- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of 3D Cellular Organization of Fixed Plant Tissues Using a User-guided Platform for Image Segmentation
Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2355 Views: 12482
Reviewed by: Carsten AdeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2008
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The advent of non-invasive, high-resolution microscopy imaging techniques and computational pipelines for high-throughput image processing has contributed to gain insights in plant organ morphogenesis at the cellular level. Confocal scanning laser microscopy (CSLM) allows the generation of three dimensional images constituted of serial optical sections reporting on stained subcellular structures. Fluorescent labels of cell walls or cell membranes, either chemically or through reporter proteins, are particularly useful for the analyses of tissue organization and cellular shapes in 3D. Image segmentation based on cell boundary signals is used as an input to generate 3D-segments representing cells. These digitalized, 3D objects provide quantitative data on cell shape, size, geometry, position or on (intercellular) intensity signals if additional reporters are used. Herein, we report a detailed, annotated workflow for image segmentation using microscopic data. We used it in the context of a study of tissue patterning during ovule primordium development in Arabidopsis thaliana. Whole carpels are stained for cell boundaries using a modified pseudo-Schiff propidium iodide (mPS-PI) protocol, 3D images are acquired at high resolution by CSLM, segmented and annotated for individual cell types using ImarisCell. This allows for quantitative analyses of cell shape and cell number that are relevant for tissue morphodynamic studies.
Keywords: High-resolution 3D imagingBackground
Organ and tissue morphodynamic studies in plants rely on the analysis of the three-dimensional process of growth along development progression. The evolution of cell number, cell size and cell shape allows interpreting events of proliferation, cellular expansion and anisotropy, respectively (Roeder et al., 2011; Barbier de Reuille et al., 2015; Bassel and Smith, 2016; Coen and Rebocho, 2016). While time-lapse imaging appears in principle as the method of choice, it is not easily applicable to all plant organs, sometimes embedded in inaccessible structures, and image quality often compromise on robust quantitative analyses over a large number of samples and at the cellular level. A complementary, robust alternative is to stain and record three dimensional images of optically cleared tissue/organ at high-resolution, at consecutive time points in order to reconstruct a developmental progression. These 3D images can then be digitally segmented into individual cell objects, from which measurements of number, different descriptors of cell shape and cell size can be extracted. Whole-mount tissue clearing and staining of the cell wall using the modified pseudo-Schiff propidium iodide (mPS-PI) protocol (Truernit et al., 2008) is widely used in the plant community for 3D shape analyses (Sankar et al., 2014; Yoshida et al., 2014; Hervieux et al., 2016). It is described here with only minor modifications and specific notes enabling high-quality sample preparation for the delicate carpel structures. In addition, while plant tissue imaging using confocal scanning laser microscopy became common practice in many labs, specific knowledge on how to fine-tune the optical and software-controlled image recording remains elusive and often kept ‘in house’. Here we provide detailed recommendations aimed at guiding the user towards producing high-quality, high-resolution 3D images suitable for robust qualitative and quantitative analyses. For image segmentation, different open-source algorithms proved invaluable for tissue morphodynamic studies in plants and Drosophila, namely: MARS-ALT (Fernandez et al., 2010), MorphographX (Barbier de Reuille et al., 2015) and RACE (Stegmaier et al., 2016). Yet, these interfaces usually require computational skills to fine-tune segmentation parameters, correct manually for wrongly segmented objects, customize cell labelling when tissue models are not available in the software and export quantitative data for downstream analyses. An alternative solution for biologists lacking this expertise lies in the use of commercially available software with a streamlined user interface. We present here one option with Imaris, a software for 3D visualization and image processing. We report a detailed, annotated workflow applied to ovule tissue analyses but that is of broad application to analyze various plant tissues.
Materials and Reagents
- Microscope slides (76 x 26 mm) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 10143562CEF )
- Microscope cover slips for confocal imaging: 18 x 18 mm, 0.17 ± 0.01 mm thickness (Hecht Assistant, catalog number: 41014509 )
- Dissecting needles (Tungsten, diameter: 0.75 and 0.35 mm)
- Round Petri dishes 35 mm (Greiner Bio One International, catalog number: 627102 )
- 2 ml micro tubes (SARSTEDT, catalog number: 72.695.500 )
- Dust-free paper
- Glass wool
- Plant material: Flowering Arabidopsis thaliana plants
- Sodium dodecyl sulfate, sodium salt (SDS) (Sigma-Aldrich, catalog number: 71729 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 71690 )
- Ethanol (70%, 80%) (Fisher Scientific, catalog number: 10428671 )
- Periodic acid (Sigma-Aldrich, catalog number: 375810 )
- Nail polish
- Mounting medium
- Methanol (Sigma-Aldrich, catalog number: 34860 )
- Acetic acid (Merck, catalog number: 100063 )
- Sodium metabisulphite (Na2S2O5) (Sigma-Aldrich, catalog number: 31448 )
- Hydrochloric acid fuming, 37% (HCl) (Carl Roth, catalog number: 4625.1 )
- Propidium iodide (PI) (Sigma-Aldrich, catalog number: P4864 )
- Chloral hydrate (Sigma-Aldrich, catalog number: 15307 )
- Glycerol (Carl Roth, catalog number: 3783.1 )
- Gum arabic (Sigma-Aldrich, catalog number: 51198 )
- Modified pseudo-Schiff propidium iodide (mPS-PI) solution (see Recipes)
- Fixative solution
- Pseudo-Schiff reagent with propidium iodide (PI)
- Chloral hydrate solution
- Hoyer’s solution
Equipment
- Stereomicroscope (e.g., Leica Microsystems, model: Leica M60 )
- Incubator (e.g., Eppendorf, model: Thermomixer® C )
- Diamond- or carbide-tip pen (Sigma-Aldrich, catalog number: Z225568 )
- Confocal scanning laser microscope, resonant scanner with laser line 561 nm and APO PL objectives lenses 20x (NA 0.7) and 63x (NA 1.4) suitable for glycerol immersion (e.g., Leica Microsystems, model: Leica TCS SP5 )
- Computer ideally with high-end processor, memory and graphic environment as recommended (http://www.bitplane.com/systemrequirements.aspx). Lower-end settings are possible but will result in slower processing speed
Software
- Imaris 8.3.1 (www.bitplane.com, Oxford Instruments, UK)
- Data analysis software (e.g., R, www.r-project.org)
Procedure
- Sample preparation
- Collect floral buds from A. thaliana apical inflorescences at the desired stage and place them on a glass slide with a drop of sterile water.
- Isolate the pistils under the stereomicroscope with dissecting needles.
- Transfer the pistils to a small Petri dish with 3 ml fixative solution (see Recipes).
Note: Verify that the pistils are completely immersed in the fixative solution. - You can keep the samples at 4 °C up to 1 month at this stage.
- The pseudo-Schiff PI staining follows a protocol previously published (Truernit et al., 2008; Yoshida et al., 2014) and detailed below.
- Transfer the pistils into a 2 ml micro tube with 1 ml solution of 1% SDS, 0.2 N NaOH for an overnight treatment at room temperature (RT).
- Discard the 1% SDS, 0.2 N NaOH solution and add 1 ml of 80% ethanol. Incubate at 80 °C at 350 rpm for 5 min.
- Remove the 80% solution leaving behind a minimum of 10 µl in the tube. Add 1 ml of fixative solution. Incubate at RT for 1 h.
- Rinse the tissues once with water and incubate in 1% periodic acid at RT for 1 h.
- Rinse the tissues once with water and incubate in the modified pseudo-Schiff PI solution (see Recipes) (10 µg/ml) at RT for 1 h or 2 h.
Note: Tissues are stained if they appear reddish under eye inspection. - Rinse three times with water and add 300 µl of chloral hydrate solution. Leave overnight at RT for further clarification.
Note: Leave tissues in chloral hydrate solution at RT for a maximum of two days.
- Collect floral buds from A. thaliana apical inflorescences at the desired stage and place them on a glass slide with a drop of sterile water.
- Mounting for microscopy imaging
- Clean a slide and a coverslip with 70% ethanol using dust-free paper and let it dry.
- Cut a coverslip in 5-6 small pieces (ca. 3 x 18 mm per piece) using a diamond tip pen (Figures 1A and 1B).
- Place a drop of Hoyer’s solution or medium of choice on a slide between the two pieces of glass coverslip leaving a ca. 10-12 mm width.
- Place one pistil in the drop (Figure 1D).
- Place a coverslip on top without applying pressure (Figure 1E). This is very important to maintain the three dimensional structure of the pistil.
- Seal the borders of the coverslip with nail polish. Leave the slide positioned horizontally to allow the medium to solidify.
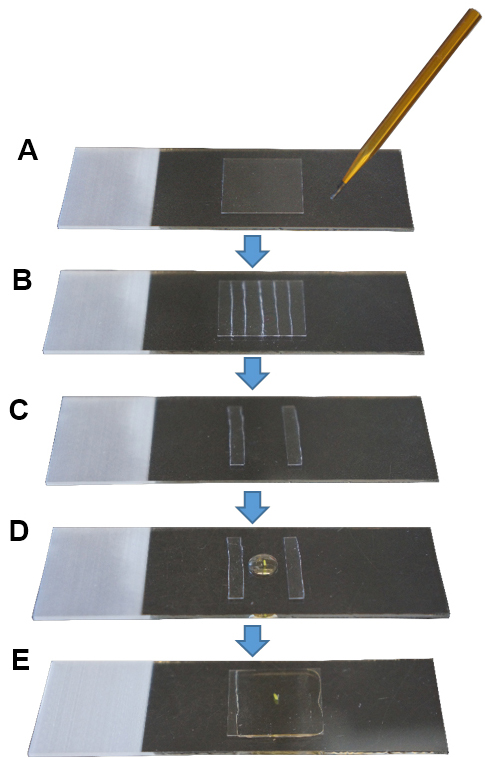
Figure 1. Sample mounting for microscopy imaging. A. Cleaned coverslip; B. With a diamond tip pen, cut the coverslip in small pieces; C. Use two pieces as pillars, leaving a space in between; D. Place a pistil in a drop of mounting medium; E. Place a coverslip on top and seal its borders with nail polish.
- Clean a slide and a coverslip with 70% ethanol using dust-free paper and let it dry.
- Image acquisition
- Find the sample with the 20x objective using transmission light or epifluorescence (green excitation filter, red emission-long bandpass range) and then switch to higher magnification for acquisition.
- Set up parameters for image acquisition
- Excitation: 561 nm.
- Emission: 570-616 nm.
- Laser Transmission: 30-40%.
Note: This can vary depending on laser type/age. - Detection: ideally a new generation detector (e.g., HyD), Bright Contrast Gain mode.
Note: The gain should be between 20 and 30% for good quality images. - Scanning speed: Resonance mode, 8,000 Hz.
- Pinhole diameter: 1 AU (Airy Unit).
- Average frame: 4.
Note: If the fluorescence signal is lower and laser transmission/gain have to be increased, higher averaging will increase image quality–this has to be tested. - Objective: ideally 63x, plan + apochromatic corrected (PL APO), glycerol immersion lens (GLY), NA 1.4, working distance (WD) 0.3 mm.
Note: Samples are mounted in a glycerol-based medium; hence a glycerol immersion lens allows for best match of the refractive index. - Image format: 512 x 512 pixels.
- Image size: the field of view is cropped at around 80 to 85 µm to obtain x and y ≈ 85 nm.
Note: This allows for a 2x oversampling using a 63x NA 1.3 objective at 561 nm (rx,y = 176 nm using rx,y = 0.4 x λexc/NA). - Set the z-series to capture the entire pistil/object of interest (e.g., ovule).
Note: Depending on the stage of ovule development, z-series range from 300 to 500 sections (0.02 to 0.04 GB with the image format described above, respectively). - Set the z-steps to ~80 nm.
Note: This allows for a near cubic voxel in the final image with sufficient over-sampling (Nyquist-Shannon theorem) for downstream image processing (deconvolution or segmentation).
- Excitation: 561 nm.
- Acquire the image stack.
- Save the dataset. There is no need of a specific file format. Imaris can read files from most microscopy systems.
Note: We strongly recommend to name the dataset according to a serial number determined by a hierarchical database (for instance Author_Initials_3or4digits, e.g., EM_023, EM_572, etc.). This allows for easy dataset calling and batch processing. - Immediately update the image description in a customized database.
Note: We strongly recommend to elaborate an image database (e.g., Excel spreadsheet or other specialized program) gathering information such as sample type (biological material, stage, genotype, species, staining batch, etc.), image type and user-based assessment of image/staining quality. This allows for efficient tracking and handling of replicate datasets for downstream processing.
- Find the sample with the 20x objective using transmission light or epifluorescence (green excitation filter, red emission-long bandpass range) and then switch to higher magnification for acquisition.
- 3D reconstruction and image pre-processing
For the basic usage of Imaris described here (3D view, section view, create contour surface/create mask), first users are referred to free online tutorials (http://www.bitplane.com/learning).- Upload a lif file in Imaris 8.3.1.
Note: Imaris Batch allows the user to convert all series in a lif file to ims files. - Inspect the quality of the image (Figure 2 and Video 1) using the ‘Section’ mode in the tab ‘View’. Cell boundaries have to be clearly resolved in all dimensions (viewed in the XY, XZ, YZ panels).
Note: Insufficient sampling in xy (image format too low) or along the z dimension (too little optical sections), unbalanced signal acquisition and/or refractive index in the mounting and immersion medium, unstable fluorescent probe and/or steep gradient of signal loss in depth will produce images with low signal to noise ratio impairing on robust segmentation.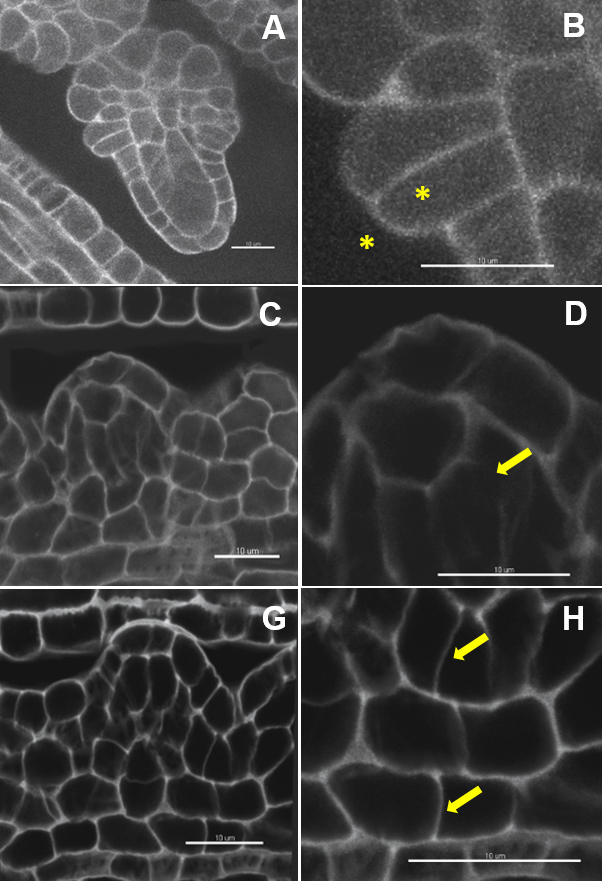
Figure 2. Image quality: the ugly, the bad and the good. Image dataset with a very low (A, B) and low signal to noise ratio (C, D). Weak contrast between the outer and inner cell space (B), interrupted and fuzzy boundary signals (D). Image dataset with a high signal-to-noise ratio (G) and clear cell boundaries (H). Scale bars = 10 µm. Video 1. Short tutorial for membrane-based 3D image segmentation using Imaris Cell. Selected points of the protocol are presented as follows: D. 3D-reconstruction and image processing: 2. Inspect the image quality, 3. Down-sampling, 5. in silico dissection, 6. Free rotate; E. Image segmentation and labelling: 1. Automated segmentation, 4. Cell labelling.
Video 1. Short tutorial for membrane-based 3D image segmentation using Imaris Cell. Selected points of the protocol are presented as follows: D. 3D-reconstruction and image processing: 2. Inspect the image quality, 3. Down-sampling, 5. in silico dissection, 6. Free rotate; E. Image segmentation and labelling: 1. Automated segmentation, 4. Cell labelling. - Down-sampling (optional) (Video 1): Edit/Resample 3D/Change the values in X, Y, Z and tick the option ‘Fixed ratio X/Y/Z’.
Note: Segmentation speed depends on the computer environment. As an example, an 8-bit, grey scale, 0.1 GB image (512 x 512 x 385 pixels) was segmented in 4 min in Imaris 8.3.1 run on a PC with an Intel Xeon E5 v3 clocked at 3.20 GHz processor, Nvidia Geforce GTX Titan X graphics card and an SSD 850 PRO 1TB. Down-sampling by 2x in each dimension reduces the time to 2 min without quality loss in our case but this should be verified by the user. - Crop the dataset to the informative region (optional): ‘Edit/Crop 3D’, move the sliders to create a 3D box capturing the region of interest (Figure 3).
Note: This operation is recommended if the informative region represents a well-defined subset of the image. If the object has a complicated shape, in silico dissection (see step D5) is recommended instead.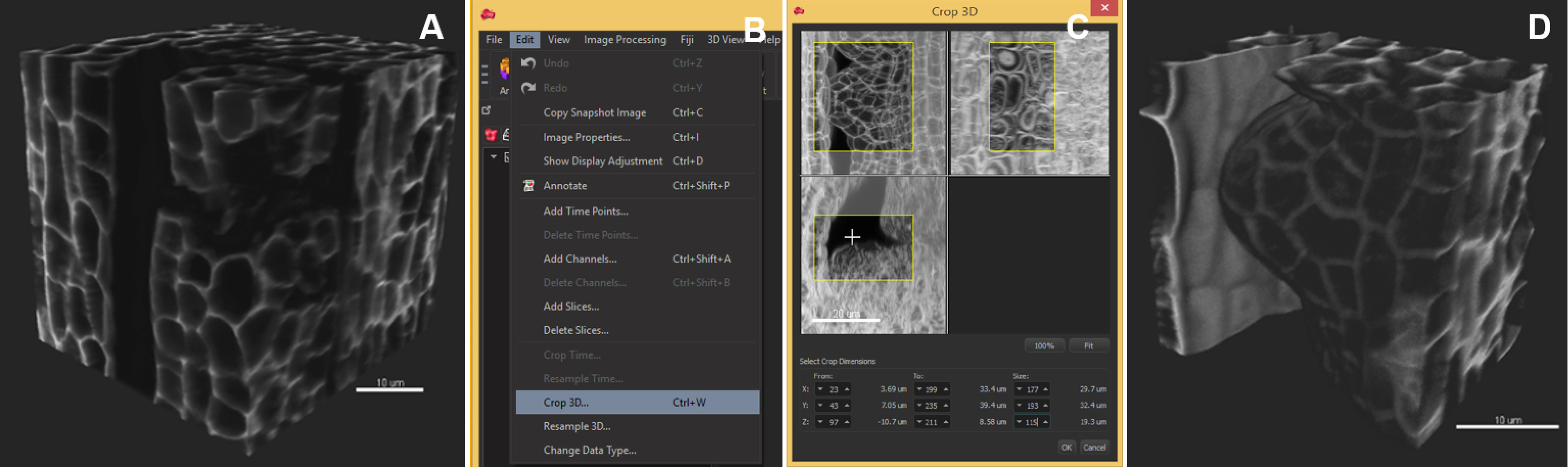
Figure 3. Image pre-processing–crop in 3D. The region of interest occupies a volume smaller than the original image. The image is cropped in 3D around the ovule primordia. A. Original image, 3D volume rendering, ‘Blend’ mode; B. In the ‘Edit’ tab, choose ‘Crop in 3D’; C. Adjust the three slider boxes in the pop-up window; D. Crop (Apply): the cropped stack is smaller and will be segmented faster. Scale bars = 10 µm. - Select an object of interest by in silico dissection (optional) (Video 1):
Notes:- This step is relatively quick (few minutes) and highly recommended for images of high complexity, i.e., capturing several and/or intermingled objects and for images where only a subset is informative for downstream analyses. Typically, this step is particularly useful to dissect individual ovules away from neighboring ones and surrounding carpel tissue. It also allows resolving problems due to touching tissues that create inaccurate cell contours.
- If several objects are isolated using this procedure, name them for ease of downstream steps (click on the channel name in the Edit/Display adjustment tab).
- Create a Surface using the ‘Add new surfaces’ function and edit manually.
- In the wizard, select ‘Skip automatic creation, edit manually’. In the ‘Contour/Autofit’ tab, adjust the sliders to smooth shape and weak impact (leftmost position).
- Draw the contours: under the tab Contour/Mode select the drawing mode (not shown), under the tab Contour/Board, select the orthogonal plane, activate ‘Draw’ or Ctrl + Space bar.
- Only few contours are sufficient to create an approximate surface around the region of interest; take care to define precisely the beginning, end and median planes.
- In the wizard, proceed to ‘Create surface’, a surface object (yellow) is created (Figure 4B).
- In the ‘Edit’ tab, click on ‘Mask all’ and tick the box ‘Duplicate channel before applying mask’ and ‘Set voxels outside surface to 0.000’. A new channel (mask) is created comprising the voxels captured by the surface–this channel is used for segmentation.
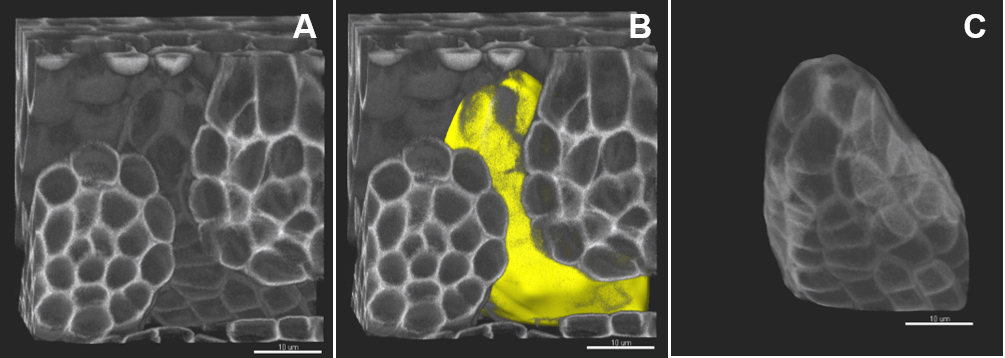
Figure 4. Image pre-processing–in silico dissection of an object of interest. A. The original stack is volume-rendered. B. The created surface is presented in yellow. C. A new channel (mask) is created comprising the voxels captured by the surface–this channel is used for segmentation. Scale bars = 10 µm.
- This step is relatively quick (few minutes) and highly recommended for images of high complexity, i.e., capturing several and/or intermingled objects and for images where only a subset is informative for downstream analyses. Typically, this step is particularly useful to dissect individual ovules away from neighboring ones and surrounding carpel tissue. It also allows resolving problems due to touching tissues that create inaccurate cell contours.
- Rotate the sample in space to align the main growth axis along the YZ dimension (Figure 5 and Video 1).
Note: This step is optional. It is however useful to have the sample aligned along one of the orthogonal axis for easier navigation with the slicer and pointer at later stages during cell identification and labelling. - In the ‘Edit’ tab, click on ‘Free rotate’. A window shows the new coordinates automatically entered.
- Click on ‘Apply’ and the object is thus reframed. Repeat if necessary.
- Otherwise, choose another alignment in which tissue layers/cells of interest are in one of the orthogonal planes.
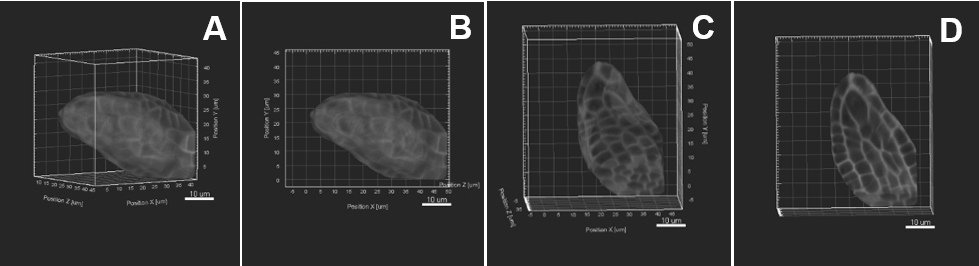
Figure 5. Image pre-processing–Sample rotation. A. The object of interest is tilted to the right side. Sample rotation is performed using «Free rotate», the object is therefore aligned straight along the y z axis (B, C). D. An active clipping plane shows a mid-longitudinal section. Scale bars = 10 µm.
- Upload a lif file in Imaris 8.3.1.
- Image segmentation and labelling
- Automated segmentation (Video 1)
- Choose the function ‘Add new cells’ to create a ‘Cell’ object. This opens a wizard guiding you through the steps of automated segmentation (Figures 6A and 6B). Each step must be validated by the blue arrow. The green arrow finalizes the process.
- In ‘Select detection type’, choose ‘Detect cell only’ (Figure 6C).
Note: There is the possibility to segment only a region of interest as alternative to pre-processing steps (see step D4). - Next, choose ‘Detect cell boundary from cell membrane’. Select the source channel (Figure 6D), i.e., the masked channel if relevant (see step D5).
- For the next step, it is needed to measure two parameters: the ‘cell smallest diameter’ and ‘membrane detail’ (corresponding to the membrane’s thickness): the software proposes default values that are usually good suggestions but real values should be measured: this can be done in the Slice View mode using the line measurement tool (drawn by positioning two crosses in two mouse clicks; values are indicated on the right panel) across the smallest cell aimed in segmentation (Figure 6E), and across the membrane (Figure 6F).
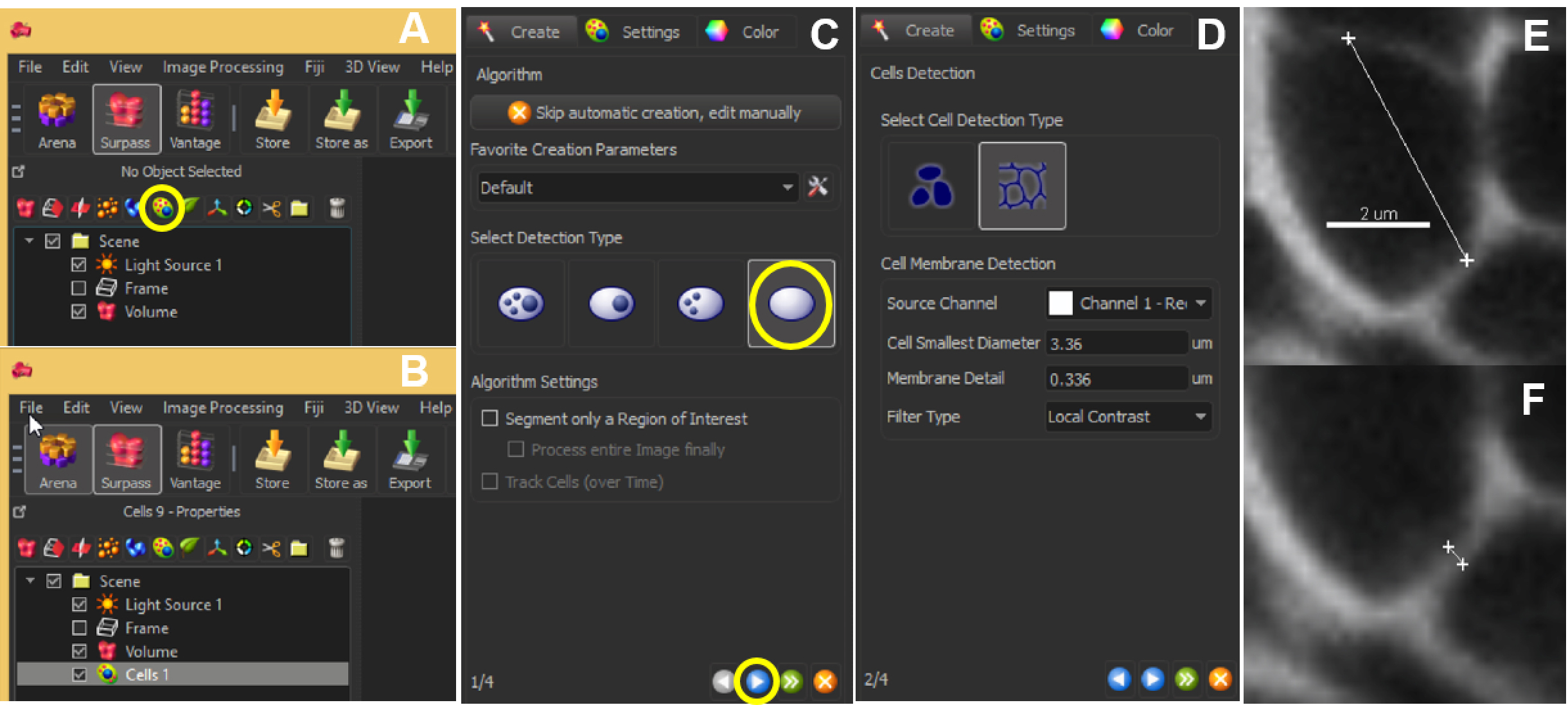
Figure 6. Automated segmentation–parameters. The ‘Add new cells’ icon allows the user to start off the segmentation (A). The created cell will show up in the list of other parameters listed within Scene (B). At the same time, a panel will appear on the left bottom corner of the Imaris interface. Choose ‘Detect cell only’ and hit the blue button to proceed (C). Next, choose ‘Detect cell boundary from cell membrane’ and enter the optimal ‘Cell smallest diameter’ and ‘Membrane detail’ values (D), which can be obtained by clicking 2 points across the cell (E) or the membrane (F). Scale bar = 2 µm. - With this information, adjust the parameters ‘Cell smallest diameter’ and ‘Membrane detail’ in the Cells creation wizard.
Note: Values proposed by default are based on signal distribution and voxel size. They do not necessarily correspond to the optimal, biologically-relevant structures. When doing segmentation for the first time, it is advised to try different values. Reducing the smallest cell diameter or membrane detail allows capturing more cell segments with higher accuracy. Yet, too small or too large values result in over (Figure 7A) and under-(Figure 7B) segmentation, respectively.
Figure 7. Effect of ‘Cell smallest diameter’ and ‘Membrane details’ on segmentation accuracy. ‘Cell smallest diameter’ (CSD) and ‘Membrane details’ (MD) are critical parameters for an optimal segmentation. Note that MD represents the thickness of the cell boundary signal but is not an actual biological measure of this cellular structure which cannot be resolved in conventional CSLM imaging. In this representative example CSD and MD measured values ranged from 1.5 to 3.36 µm, and 0.3 to 0.6 µm, respectively. At fixed MD (here 0.30 µm), smaller CSD values produce over-segmentation shown in yellow (A) whereas under-segmentation (yellow) occurs at higher MD values and fixed CSD (here 3.36 µm, B). In this example, optimal values are for CSD = 3.36 µm and MD = 0.30 µm. Scale bars = 10 µm. - Select a ‘Filter type’ for the Cell membrane detection. ‘Local contrast’ proved robust for images created as described here.
Note: This filter applies a pre-processing step prior to the segmentation algorithm. The smooth method applies a Gaussian filter whereas the local contrast method uses a combination of Gaussian smoothing and baseline subtraction (Imaris 8.3 reference manual). The latest uses more memory but provides more fine-tuning possibilities in downstream steps. - The next step launches the segmentation algorithm. The processing time depends on the image size and computational environment (e.g., 4 min for a 100 MB in Imaris 8.3.1 on our PC, see step D3).
- The raw segmentation result is displayed for one section plane in a transparency mode showing the original image signal. Inspect the segmentation quality by moving the slicer along the image. Check it in other orthogonal planes or oblique planes depending on the orientation/shape of your object of interest.
- From this inspection, estimate if there is a typical failure, i.e., over- or under-segmentation and identify a few typical planes where this should be corrected.
- Adjust the merging thresholds using signal intensity and quality filter criteria (local contrast method) or only intensity (smooth method). Dynamically verify the segmentation accuracy on several planes until best-fit (Figures 8C and 8F).
Note: This step aims at merging neighboring segments into realistic cell objects and takes into account the intensity of the small segments boundaries (‘intensity’) and of merged regions (‘quality’) hence the latest are interdependent (Imaris 8.3 reference manual). - Proceed to the next step: 3D cell objects are created.
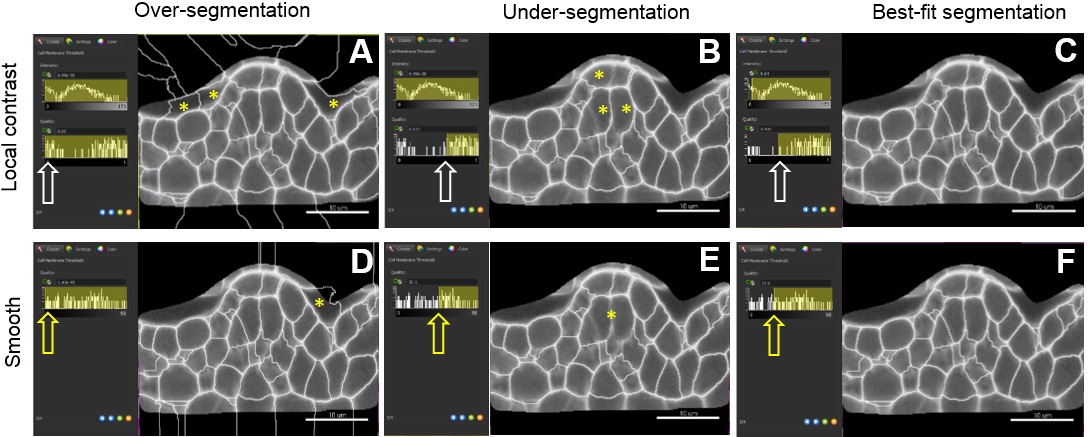
Figure 8. Choosing a pre-processing method and fine-tuning merging thresholds. The local contrast filter possesses two values which can be adjusted by the user (intensity and quality values) whereas the smooth filter has only the latter one. Sliding the window to different positions (white and yellow arrows) give three different outputs: over-(A, D), under-(B, E) and best-fit segmentation (C, F). For both methods, yellow asterisks indicate over-segmentation of the background inside and outside the masked channel. They also pinpoint under-segmented cells. Scale bars = 10 µm. - A last step (‘Classify cells’) allows removing outliers according to user-based criteria. (Figure 8). By default, ‘Cell number of voxels’ is chosen as a filter. This enables to rapidly remove small or large segments created around the object of interest and corresponding to intercellular space or tissue cavities.
Note: The filtering mode can be customized to all ‘Cell statistics’ available in Imaris alone or in combinations. This includes ‘Cell position’ (absolute or relative to the image border), cell volume, area, and other geometric parameters. An example of filtering against cells close to the image border is shown in Figures 9E and 9F.
- Finalize the segmentation (green button ‘Finish’). The 3D segmentation can be visualized in 3D or along 2D slicers (‘Slicer display’ in the tab ‘Settings’). Cells can be rendered in different coloring modes and transparency (mapped color, random, statistics based, etc., see Imaris 8.3 reference manual).
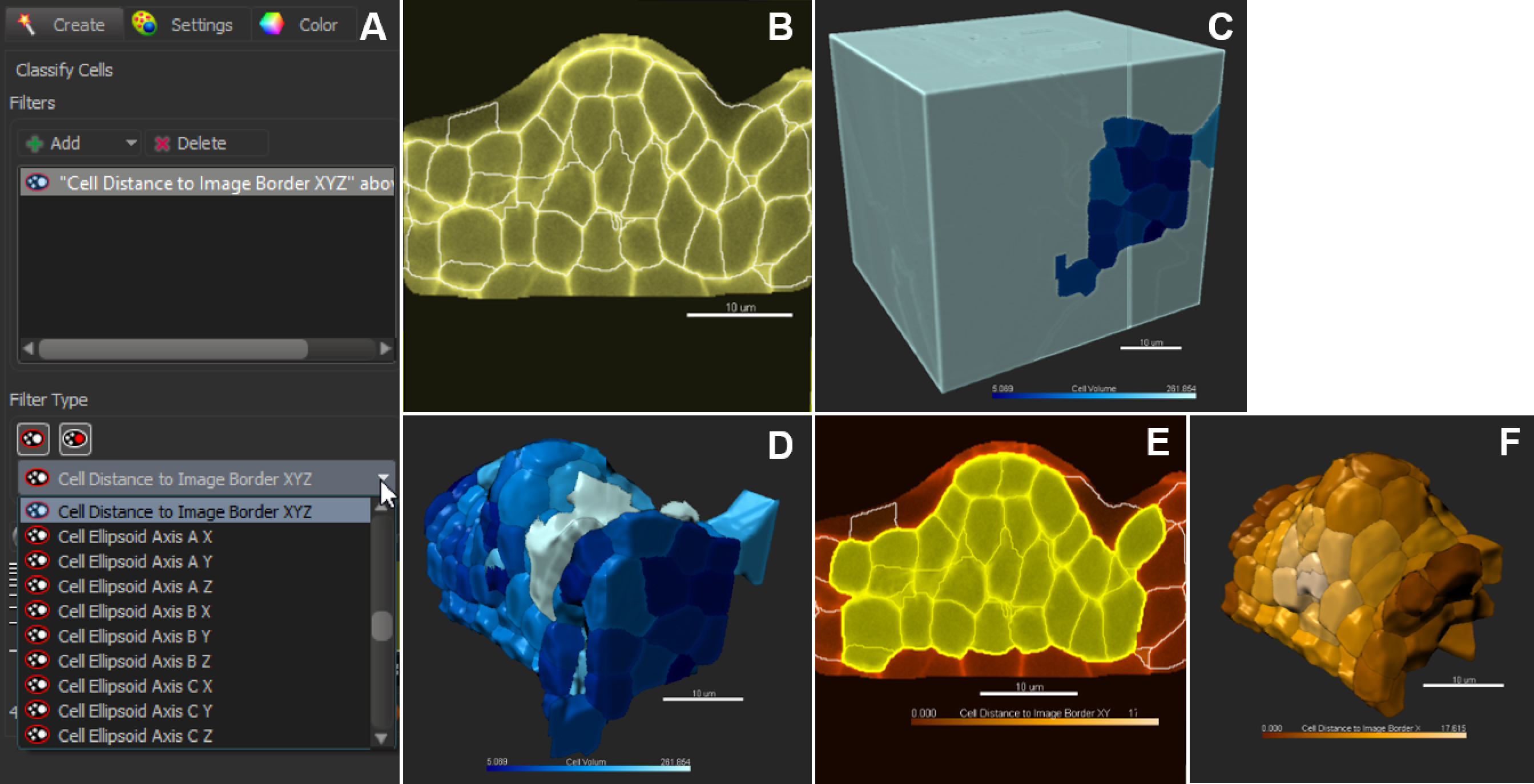
Figure 9. Classification and filtering for customized selection of cells. A. Pull-down menu showing the different cell statistics available for filtering. Click on + to add several filters in combination. B. In the slice view mode, all cells are selected (no filter applied); 3D coloring mode follows the filter type ‘Cell Volume’ (C). D. Cells are sorted against the filter ‘Cell volume’. E and F. Cells are sorted against the filter ‘Cell distance to image border XYZ’. Scale bars = 10 µm. - Manual Curation–Delete, Merge and Split function
The automated segmentation is generally robust with 6-15% inaccurate segmentation on image datasets as produced according to the protocol described here. Over- and under-segmentation largely results from heterogeneous boundary signal throughout the image. Some cells will remain over- or under-segmented despite merging threshold adjustments. A set of methods is described below that allow for a rapid curation.- Inspection of the segmentation is best achieved in the slicer mode. Use orthogonal or oblique planes.
Note: If the image has been rotated (see step D6), the axis with the lowest resolution is no longer along the z axis as in the original image. - Curate the erroneous regions using the ‘Delete’, ‘Merge’ or ‘Split’ functions in the ‘Edit’ tab to correct for irrelevant, under- and over-segmentation, respectively. For instance, to correct an over-segmented region, select the two erroneous segments belonging to the same cell and use the ‘Merge’ function. By contrast, to correct an under-segmented region, select the segment capturing two (or more cells) and re-segment using the ‘Split’ function. This action often solves the issue. If it generates over-segmented fragments; however, those can quickly be merged again as before (Figure 10). If this action does not correct the problem, follow step 3.
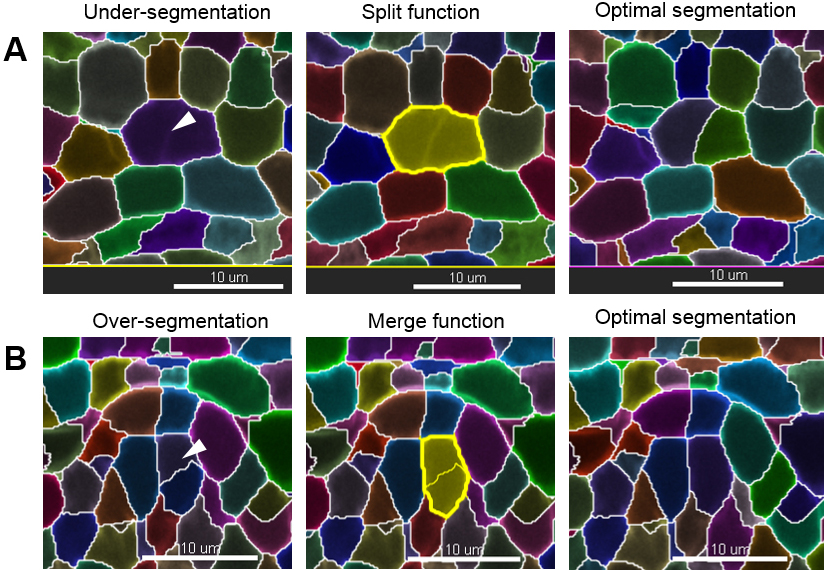
Figure 10. Manual curation–merge and split functions. Scale bars = 10 µm.
- Inspection of the segmentation is best achieved in the slicer mode. Use orthogonal or oblique planes.
- Manual Curation–Re-segmentation of a small region of interest (ROI)
This approach is particularly useful when split and merge functions fail to correct the segmentation. The idea is to re-segment locally a difficult region, for instance with cell boundaries of disrupted or varying signal intensity. For this, a new cell object is created. The new (correct) cells can, however, not be directly imported into the first Cell object to curate (at least in Imaris 8.3). They first have to be exported as Surface object. The procedure is described below. The user is advised to check the latest software version, as future improvements may provide a direct Cell import option.- Delete the badly segmented cells (select in the slicer mode, ‘Edit/Delete’).
- Create a new Cell Object (‘Add new cells’).
- Choose ‘Segment only a region of interest’ and define the box around the region to be re-segmented. To better locate the ROI, keep the former Cell object visible in the background (Slicer mode).
- Follow the wizard for automated segmentation but try different parameters. Decreasing the membrane details or smallest cell diameter usually solves the problem.
- If the ROI also captured cells that were previously properly corrected, delete them (see step E2b). Or, specifically select the new cells to be imported into the first segmentation object for correction.
- Export the selected, correct Cells: Tab ‘Edit/Export cells to surfaces’. Check the box ‘Selection’. A Surface object ‘Cell Export’ is created.
- Import the Surface object into the first Cells object to be curated: ‘Edit/Import surface to cell’. Follow the wizard. Verify the result of the import in the slicer display–the badly segmented region is now corrected.
- Delete the badly segmented cells (select in the slicer mode, ‘Edit/Delete’).
- Cell Labelling–create tissue or cell type-specific categories (Video 1)
In order to analyze the statistics of the cells (size, geometric description, position, etc.) according to their identity, or position in tissue layers, it is possible to give cells a label, corresponding to a category defined by the user. For instance, a tissue layer (epidermis, cortex, L1, L2, etc.), cell type (guard cell, root hair, precursor, etc.) or position relative to an axis (radial, apico-basal, etc.). - Select the Cell object, which contains the segmented region. To tag a subset of cells, go to the panel on the right of the main display frame. First, delete the default labels if any.
- Create a new label group by clicking on +. All created labels will appear in the drop-down menu.
- A given label group can comprise different labels. For that, select the label group and click on ‘Add a label’ (Figure 11A). Enter the name of the new label and edit the color if needed.
- Once all labels groups and their corresponding labels are created, select the cells belonging to one category using either the slicer or volume display in the ‘Settings’ tab (for peripheral cells). Double-click to assign a label to the cell subset. To remove the current label, double-click again.
- In the ‘Color’ tab, choose ‘Labels’ and tick off all or selected labels groups to be visualized (Figure 11E).
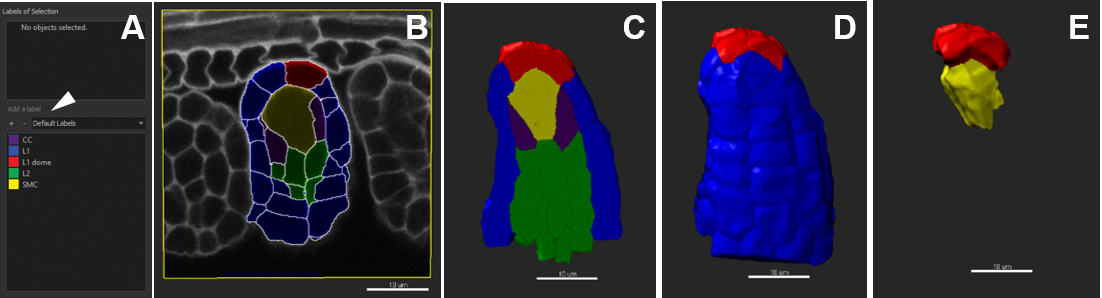
Figure 11. Customized cell labelling. Scale bars = 10 µm.
Data analysis
- Quantitative data: visualization and export
Cell labelling allows classifying the quantitative data associated with each cell object per category. The variables can be selectively exported (one txt file per variable) or in batch (one txt file for all variables per cell). Common cell variables include area, volume, sphericity, ellipticity (oblate and prolate), x, y, z coordinates (center of mass). The full list includes approximately 100 variables (Imaris 8.3 reference manual). The data can be plotted and visualized using a third-source data analysis software (e.g., R, www.r-project.org) or Vantage in Imaris as described below.- Visualization using Vantage Plot
- Select the Cell object to plot.
- Go to the ‘Vantage’ plot application (Top Menu).
- Choose the viewing mode (e.g., ‘1D View’), select the variables to plot (‘Type’ in the drop-down menu).
- Choose the color display in the ‘Draw style’ drop-down menu. An example is presented in Figures 12A and 12B for two variables.
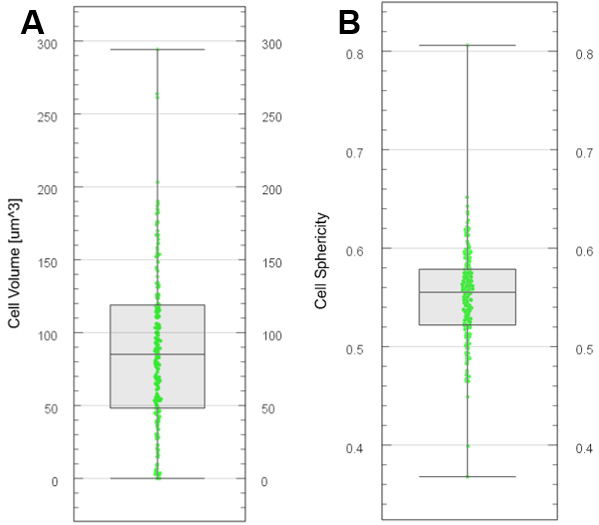
Figure 12. Plot generation with Vantage. Boxplots generated for cell volume (A) and cell sphericity (B) from segmented and labelled cells. - Export various descriptors of cell segments
- In ‘File/Preferences/Statistics’, choose the statistic values to be exported. Otherwise, all statistics will be included.
Note: The term ‘Statistics’ in ImarisCell refers to different quantitative descriptions associated with each cell object (e.g., area, volume, sphericity, fluorescence intensity etc.). It however does not relate to a statistical treatment of the data. - In the ‘Statistics’ tab of the cell object, select the category ‘Detailed’. In the drop-down menu, choose all values, average or specific values.
- Export all values by clicking on the floppy disk icon ‘Export statistics on tab display to file’. Browse the location in which the data will be stored.
- By default, the file name is saved as .csv. However, it is also possible to save as .xls and .xml.
- Exported data can be used to plot specific cell categories and quantitative measurements in third-source data analysis software (e.g., R, www.r-project.org).
Note: Each study has its own requirement in terms of cellular and tissue layer comparison. Data plotting and statistical analyses can be done using standard software and approaches recommended in biological sciences (http://www.nature.com/collections/qghhqm). One recommendation, however, is to base the study on a minimum of fifteen replicate images for the same sample stage. This number takes into account the technical difficulty and time required to process images, data and variability between sample preparations.
- In ‘File/Preferences/Statistics’, choose the statistic values to be exported. Otherwise, all statistics will be included.
Notes
- This protocol is applicable to a wide variety of tissues. We successfully imaged and segmented tissues from maize, rice, petunia, wheat and Marchantia (data not shown).
- Reproducibility of the staining quality depends on the health state and age of the plant. Stressed and/or old plant tissue will stain poorly and show higher light scattering (hence noise).
- The robustness of the segmentation strongly depends on the homogeneity of the signal throughout the 3D image, as commented in the section ‘segmentation’.
- The variability in quantitative measurements on cell objects depends on the quality of sample preparation: if the tissue is slightly squashed under the coverslip and does not show a native 3D shape, the image should not be used for segmentation.
Recipes
- Modified pseudo-Schiff propidium iodide reagent
Note: Prepare the following solutions under the fume hood. - Fixative
50% (v/v) methanol-water
10% (v/v) acetic acid-water - Pseudo-Schiff reagent with propidium iodide (PI)
100 mM sodium metabisulphite (Na2S2O5)
0.15 M HCl
10 µg/ml PI - Chloral hydrate solution
4 g chloral hydrate
1 ml glycerol
2 ml distilled water - Hoyer’s solution
30 g gum arabic
50 ml distilled water
200 g chloral hydrate
20 ml glycerol
Dissolve the gum arabic in water at 30 °C by stirring overnight. Add the chloral hydrate; dissolve by stirring further, and finally add 20 ml glycerol. Filter the solution through glass wool. Store at RT and protect from the light in a tightly sealed glass bottle
Acknowledgments
This work was supported and funded by the Commission for Technology and Innovation (CTI grant 16997), and the Baugarten Stiftung Zürich, the University of Zürich and the Swiss National Science Foundation (SNF grants to CB and UG). We acknowledge Peter Majer and Xuan Zhang (Bitplane R&D, Zürich) for insightful discussions and technical assistance with ImarisCell, Professor Ueli Grossniklaus (UG) for scientific support and insightful discussions and technical assistance of the department for organisational support and assistance with microscopy imaging (Christoph Eichenberger, Valeria Gagliardini, Arturo Bolaños, Peter Kopf). The staining protocol was performed based on previously published work of Truernit et al. (2008) and Yoshida et al. (2014).
References
- Barbier de Reuille, P., Routier-Kierzkowska, A. L., Kierzkowski, D., Bassel, G. W., Schupbach, T., Tauriello, G., Bajpai, N., Strauss, S., Weber, A., Kiss, A., Burian, A., Hofhuis, H., Sapala, A., Lipowczan, M., Heimlicher, M. B., Robinson, S., Bayer, E. M., Basler, K., Koumoutsakos, P., Roeder, A. H., Aegerter-Wilmsen, T., Nakayama, N., Tsiantis, M., Hay, A., Kwiatkowska, D., Xenarios, I., Kuhlemeier, C. and Smith, R. S. (2015). MorphoGraphX: A platform for quantifying morphogenesis in 4D. Elife 4: 05864.
- Bassel, G. W. and Smith, R. S. (2016). Quantifying morphogenesis in plants in 4D. Curr Opin Plant Biol 29: 87-94.
- Coen, E. and Rebocho, A. B. (2016). Resolving conflicts: modeling genetic control of plant morphogenesis. Dev Cell 38(6): 579-583.
- Fernandez, R., Das, P., Mirabet, V., Moscardi, E., Traas, J., Verdeil, J. L., Malandain, G. and Godin, C. (2010). Imaging plant growth in 4D: robust tissue reconstruction and lineaging at cell resolution. Nat Methods 7(7): 547-553.
- Hervieux, N., Dumond, M., Sapala, A., Routier-Kierzkowska, A-L., Kierzkowski, D., Roeder, A.H.K., Smith, R.S., Boudaoud, A. and Hamant, O. (2016). A mechanical feedback restricts sepal growth and shape in Arabidopsis. Curr Biol 26(8): 1019-1028.
- Imaris 8.3 reference manual.
- Roeder, A. H., Tarr, P. T., Tobin, C., Zhang, X., Chickarmane, V., Cunha, A. and Meyerowitz, E. M. (2011). Computational morphodynamics of plants: integrating development over space and time. Nat Rev Mol Cell Biol 12(4): 265-273.
- Sankar, M., Nieminen, K., Ragni, L., Xenarios, I. and Hardtke, C. S. (2014). Automated quantitative histology reveals vascular morphodynamics during Arabidopsis hypocotyl secondary growth. Elife 3: e01567.
- Stegmaier, J., Amat, F., Lemon, W. C., McDole, K., Wan, Y., Teodoro, G., Mikut, R. and Keller, P. J. (2016). Real-time three-dimensional cell segmentation in large-scale microscopy data of developing embryos. Dev Cell 36(2): 225-240.
- Truernit, E., Bauby, H., Dubreucq, B., Grandjean, O., Runions, J., Barthelemy, J. and Palauqui, J. C. (2008). High-resolution whole-mount imaging of three-dimensional tissue organization and gene expression enables the study of Phloem development and structure in Arabidopsis. Plant Cell 20(6): 1494-1503.
- Yoshida, S., Barbier de Reuille, P., Lane, B., Bassel, G. W., Prusinkiewicz, P., Smith, R. S. and Weijers, D. (2014). Genetic control of plant development by overriding a geometric division rule. Dev Cell 29(1): 75-87.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mendocilla Sato, E. and Baroux, C. (2017). Analysis of 3D Cellular Organization of Fixed Plant Tissues Using a User-guided Platform for Image Segmentation. Bio-protocol 7(12): e2355. DOI: 10.21769/BioProtoc.2355.
Category
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Confocal microscopy
Cell Biology > Tissue analysis > Tissue staining
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.