keeping it in a sterile environment.
- Place the tube upright for a few minutes to allow the cortices to sediment at the bottom of the tube.
- Without disturbing the cortices, slowly and carefully remove the papain/DNase solution using a 1 ml micropipette (be sure to remove as much papain/DNase solution as possible) and add 1 ml of plating medium.
- Dissociate the cells with a gentle and consistent motion of pipetting up and down several times through a cotton-plugged, fire-polished Pasteur pipet using a pipet-aid adjusted to the lowest speed. It is important to avoid the formation of bubbles. Triturate the tissue until the medium turns slightly cloudy, and no remaining tissue clumps are visible (Video 2). Video 2. Trituration of cortices
- Transfer the cells to a 15 ml conical tube and add 4 ml of plating medium. To ensure small clumps of cells are broken up, perform an additional trituration by gently and consistently pipetting up and down several times through a cotton-plugged, fire-polished Pasteur pipet using a pipet-aid adjusted to the lowest speed.
- Let the solution sit for 2 min to allow any debris to settle to the bottom of the 15 ml tube
- Transfer the supernatant containing the single cell neuronal suspension to a new 15 ml tube.
- Transfer a small drop of cells to a tissue culture treated dish and, using a tissue culture microscope (10x magnification), observe if there is a single cell suspension (Figure 4). If not, perform another trituration using a cotton-plugged, fire-polished Pasteur pipet, as previously described.
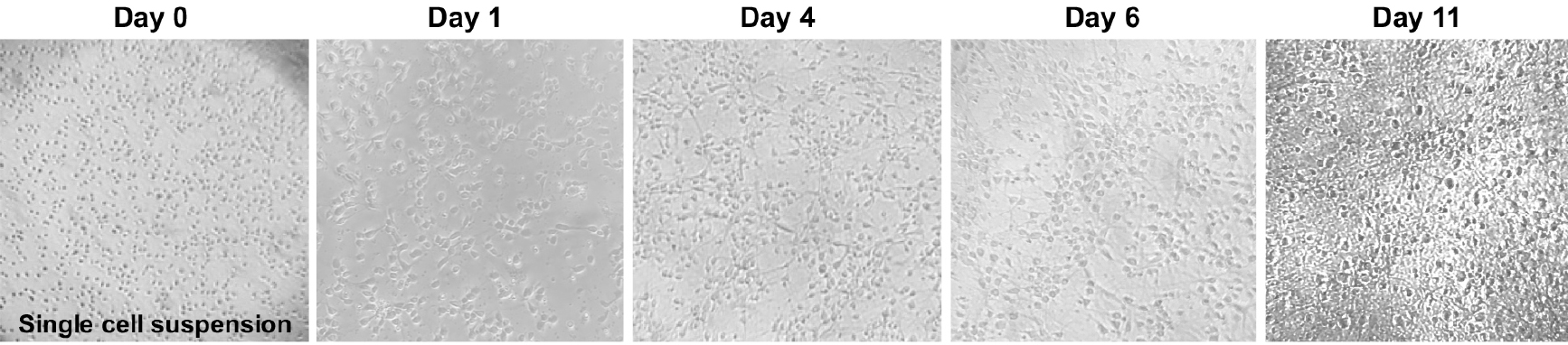
Figure 4. Primary cortical neuron morphology. Phase contrast images (4x objective) of mouse primary cortical neurons showing the neuronal morphology acquisition over time. - Using trypan blue stain solution and a hemocytometer, count the live cells present in the cell suspension. Add 10 μl of cells to 90 μl of trypan blue stain solution and mix gently by pipetting. Transfer a 10 μl aliquot to the hemocytometer and count the live cells from all four quadrants of the hemocytometer. Average the four numbers and multiply by 1 x 105. This number corresponds to the number of cells per ml of medium. On average, each pup will yield ~5-10 x 106 cells.
- Transfer the cells to a 50 ml conical tube and adjust the density of viable cells with pre-warmed plating medium to 1 x 106 cells/ml. For example, if the cell concentration is 5 x 106 cells/ml in 5 ml, add 20 ml of plating medium to obtain a final cell concentration of 1 x 106 cells/ml in 25 ml final volume.
- Close and invert the tube to ensure a thorough mixture of the cell suspension. Transfer the cells to a 50 ml reagent reservoir.
- Using a 200 μl multichannel micropipette, transfer 100 μl of cells (1 x 105 cells) to each well of a poly-L-lysine pre-coated 96-well plate. To seed an equal number of cells in each well, mix the cells in the reservoir by pipetting up and down before each transfer into the 96-well plate.
- Incubate the plates overnight at 37 °C, 5% CO2 in a tissue culture incubator.
- The following day, using a multichannel micropipette, slowly add 100 μl of feeding medium (containing anti-mitotics) to each well to prevent the growth of non-neuronal cells. Be careful not to disturb the cells.
- Every 3-4 days, replace half of the volume of media with freshly prepared feeding medium. Using a multichannel micropipette, slowly remove 100 μl of medium from each well without touching the bottom of the well to avoid disturbing the cells. Very gently, add back 100 μl of freshly prepared and pre-warmed feeding medium without disturbing the cells.
- Store in the tissue culture incubator at 37 °C. Repeat feeding step as necessary.
- Euthanize pregnant mice with CO2 followed by cervical dislocation.
- Treatment of primary neurons with oligonucleotide therapeutics
Note: Oligonucleotides are stored at -20 °C. Prior to any experiment, thaw the compounds on ice. This procedure is performed in a biological biosafety cabinet.
Prepare the oligonucleotide dilutions in feeding medium. Carefully design your plate map to include the oligonucleotides of interest (hsiRNATarget), the non-targeting control (hsiRNANTC) and the untreated cells. Compound is prepared at two times the final concentration so that 50 µl compound solution can be added to 50 µl of the conditioned medium. Be sure to prepare enough compound to treat cells in the number of desired replicates. For the purposes of this protocol, we describe a preparation for three replicate wells per compound and a final concentration of 1.5 µM hsiRNA.- In a deep 96-well plate, prepare a 2x hsiRNA master mix by diluting the stock hsiRNA in fresh feeding medium containing anti-mitotics to a final concentration of 3 µM (two times the final concentration of 1.5 µM) in a final volume of 200 µl.
- Mix gently up and down with the micropipette.
- Using a micropipette, gently remove the entire conditioned medium from the neurons and immediately add back 50 µl of conditioned medium to each well (you can combine the conditioned medium into a sterile reagent reservoir for easy pipetting back into the plate). Only remove medium from one or two rows at a time to prevent the cells from drying. To avoid disturbing the cells, pipet down the medium slowly with the tips touching the edge of the wells. This step is intended to control for any evaporation that may have happened during incubation at 37 °C, and to ensure that all cells are treated with the same concentration of hsiRNA.
- Using a multichannel micropipette and clean tips for each row, transfer 50 μl of the 2x hsiRNA master mix from the deep 96-well plate into each of the replicate wells of the cell culture plate for a final volume of 100 µl per well.
- Incubate the plate in a tissue culture incubator at 37 °C. For a one week long incubation, feed the cells once with 100 µl freshly prepared feeding medium (3-4 days after treatment). Then lyse the cells one week after treatment. This step ensures that no hsiRNA will be removed from the wells during the experiment. For longer term experiments, where cells need to be fed more than once, remove 100 µl of conditioned medium from the 200 µl in the cell culture plate and add back 100 µl of fresh medium. Continue this pattern of feeding every 3-4 days.
- In a deep 96-well plate, prepare a 2x hsiRNA master mix by diluting the stock hsiRNA in fresh feeding medium containing anti-mitotics to a final concentration of 3 µM (two times the final concentration of 1.5 µM) in a final volume of 200 µl.
- mRNA quantification
mRNA is quantified using the QuantiGene 2.0 bDNA Assay (Affymetrix). All the volumes used in the following steps are for one plate. Prepare all the solutions used for this procedure in clean 50 ml conical tubes using clean serological pipettes.- In a 50 ml conical tube, prepare the diluted lysis mixture (DLM; 1:2 v/v, lysis mixture/water, e.g., 10 ml lysis mixture:20 ml water) complemented with 100 μl of 50 μg/μl Proteinase K (provided with the kit) and pour it into a 50 ml reservoir. Diluted lysis mixture should be prepared fresh for every experiment. Storing the diluted lysis mixture in the fridge will cause precipitate to form.
- Remove all growth medium from the plate.
- Using a 300 μl multichannel pipette, add 250 μl of diluted lysis mixture in each well of the 96-well culture plate. There is enough lysate to perform the quantification of the target and the control mRNA.
- To lyse the cells, gently pipette up and down avoiding the formation of bubbles. Use clean tips between each row.
- Incubate for 30 min at 55 °C in a forced air oven.
- Using a 300 μl multichannel pipette setup at 150 µl, mix the cell lysate thoroughly by gently pipetting up and down avoiding the formation of bubbles.
- Add 20 μl of probe set, diluted according to the kit protocol to each well of the capture plate (provided with the kit).
- Using a 100 μl multichannel pipette, transfer the appropriate amount of each lysate to the capture plate. Avoid touching the bottom of the coated plate. For background wells, simply add DLM (no Proteinase K) instead of cell lysate. Prior to testing a new gene using the QuantiGene 2.0 kit, you will need to validate the correct volume of lysate to obtain the proper degree of luminescent signal (at least a 5 fold signal to noise ratio, and within the linear range of detection for the plate reader you are using). This can be tested by measuring signal from different amounts of lysate (between 5 and 80 μl) and plotting the corresponding luminescent values. Housekeeping and target genes may require different volumes of lysate in the same experiment.
- Top off each well to 100 μl with DLM (no Proteinase K).
- Seal the plate very tightly with an aluminum plate seal (provided with the kit) and centrifuge for 1 min at 240 x g at room temperature.
- Incubate over night at 55 °C in a forced air oven.
- The next day perform signal amplification as recommended by Affymetrix.
- Read the luminescence using a microplate reader setup for a 200-500 msec reading time per well.
- Analyze the data.
- In a 50 ml conical tube, prepare the diluted lysis mixture (DLM; 1:2 v/v, lysis mixture/water, e.g., 10 ml lysis mixture:20 ml water) complemented with 100 μl of 50 μg/μl Proteinase K (provided with the kit) and pour it into a 50 ml reservoir. Diluted lysis mixture should be prepared fresh for every experiment. Storing the diluted lysis mixture in the fridge will cause precipitate to form.
Data analysis
Note: Data are processed using Microsoft Office Excel and analyzed using GraphPad Prism 7 software.
- For each plate and each individual probeset, average the values obtained in the DLM (blank) wells and subtract the values from all sample wells.
- To normalize the target gene expression, divide the target gene luminescent value by the housekeeping gene luminescent value.
- Average the normalized untreated cell values.
- To calculate the percent of target gene expression relative to untreated cells, divide each individual normalized well by the average of the untreated wells and multiply by 100.
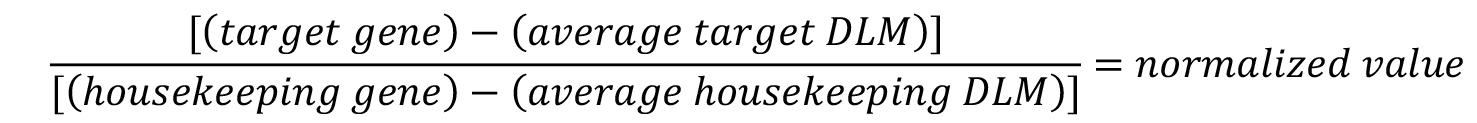
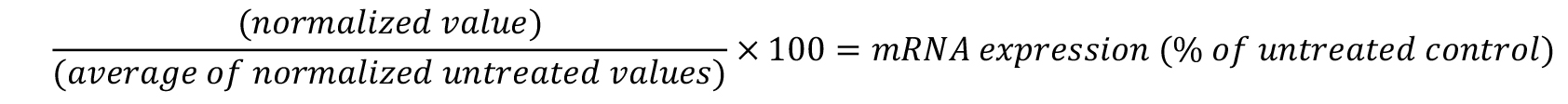
- If compounds are tested in replicate, average the replicates and calculate the standard deviation.
- For dose response analysis, graph concentration-dependent IC50 curves using a non-linear regression curve fit, log(inhibitor) vs. response–variable slope (four parameters). If necessary, set the lower limit of the curve at zero, and the upper limit of the curve at 100.
- Compare efficacy of target sequences to the non-targeting control (NTC) to determine statistical significance. Differences in all comparisons are considered significant at P-values less than 0.05 compared with the NTC group.
Notes
- After dissociation, cells look round if the trituration was gentle.
- Cells should show signs of making neurites by two hours after plating. If they are still round without neurites on the second day, they will probably not grow.
- Cells should be plated as a single cell suspension. If cells are doublets or triplets, they tend to grow in clumps, which will affect the data of the experiment, as well as the cell growth.
- Pay attention to the neuronal medium lot. Lot to lot variability can affect the clumpiness, growth, and viability of the cells.
- Particular attention should be given to the hsiRNA modification pattern. Functionally efficient hsiRNA requires extensive chemical stabilization. Chemical modification of the ribose with 2’-O-methyl and 2’-fluoro results in a significant increase of the hsiRNA-resistance to nuclease degradation, providing higher stability in vitro and in vivo (Byrne et al., 2013; Alterman et al., 2015). Unmodified or partially modified hsiRNAs will undergo nuclease degradation, affecting functional efficacy in culture.
- We have observed that at high concentrations of hsiRNA (2 µM and up) induces the formation of cell clumps.
- If the cells are clumping or pulling away from the edge of the well (aggregating at the center of the well) later in the hsiRNA incubation period, this is a sign of unhealthy treatment or plating conditions.
Recipes
Note: For all recipes and at any step of the procedure use Milli-Q-purified water.
- Papain/DNase solution
- Dissolve Papain in 2 ml Hibernate E and 1 ml PBS and mix gently by inverting the vial 2-3 times (Do not vortex)
- Separately, suspend DNase in 0.5 ml Hibernate E and gently invert the vial 2-3 times
- The final Papain solution contains 0.25 ml of suspended DNase mixed in 3 ml of Papain solution
- Plating medium
NbActiv4 supplemented with 2.5% FBS
For 50 ml of plating medium, add 1.25 ml of 100% FBS in 48.75 ml of NbActiv4
Alternatively, use NeuralQTM medium supplemented with GS21TM supplement (50x), 0.5 mM dipeptide Ala-Gln and 2.5% FBS - Feeding medium
NbActiv4 supplemented with anti-mitotics, 0.484 μl/ml of 5’UTP and 0.2402 μl/ml of 5’FdU
For 50 ml of NbActiv4, add 24.2 μl of 5’UTP and 12 μl of 5’FdU
Alternatively, use NeuralQTM medium supplemented with GS21TM supplement (50x), 0.5 mM dipeptide Ala-Gln, and anti-mitotics 0.484 μl/ml of 5’UTP and 0.2402 μl/ml of 5’FdU
Acknowledgments
We thank all the members of the Khvorova and Aronin Laboratories, NIH and CHDI Foundation Inc. for helpful discussions. Specifically, we would like to thank Kathryn Chase in the Aronin Laboratory, from whom the primary neuron isolation protocol was originally adapted. This work is supported in part by grants from the NIH UH2-TR000888 and UH3-4UH3TR000888-03, NIH NS38194 and CHDI Foundation (Research Agreement A-6119). The authors declare no conflict of interest.
References
- Alterman, J. F., Hall, L. M., Coles, A. H., Hassler, M. R., Didiot, M. C., Chase, K., Abraham, J., Sottosanti, E., Johnson, E., Sapp, E., Osborn, M. F., Difiglia, M., Aronin, N. and Khvorova, A. (2015). Hydrophobically modified siRNAs silence huntingtin mRNA in primary neurons and mouse brain. Mol Ther Nucleic Acids 4: e266.
- Byrne, M., Tzekov, R., Wang, Y., Rodgers, A., Cardia, J., Ford, G., Holton, K., Pandarinathan, L., Lapierre, J., Stanney, W., Bulock, K., Shaw, S., Libertine, L., Fettes, K., Khvorova, A., Kaushal, S. and Pavco, P. (2013). Novel hydrophobically modified asymmetric RNAi compounds (sd-rxRNA) demonstrate robust efficacy in the eye. J Ocul Pharmacol Ther 29(10): 855-864.
- Canales, R. D., Luo, Y., Willey, J. C., Austermiller, B., Barbacioru, C. C., Boysen, C., Hunkapiller, K., Jensen, R. V., Knight, C. R., Lee, K. Y., Ma, Y., Maqsodi, B., Papallo, A., Peters, E. H., Poulter, K., Ruppel, P. L., Samaha, R. R., Shi, L., Yang, W., Zhang, L. and Goodsaid, F. M. (2006). Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol 24(9): 1115-1122.
- Coles, A. H., Osborn, M. F., Alterman, J. F., Turanov, A. A., Godinho, B. M., Kennington, L., Chase, K., Aronin, N. and Khvorova, A. (2015). A high-throughput method for direct detection of therapeutic oligonucleotide-induced gene silencing in vivo. Nucleic Acid Ther 26: 86-92.
- Collins, M. L., Irvine, B., Tyner, D., Fine, E., Zayati, C., Chang, C., Horn, T., Ahle, D., Detmer, J., Shen, L. P., Kolberg, J., Bushnell, S., Urdea, M. S. and Ho, D. D. (1997). A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml. Nucleic Acids Res 25(15): 2979-2984.
- Dalby, B., Cates, S., Harris, A., Ohki, E. C., Tilkins, M. L., Price, P. J. and Ciccarone, V. C. (2004). Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 33(2): 95-103.
- Didiot, M. C., Hall, L. M., Coles, A. H., Haraszti, R. A., Godinho, B. M., Chase, K., Sapp, E., Ly, S., Alterman, J. F., Hassler, M. R., Echeverria, D., Raj, L., Morrissey, D. V., DiFiglia, M., Aronin, N. and Khvorova, A. (2016). Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol Ther 24(10): 1836-1847.
- Gresch, O., Engel, F. B., Nesic, D., Tran, T. T., England, H. M., Hickman, E. S., Korner, I., Gan, L., Chen, S., Castro-Obregon, S., Hammermann, R., Wolf, J., Muller-Hartmann, H., Nix, M., Siebenkotten, G., Kraus, G. and Lun, K. (2004). New non-viral method for gene transfer into primary cells. Methods 33(2): 151-163.
- Hughes, S. M., Moussavi-Harami, F., Sauter, S. L. and Davidson, B. L. (2002). Viral-mediated gene transfer to mouse primary neural progenitor cells. Mol Ther 5(1): 16-24.
- Janas, J., Skowronski, J. and Van Aelst, L. (2006). Lentiviral delivery of RNAi in hippocampal neurons. Methods Enzymol 406: 593-605.
- Kern, D., Collins, M., Fultz, T., Detmer, J., Hamren, S., Peterkin, J. J., Sheridan, P., Urdea, M., White, R., Yeghiazarian, T. and Todd, J. (1996). An enhanced-sensitivity branched-DNA assay for quantification of human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 34(12): 3196-3202.
- Ly, S., Navaroli, D. M., Didiot, M. C., Cardia, J., Pandarinathan, L., Alterman, J. F., Fogarty, K., Standley, C., Lifshitz, L. M., Bellve, K. D., Prot, M., Echeverria, D., Corvera, S. and Khvorova, A. (2017). Visualization of self-delivering hydrophobically modified siRNA cellular internalization. Nucleic Acids Res 45(1): 15-25.
- Mertz, K. D., Weisheit, G., Schilling, K. and Luers, G. H. (2002). Electroporation of primary neural cultures: a simple method for directed gene transfer in vitro. Histochem Cell Biol 118(6): 501-506.
- Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F. H., Verma, I. M. and Trono, D. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272(5259): 263-267.
- Ohki, E. C., Tilkins, M. L., Ciccarone, V. C. and Price, P. J. (2001). Improving the transfection efficiency of post-mitotic neurons. J Neurosci Methods 112(2): 95-99.
- Sharma, P., Ando, D. M., Daub, A., Kaye, J. A. and Finkbeiner, S. (2012). High-throughput screening in primary neurons. Methods Enzymol 506: 331-360.
- Zhang, X. S., Huang, J., Zhan, C. Q., Chen, J., Li, T., Kaye, A. D., Wu, S. X. and Xiao, L. (2016). Different influences of lipofection and electrotransfection on in vitro gene delivery to primary cultured cortex neurons. Pain Physician 19(3): 189-196.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Alterman, J. F., Coles, A., Hall, L. M., Aronin, N., Khvorova, A. and Didiot, M. (2017). A High-throughput Assay for mRNA Silencing in Primary Cortical Neurons in vitro with Oligonucleotide Therapeutics. Bio-protocol 7(16): e2501. DOI: 10.21769/BioProtoc.2501.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Neuroscience > Nervous system disorders > Animal model
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.