- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Liposome Disruption Assay to Examine Lytic Properties of Biomolecules
Published: Vol 7, Iss 15, Aug 5, 2017 DOI: 10.21769/BioProtoc.2433 Views: 14270
Reviewed by: Venkatasalam ShanmugabalajiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Proteins may have three dimensional structural or amino acid features that suggest a role in targeting and disrupting lipids within cell membranes. It is often necessary to experimentally investigate if these proteins and biomolecules are able to disrupt membranes in order to conclusively characterize the function of these biomolecules. Here, we describe an in vitro assay to evaluate the membrane lytic properties of proteins and biomolecules. Large unilamellar vesicles (liposomes) containing carboxyfluorescein at fluorescence-quenching concentrations are treated with the biomolecule of interest. A resulting increase in fluorescence due to leakage of the dye from liposomes and subsequent dilution in the buffer demonstrates that the biomolecule is sufficient for disrupting liposomes and membranes. Additionally, since liposome disruption may occur via pore-formation or via general solubilization of lipids similar to detergents, we provide a method to distinguish between these two mechanisms. Pore-formation can be identified and evaluated by examining the blockade of carboxyfluorescein release with dextran molecules that fit the pore. The methods described here were used to determine that the malaria vaccine candidate CelTOS and proapoptotic Bax disrupt liposomes by pore formation (Saito et al., 2000; Jimah et al., 2016). Since membrane lipid binding by a biomolecule precedes membrane disruption, we recommend the companion protocol: Jimah et al., 2017.
Keywords: MembraneBackground
This protocol presents the procedure to evaluate the membrane lytic properties of proteins and other biomolecules. This protocol aims to provide a clear description of the various experimental steps necessary to study the membrane disrupting properties of biomolecules. Finally, the protocol describes a quantitative measurement of membrane disruption and can be applied to provide insight into the kinetics and mechanism of liposome disruption. This protocol was successfully used to study the malaria vaccine candidate CelTOS that provided a clear description of the first in vitro functional assay for CelTOS. The application of this protocol revealed that CelTOS (cell traversal protein for ookinetes and sporozoites) disrupts membranes containing phosphatidic acid. This insight suggests that CelTOS is secreted by malaria parasites within invaded host cells to disrupt the host cell membrane and enable the exit of parasites (Jimah et al., 2016). Finally, this assay can be readily applied to investigate the inhibition of CelTOS-mediated liposome disruption by small molecules, antibodies or peptides. While designed for CelTOS, this protocol is readily generalizable and applicable to any other biomolecule of interest.
Materials and Reagents
- Centrifuge tubes, 50 ml (Genesee Scientific, catalog number: 21-106 )
- Microcentrifuge tubes, 1.7 ml (Midsci, catalog number: AVSS1700RA )
- Parafilm (Bemis, catalog number: PM996 )
- Glass Pasteur pipets (Fisher Scientific, catalog number: 13-678-20C )
- Gravity sizing column (Bio-Rad Laboratories, catalog number: 7371032 )
- Glass tubes with plain end (Fisher Scientific, catalog number: 14-961-27 )
- Ring stand (Fisher Scientific, catalog number: S13747 )
- Clamps (Fisher Scientific, catalog number: 02-217-005 )
- Whatman filter (GE Healthcare, catalog number: 230300 )
- 200 nm polycarbonate Track-Etched filters (GE Healthcare, catalog number: 800281 )
- Carboxyfluorescein (Sigma-Aldrich, catalog number: C0662 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: S8045 )
- Potassium hydroxide (KOH) (Sigma-Aldrich, catalog number: P5958 )
- Sephadex G® G-25 (Sigma-Aldrich, catalog number: G25300 )
- Phospholipids (dissolved in chloroform, as supplied by manufacturer). Commonly used phospholipids are:
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids, catalog number: 850375C )
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, catalog number: 850457C )
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA) (Avanti Polar Lipids, catalog number: 840857C )
1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS) (Avanti Polar Lipids, catalog numbers: 840034C ) - Nitrogen gas tank (Airgas, catalog number: NI HP300 )
- Triton-X 100 (Sigma-Aldrich, catalog number: X100 )
- Dextran molecules of different molecular weights and radii of gyration:
5 kDa (Sigma-Aldrich, catalog number: 31417 )
9 kDa (Sigma-Aldrich, catalog number: D9260 )
39 kDa (Sigma-Aldrich, catalog number: D4133 )
Note: This product has been discontinued.
66.9 kDa (Sigma-Aldrich, catalog number: D1537 )
Note: This product has been discontinued.
148 kDa (Sigma-Aldrich, catalog number: D4876 )
500 kDa (Sigma-Aldrich, catalog number: D5251 )
Note: This product has been discontinued.
1,500-2,800 kDa (Sigma-Aldrich, catalog number: D5376 ) - HEPES (Sigma-Aldrich, catalog number: H3375 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9333 )
- Ethyl ether (Fisher Scientific, catalog number: E138-500 )
- Ferric(III) chloride hexahydrate (Sigma-Aldrich, catalog number: 236489 )
- Ammonium thiocyanate (Sigma-Aldrich, catalog number: 221988 )
- Buffer-KCl (see Recipes)
- Buffer-NaCl (see Recipes)
- 0.1 M ammonium ferrothiocyanate (see Recipes)
Equipment
- Glass beaker (Fisher Scientific, catalog number: FB1012000 )
- pH meter (Fisher Scientific, model: AccumetTM AE150, catalog number: 13-636-AE153 )
- Stir bar
- BenchRockerTM variable 2D rocker (Benchmark Scientific, catalog number: BR2000 )
- Water sonicator (Laboratory Supplies, model: G112SP1T )
- Gas pressure regulator (Linde, catalog number: UPE-3-75-580 )
- Freeze-dry system for lyophilisation (Labconco, catalog number: 7750021 )
- Hi-vac flask (Labconco, catalog number: 7544400 )
- Vortexer (Fisher Scientific, catalog number: 02-215-414 )
- Rotary flask evaporator: Rotovap (IKA, model: RV 8 FLEX , catalog number: 0010002178)
- Round bottom flask, for rotovap (IKA, catalog number: 0003743200 )
- Mini extruder (Avanti Polar Lipids, catalog number: 610023 )
- Centrifuge (Beckman Coulter, model: J6-MI )
- Cary Eclipse fluorescence spectrophotometer (Varian Inc./Agilent, Santa Clara, CA, USA)
- UV/Vis/NIR spectrophotometer (capable of measuring optical density at 488 nm for Stewart assay) (Beckman Coulter, model: DU-640 )
Software
- Microsoft Excel
- GraphPad Prism
Procedure
- Preparing 20 mM carboxyfluorescein
- Weigh out appropriate mass of carboxyfluorescein to make 20 mM carboxyfluorescein in a desired volume.
Note: 40 ml is enough to make 80 preparations of liposomes. Each preparation is sufficient for 500 experimental assays. - Add ~70% of the target volume of buffer to the carboxyfluorescein powder.
Note: Commonly used buffers are buffer-KCl and buffer-NaCl (see the Recipes section). Identical buffers should also be used in all experiments, including all liposome and protein preparation steps. - While stirring the solution with a stir bar, titrate with 1 N KOH or NaOH a drop at a time.
Note: Be sure to not go above a pH of 8 as increasing the pH causes carboxyfluorescein to precipitate, and pH also affects the fluorescence spectra. - After the precipitate dissolves and the solution becomes dark, adjust the final pH to 7.4.
Note: The concentrated dark solution is fluorescence quenched. - Add appropriate volume of buffer to obtain a final concentration of 20 mM carboxyfluorescein.
- Weigh out appropriate mass of carboxyfluorescein to make 20 mM carboxyfluorescein in a desired volume.
- Preparation of the size-exclusion chromatography column
- Suspend 1 g of Sephadex® G-25 resin with 35 ml of buffer in a 50 ml conical tube.
Note: Commonly used buffers are buffer-KCl and buffer-NaCl. - Gently shake the tube at 30 rpm for 12-16 h at room temperature.
- Add about 2 ml of buffer to the gravity column, and resin slurry in aliquots frequently enough to avoid layering in the packed column.
Note: The buffer provides resistance to the resin as it settles into the column and prevents the resin from forming channels. - During aliquot addition allow the column to flow slowly and continuously.
Note: Avoid introducing air bubbles when packing the column, and maintain buffer on top of the resin to prevent it from drying or forming channels. - Fill the ~30 ml gravity column about 95% to the top.
Note: Leave room at the top to add sample (liposomes). It is important to be able to see the top and bottom meniscus of dark orange colored liposomes through the clear glass stem of the sizing column. - The column can be reused indefinitely provided it has not dried out, the resin is not broken, and there are no air bubbles trapped in the resin.
Note: The column should be washed after each use, and equilibrated 3 times with 1 column volume of buffer before use.
- Suspend 1 g of Sephadex® G-25 resin with 35 ml of buffer in a 50 ml conical tube.
- Drying phospholipids
- Test glass tubes for integrity by sonicating for 30 sec in a water sonicator.
- Aliquot 10 mg of lipid (supplied by manufacturer with lipids dissolved in chloroform) into glass vial.
- Clamp the vial to a stand while it is immersed in a beaker filled with water (Figure 1).

Figure 1. Drying phospholipids - Evaporate off all of the chloroform by flowing a nitrogen stream over the phospholipid solution until there is a thin layer of lipid.
- Cover the test tube with Parafilm, and poke a small hole in the Parafilm.
- Place the tube in a hi-vac flask, and lyophilize under vacuum for 3 h.
- After drying the lipid, fill the vial with nitrogen and cover with Parafilm.
Note: Dried lipids can usually be stored for 1-3 months at -20 °C.
- Test glass tubes for integrity by sonicating for 30 sec in a water sonicator.
- Making large unilamellar vesicles (liposomes) containing carboxyfluorescein
- Add 500 µl of ethyl ether and 500 µl of the 20 mM carboxyfluorescein solution to the glass vial containing the dried lipid.
- Cover the vial with Parafilm.
Note: Provide excess Parafilm because the Parafilm expands as the volatile gases escape from the vial. - Vortex for 5 sec to dissolve the lipids in the solution of ether and carboxyfluorescein.
Note: A layer of ether will sit on top of the green layer of carboxyfluorescein. - Sonicate the vial in a water sonicator three times for 30 sec each.
Note: Vortex sample for 15 sec between each sonication step. For optimal sonication, the vial should be at the center of the harmonic standing wave that forms in the water sonicator. - Use a Pasteur pipette to transfer the water-ether-phospholipid suspension into a round bottom flask.
- Attach the round bottom flask to a rotary flask evaporator to remove the ether.
Note: Using a water pump vacuum start the vacuum low to avoid excess bubbling and to reduce bumping. Gradually increase the vacuum, for about a minute at a time as the ether is removed. - Release the vacuum and remove the round bottom flask from the roto-vap spindle. Sniff to determine if all the ether has evaporated. Usually, all the ether has evaporated 3 min after the sample stops bubbling while under the highest vacuum.
Notes:- Use the ‘wafting technique’ when sniffing.
- At this step liposomes containing carboxyfluorescein will have typically formed as a suspension in the remaining buffer. However, it is critical to obtain liposomes of uniform size by extrusion.
- Use the ‘wafting technique’ when sniffing.
- Assemble the extruder (Avanti mini-extruder) with a 100 nm diameter cut off filter following the manufacturer’s directions (Figure 2).
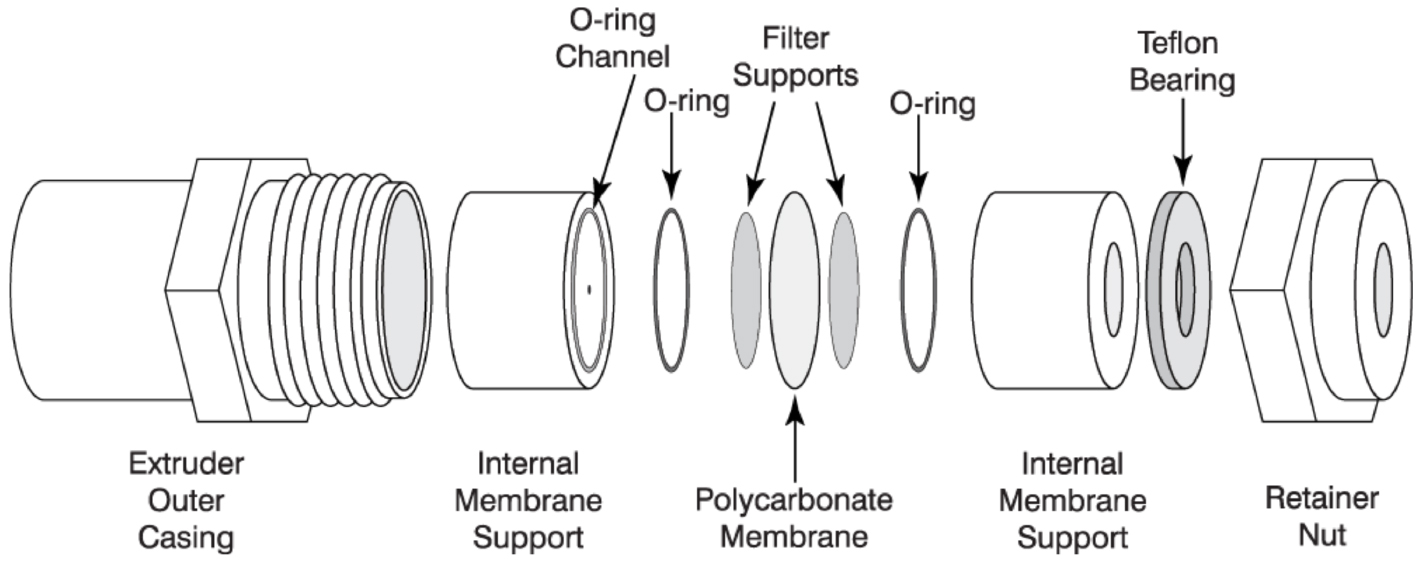
Figure 2. Assembly of Avanti mini-extruder (Avanti Polar Lipids)
Notes:- Image from ‘Mini-Extruder Assembly Instructions’. Avanti Polar Lipids. Web. 19 June 2017 https://avantilipids.com/divisions/equipment/mini-extruder-assembly-instructions/.
- All components above are included in the Avanti mini-extruder (Avanti Polar Lipids) except the ‘filter supports’ (Sigma-Aldrich) and ‘polycarbonate membrane’ (Sigma-Aldrich).
- Image from ‘Mini-Extruder Assembly Instructions’. Avanti Polar Lipids. Web. 19 June 2017 https://avantilipids.com/divisions/equipment/mini-extruder-assembly-instructions/.
- Pull buffer into one syringe and place it opposite to an empty syringe on the extrusion chamber. Gently inject the buffer across the extrusion chamber and membrane into the empty syringe.
Note: Injecting the buffer across the extruder wets and prepares it for liposomes. Empty out the receiving syringe, and place it back into the extruder. - Fill the injector syringe with the liposome suspensions and inject it through the extruder and passing it between the syringes five times. The liposome suspension will be at the receiving syringe. At this step, the solution contains liposomes that contain carboxyfluorescein and are of ~200 nm in diameter. However, the solution also contains a high concentration of carboxyfluorescein outside the liposomes, which needs to be removed.
- Equilibrate the gravity sizing column with desired final buffer, of the same molarity as the buffer used for the liposome preparation.
- Remove all of the buffer placing the meniscus on the top of the resin.
- Gently add the liposome sample to the top of the resin, while causing minimal disruption of the packed resin.
- Allow the column to flow, and stop it as soon as the entire liposome sample meniscus enters the resin.
- Gently add buffer over the top of the beads to fill up the column. The dark orange/greenish colored liposome sample and the resin should remain intact (Figure 3A).
- Allow the column to run.
Note: The liposomes, containing carboxyfluorescein, exceed the pore size of the resin and will elute in the void volume of the column. The free carboxyfluorescein molecules elute later because they enter the resin beads as they pass through the column (Figure 3B). - Collect the elution fractions containing liposomes, which are dark orange in color (Figure 1B).
Note: The free carboxyfluorescein is bright green and elutes after the liposomes. Expect about 0.5-1 ml of liposomes since liposomes were made by dissolving lipids in 500 µl of ether and carboxyfluorescein. - To determine the integrity of the liposomes, dilute the liposome fraction 1,000 fold and measure the fluorescence intensity at 512 nm upon excitation at 492 nm. Addition of 0.1% Triton-X 100 to the liposomes should result in approximately a threefold increase in fluorescence intensity.

Figure 3. Purifying liposomes containing carboxyfluorescein. A. Addition of sample to size-exclusion chromatography column. S: sample containing a mixture of liposomes containing carboxyfluorescein (L+CF), and free carboxyfluorescein outside liposomes (CF). B. Separation of liposome sample into two fractions. CF: free carboxyfluorescein that is bright green. L+CF: liposomes containing carboxyfluorescein that is orange in color.
- Add 500 µl of ethyl ether and 500 µl of the 20 mM carboxyfluorescein solution to the glass vial containing the dried lipid.
- Determining the lipid concentration
Note: If numerical studies are desired it is useful to determine the lipid content in the liposome preparation using the Stewart assay (Stewart, 1980). Lipid is lost during preparation especially during extrusion and gel filtration. The assay is based on the formation of a complex formed from the phospholipid and ammonium ferrothiocyanate, and sensitive for a lipid range of 0.01-0.1 mg.- To generate a standard curve, dissolve known amounts of lipids covering a range between 0.01 and 0.1 mg of lipid in chloroform to 2 ml. Five to ten points between 0.01 and 0.1 mg of lipid is sufficient to make a standard curve.
- To determine the concentration of lipids in the prepared liposome sample, add an appropriate amount of chloroform to obtain a final volume of 2 ml.
Note: Since 10 mg of lipid was used to prepare liposomes, ensure that the sample is diluted enough to be in the range of 0.01 and 0.1 mg. - Add 2 ml of 0.1 M ammonium ferrothiocyanate (see Recipes) to each of the 2 ml volumes of chloroform containing known amounts of lipid and the test liposome sample.
- Vortex the sample three times for 30 sec each, and centrifuge at 300 x g. With centrifugation, two layers will form.
- Collect the lower layer with a Pasteur pipette.
- Measure the optical density at 488 nm, and make a graph of the optical density versus lipid amount to obtain a standard curve.
- The concentration of lipid in the liposome sample can be determined from the standard curve.
- To generate a standard curve, dissolve known amounts of lipids covering a range between 0.01 and 0.1 mg of lipid in chloroform to 2 ml. Five to ten points between 0.01 and 0.1 mg of lipid is sufficient to make a standard curve.
- Preparing the biomolecule with potentially membrane lytic properties
- Ensure that the biomolecule is dissolved in the same buffer as the liposomes.
Notes:- Commonly used buffers are buffer-KCl and buffer-NaCl. Remove detergent from the biomolecule or include detergent containing controls.
- When testing a biomolecule for the first time, it is critical to use protein of good quality. Impurities, poor handling, and storage can lead to degradation, or aggregation of the protein which may impact the activity and result in the inaccurate quantification of protein activity. The type of purification needed to obtain homogenous protein samples, such as gravity column based affinity chromatography or gel filtration is dependent on each protein. It is recommended to use protein that elutes from a gel filtration column as a single monodisperse species. The purity of the protein can be determined by SDS-PAGE where the target protein is ~90% of the total protein sample.
- Commonly used buffers are buffer-KCl and buffer-NaCl. Remove detergent from the biomolecule or include detergent containing controls.
- Prepare a dilution series of the biomolecule. A range from low nanomolar to micromolar range can be recommended, since these are physiologically relevant concentrations for many proteins.
- Ensure that the biomolecule is dissolved in the same buffer as the liposomes.
- Liposome disruption assay (Figure 4)
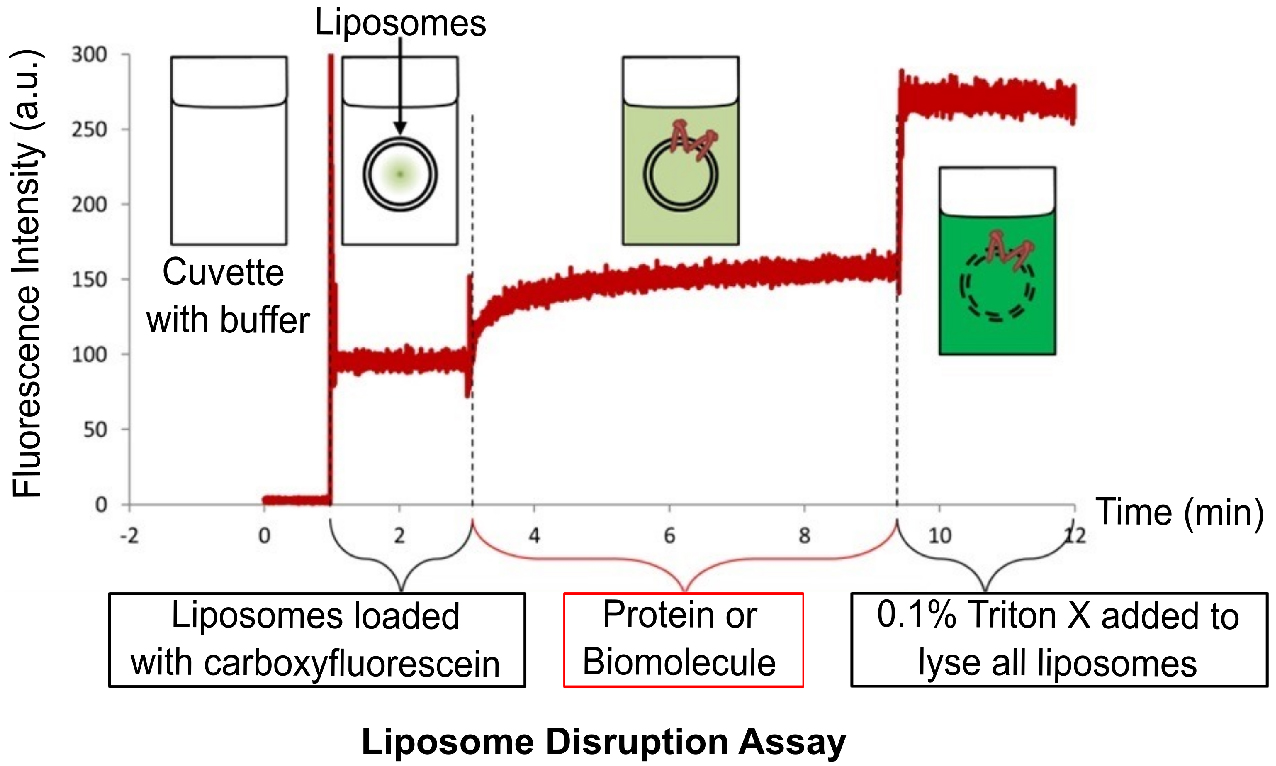
Figure 4. Overview of the liposome disruption assay. The cuvette with buffer has no fluorescence. Addition of liposomes results in a minor increase in fluorescence due to residual free carboxyfluorescein outside the liposomes. Addition of lytic protein or biomolecule disrupts liposomes resulting in the release of carboxyfluorescein and subsequent dequenching of fluorescence. Finally, addition of Triton-X disrupts all the liposomes in the cuvette.- Set the spectrofluorimeter to record the fluorescence over time in order to determine the time dependence of liposome disruption. For carboxyfluorescein, program the fluorescence spectrophotometer to record the fluorescence emission at 512 nm upon excitation at 492 nm for a time range between 2 to 30 min.
Note: It is important to empirically determine the time range appropriate for each biomolecule because they may have different rates of liposome disruption. - Add 980 µl of buffer to a cuvette, and start the kinetic program to record the fluorescence emission over time.
- Add 10 µl of liposomes from a stock to ensure a final desired lipid concentration (usually between 250 nM and 2.5 µM).
Note: Record the time that liposome sample is added. There should be a noticeable sudden increase in fluorescence mostly due to traces of free carboxyfluorescein outside liposomes remaining after previous purification steps. - Add 10 µl of the biomolecule or protein being tested.
Notes:- There will be a gradual increase in fluorescence, due to leakage of carboxyfluorescein from liposomes, if the protein disrupts liposomes.
- Increase in fluorescence due to liposome disruption typically occurs within 5-30 min.
- It is necessary to include a no protein control in order to determine the passive leakage of dye from the liposomes that is independent of a membrane lytic protein.
- There will be a gradual increase in fluorescence, due to leakage of carboxyfluorescein from liposomes, if the protein disrupts liposomes.
- Finally, add 10 µl of 10% Triton-X to disrupt all liposomes in the system and obtain the 100% release value.
- Set the spectrofluorimeter to record the fluorescence over time in order to determine the time dependence of liposome disruption. For carboxyfluorescein, program the fluorescence spectrophotometer to record the fluorescence emission at 512 nm upon excitation at 492 nm for a time range between 2 to 30 min.
- Dextran-block experiment to determine if liposome disruption occurs via pore formation.
- Prepare 20 µM solutions of dextran molecules of different sizes dissolved in the buffer.
Note: A typical liposome disruption assay has a total lipid concentration of 250 nM to 1 µM. Meaning that using the Dextran (20 µM) is in excess of the liposomes in the experimental system. - Similar to the liposome disruption assay (described in Procedure G), determine the extent of liposome disruption in the absence of dextran in the buffer.
- Determine the extent of liposome disruption with buffers containing different molecular weight dextran molecules. It is recommended to test dextran molecules ranging from 5 kDa to 2,000 kDa since the size of the pore is unknown.
- Prepare 20 µM solutions of dextran molecules of different sizes dissolved in the buffer.
Data analysis
- Liposome disruption assay data analysis
- The following formula is used to determine the percent of liposome disruption with time:
Disruptiontime = [(F512 ofliposome+protein - F512ofliposome)/(F512 ofliposome+triton - F512 ofliposome)] x 100%
Where F512 is the fluorescence intensity at 512 nm upon excitation at 492 nm. - For kinetic analyses, the time dependence of liposome disruption can be fitted to the following equation:
% LiposomesDisruptedtime = A[1 - e - (time/tau)] + m x time
where A is the maximum percentage of liposomes disrupted; Tau is the time constant for the exponential component and is the inverse of the rate constant (k); and m is the slope of the linear component.
Note: Fitting the data to this equation will provide data that can be used to obtain a hill plot as previously described (Saito et al., 2000). Additionally, examples of representative data and analysis are previously reported in Saito et al., 2000 and Jimah et al., 2016.
- The following formula is used to determine the percent of liposome disruption with time:
- Dextran-block and pore-formation assay data analysis
- A one-way ANOVA analysis using GraphPad can be used to determine if dextran molecules of a particular size block liposome disruption compared to the no dextran control.
- This dextran-block experiment may reveal the size of the pore because radius of the pore will be similar to the radius of gyration of the dextran molecule that blocks liposome disruption.
Note: Examples of representative data and analysis are previously reported in Saito et al., 2000 and Jimah et al., 2016.
- A one-way ANOVA analysis using GraphPad can be used to determine if dextran molecules of a particular size block liposome disruption compared to the no dextran control.
Notes
- Good protein purification, handling, and storage are critical because membrane active proteins are frequently unstable and subject to aggregation, in addition the presence of proteases could degrade the protein resulting in a loss of activity. It is useful to determine the activity of freshly purified protein and determine how it compares to freeze-thawed samples.
- Since proteins that disrupt membranes most likely have a solution form and membrane bound form, it might be necessary to activate the protein with detergent, or another molecule known to induce a conformational change in the protein. The amount of detergent or molecule should be enough to activate the protein without disrupting the liposomes and this must be tested with control samples.
- Ensure that the liposomes are intact before testing the ability of protein to disrupt liposomes. Liposomes can be stored for approximately two days depending on the lipid composition. The fluorescence intensity of viable liposomes containing 20 mM carboxyfluorescein should increase three fold upon addition of 0.1% Triton-X.
- The same buffers should be used in all steps to ensure there are no differences in osmolarity between the protein and liposome buffers. Differences in osmolarity may result in lysis of liposomes.
- The Stewart assay may be less accurate because the absorbance reading of the chloroform phase, containing the lipid and ammonium ferrothiocyanate, may be skewed by the leakage or presence of carboxyfluorescein in the chloroform phase. An alternative 1H NMR based method for determining the phospholipid content of liposomes is described by Hein et al., 2016.
- Exercise safety precautions when handling and storing solvents such as chloroform and ether.
Recipes
- Buffer-KCl
10 mM HEPES pH 7.4
150 mM KCl - Buffer-NaCl
10 mM HEPES pH 7.4
150 mM NaCl - 0.1 M ammonium ferrothiocyanate
Dissolve 27.03 g ferric chloride hexahydrate and 30.4 g ammonium thiocyanate in 1 L distilled water
Acknowledgments
This work was supported by the Burroughs Wellcome Fund (to NHT) and National Institutes of Health (R56 AI080792 to NHT). This protocol is adapted from Saito et al., 2000 and Jimah et al., 2016.
References
- Hein, R., Uzundal, C. B. and Henning, A. (2016). Simple and rapid quantification of phospholipids for supramolecular membrane transport assays. Org Biomol Chem 4(7): 2182-5.
- Jimah, J. R., Salinas, N. D., Sala-Rabanal, M., Jones, N. G., Sibley, L. D., Nichols, C. G., Schlesinger, P. H. and Tolia, N. H. (2016). Malaria parasite CelTOS targets the inner leaflet of cell membranes for pore-dependent disruption. Elife 5.
- Jimah, J. R., Schlesinger, H. P. and Tolia, H. N. (2017). Membrane lipid screen to identify molecular targets of biomolecules. Bio Protoc 7(15): e2427.
- Saito, M., Korsmeyer, S. J. and Schlesinger, P. H. (2000). BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat Cell Biol 2(8): 553-555.
- Stewart, J. C. (1980). Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem 104(1): 10-14.
Article Information
Copyright
![]() Jimah et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Jimah et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Jimah, J. R., Schlesinger, P. H. and Tolia, N. H. (2017). Liposome Disruption Assay to Examine Lytic Properties of Biomolecules. Bio-protocol 7(15): e2433. DOI: 10.21769/BioProtoc.2433.
- Jimah, J. R., Salinas, N. D., Sala-Rabanal, M., Jones, N. G., Sibley, L. D., Nichols, C. G., Schlesinger, P. H. and Tolia, N. H. (2016). Malaria parasite CelTOS targets the inner leaflet of cell membranes for pore-dependent disruption. Elife 5.
Category
Microbiology > Microbial biochemistry > Protein > Activity
Microbiology > Microbial biochemistry > Lipid
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.