- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Culturing Bacteria from Caenorhabditis elegans Gut to Assess Colonization Proficiency








Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2345 Views: 16289
Reviewed by: Jyotiska ChaudhuriJian ChenLeonardo G. Guilgur
Original research article
The authors used this protocol in:

Jan 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Determining an accurate count of intestinal bacteria from Caenorhabditis elegans is one critical way to assess colonization proficiency by a given bacteria. This can be accomplished by culturing appropriate dilutions of worm gut bacteria on selective or differential agarized media. Because of the high concentration of bacteria in gut worm, dilution is necessary before plating onto growth media. Serial dilutions can reduce the concentration of the original intestinal sample to levels low enough for single colonies to be grown on media plates, allowing for the calculation of the initial counts of bacteria in the intestinal sample.
Keywords: Caenorhabditis elegansBackground
Animals rarely live in isolation but rather exist in association with microorganisms. The more characteristic host-microbe interaction in nature is the symbiotic relationship between host and intestinal microbiota (Rosenberg and Zilber-Rosenberg, 2011). In mammals, host-microbe symbiotic interactions mainly occur along mucosal surfaces, with the most important one being the intestinal mucosa. When freshly isolated from the wild, C. elegans often harbours a diverse bacterial flora in its gut lumen, reminiscent of the microbial communities of higher organisms (Duveau and Felix, 2012; Bumbarger et al., 2013). By contrast, in the laboratory C. elegans is typically maintained in the presence of single bacterial strain (Brenner, 1974). Most often, this is the Gram-negative bacterium Escherichia coli. However, other species are sometimes used, such as the Gram-positive Bacillus subtilis (Garsin et al., 2003). An adult worm contains approximately 10,000 bacterial cells, a number 10-times greater than that of host worm somatic cells (Portal-Celhay and Blaser, 2012): perhaps coincidentally, this microbiota-to-host cell ratio is similar to that found in humans.
Different strategies are being used to measure the colonization proficiency of C. elegans gut by a bacterium such as fluorescein isothiocyanate (FITC)-labelled bacteria, bacteria expressing a fusion reporter (green fluorescent protein [GFP] or β-galactosidase). However, the more accurate method is to measure the number of CFU isolated from the worm intestine. In this protocol, we show how to isolate and count E. coli and B. subtilis strains from C. elegans gut. In the case of B. subtilis, we also show how to distinguish vegetative forms from highly resistant spores formed by this bacterium inside the C. elegans gut.
Materials and Reagents
- Pipette tips 2-200 µl Eppendorf® epT.I.P.S. (Eppendorf, catalog number: 022492039 )
- Pipette tips 50-1,000 µl Eppendorf® epT.I.P.S. (Eppendorf, catalog number: 022492055 )
- Petri dishes 60 x 15 mm 500/cs (Fisher Scientific, catalog number: FB0875713A )
- Petri dishes 35 x 10 mm 500/cs (Fisher Scientific, catalog number: FB0875711YZ )
- Corning® 15 ml centrifuge tubes (Corning, catalog number: 430791 )
- Eppendorf® Safe-Lock 1.5 ml microcentrifuge tubes (Eppendorf, catalog number: 022363204 )
- Toothpick
- 99.95 % Platinum, 0.05 % Iridium wire (3 ft/pk) (Tritech Research, catalog number: PT-9901 )
- OP50 E. coli bacteria (University of Minnesota, C. elegans Genetics Center, MN)
- Experimental and control C. elegans strains (University of Minnesota, C. elegans Genetics Center, MN)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 )
- Bacto peptone (BD, BactoTM, catalog number: 211677 )
- Agar (Sigma-Aldrich, catalog number: A1296 )
- Hypochlorite (Sigma-Aldrich, catalog number: 13440 )
- Commercial Bleach 60 g/L (DROGUERÍA INDUSTRIAL SAN JUAN, http://www.sanjuandrogueria.com)
- Triton X-100 (Sigma-Aldrich, catalog number: X100 )
- Luria broth (Sigma-Aldrich, catalog number: L3522 )
- Luria broth with agar (Sigma-Aldrich, catalog number: L2897 )
- Lysozyme from chicken egg white (Sigma-Aldrich, catalog number: L6876 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S3264 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5655 )
- Cholesterol (Sigma-Aldrich, catalog number: C8667 )
- 100% ethanol (Sigma-Aldrich, catalog number: E7023 )
- Magnesium sulfate heptahydrate (MgSO4·7H2O) (Sigma-Aldrich, catalog number: M1880 )
- Calcium chloride dihydrate (CaCl2·2H2O) (Sigma-Aldrich, catalog number: C3881 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: P2222 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: S8045 )
- Levamisole hydrochloride (Sigma-Aldrich, catalog number: L0380000 )
- Nematode growth medium (NGM) (see Recipes)
- M9 buffer (see Recipes)
- 5 mg/ml cholesterol (see Recipes)
- 1 M MgSO4 (see Recipes)
- 1 M CaCl2 (see Recipes)
- Phosphate buffer (see Recipes)
- 1 N NaOH (see Recipes)
- 25 mM Levamisole (see Recipes)
Equipment
- Erlenmeyer flask (Fisher Scientific, catalog number: FB5006000 )
- Pipettor (Gilson, catalog number: F167300 )
- Worm pick. Worm picks can either be purchased (Genesee Scientific, catalog number: 59-AWP ) or made in the lab as described in Wollenberg et al., 2013
- Pasteur glass pipette (Fisher Scientific, catalog number: 22-378893 )
- Autoclave (Tuttnauerusa, model: 6690 )
- Stirring hotplate (Corning, catalog number: 6795-620 )
- Centrifuge (Eppendorf, model: 5430 )
- Tabletop centrifuge (Eppendorf, model: 5424 )
- Pellet pestle (Kimble Chase Life Science and Research Products, catalog number: 7495211590 )
- Refrigerated incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: HerathermTM General Protocol Microbiological Incubators , catalog number: 51028064)
- Bunsen burner (Humbolt, catalog number: H-5870 )
- Dissecting stereomicroscope (Olympus, model: SMZ645 )
- Incubators for stable temperature (AQUA® LYTIC incubator 20 °C)
- Freezers (-20 °C; So-Low Environmental Equipment) (Siemens, model: C85-22 )
- Water bath
Procedure
- Preparation of a synchronized nematode population
- For NGM plate’s preparation, mix 3 g NaCl, 2.5 g Bacto peptone and 17 g agar in Erlenmeyer flask. Add to 1 L of dH2O. Autoclave 121 °C for 20 min. Cool flask in 55 °C water bath for 15 min. Add 1 ml 1 M CaCl2, 1 ml 5 mg/ml cholesterol in ethanol, 1 ml 1 M MgSO4 and 25 ml 1 M KPO4 (Phosphate) buffer (see Recipes). Swirl to mix well. Using sterile procedures dispense the NGM solution into 60 x 15 mm Petri plates. Fill plates 2/3 full of agar.
- Pick 10 young adults onto an E. coli OP50 seeded 60 mm NGM plate (see Recipes). Allow the worms to grow 2-3 days to ensure at least 100 gravid adult worms and an adequate number of eggs on the plate.
- Once reached this worm number, pour 3 ml of M9 buffer (see Recipes) onto the plate and gently swirl it to dislodge the worms. Repeat this procedure one more time using 2 ml of M9 buffer. One plate is enough for bleaching.
- Using a pipette, transfer the worms to a 15 ml conical tube.
- Centrifuge for about 1 min at 252 x g to pellet the worms.
- Aspirate most of the M9 without disturbing the worm pellet.
- Add about 1 ml of 3% hypochlorite solution and 2.5 ml of 1 N NaOH (see Recipes) to the tube.
- Vortex the tube for approximately 5 min or until you see a decrease in the number of intact adult worms. Do not bleach for much longer than this or you will kill the eggs.
- Once most of the bodies have dissolved, neutralize the alkaline hypochlorite by adding M9 buffer until 15 ml of the conical tube and centrifuge at 252 x g for 1 min.
- Aspirate most of the supernatant solution without disturbing the egg pellet.
- Add about 15 ml of M9 to the tube and mix well.
- Centrifuge again at 252 x g for 1 min.
- Aspirate most of the M9 without disturbing the egg pellet.
- Repeat steps A11-A13 at least one more time.
- Add about 5 ml of fresh M9 and agitate to resuspend the egg-enriched pellet. Incubate overnight at room temperature with gentle rocker stirrer. The eggs will hatch and the animals will arrest as starved L1 larvae.
- Transfer between 100 and 200 starved L1 larvae to a 60 mm NGM plate with E. coli OP50 cells (1 x 105 cells per plate) or spores (1 x 105 spores per plate) (see Note 2) of each B. subtilis strain and allow the worms to grow 2 days until they reach L4/adult stage.
- For NGM plate’s preparation, mix 3 g NaCl, 2.5 g Bacto peptone and 17 g agar in Erlenmeyer flask. Add to 1 L of dH2O. Autoclave 121 °C for 20 min. Cool flask in 55 °C water bath for 15 min. Add 1 ml 1 M CaCl2, 1 ml 5 mg/ml cholesterol in ethanol, 1 ml 1 M MgSO4 and 25 ml 1 M KPO4 (Phosphate) buffer (see Recipes). Swirl to mix well. Using sterile procedures dispense the NGM solution into 60 x 15 mm Petri plates. Fill plates 2/3 full of agar.
- Preparation of nematode samples for intestinal bacterial counting
- Use an eyebrow hair and collect 50 adult animals in M9 buffer in a 1.5 ml tube (see Note 1).
- Use the same number of animals (worms) fed on each bacterial strain during sample preparation.
- Treat the worms with 0.25 mM levamisole to induce temporal paralysis.
- Superficially sterilize with 3% commercial Bleach for 5 min (see Note 5).
- Wash at least three times with M9 buffer.
- After the worms were surface sterilized, worms devoid of outside bacteria are disrupted using a pellet pestle and 1% Triton X-100 (see Note 6).
- Centrifuge at 14,800 x g for 10 min, remove the supernatant and resuspend the pellet of each tube in 500 µl M9 buffer (see Note 4).
- Use an eyebrow hair and collect 50 adult animals in M9 buffer in a 1.5 ml tube (see Note 1).
- Preparation of sample dilutions (Figure 1)
- To begin the procedure, take 50 µl of the cell suspensions and add to 450 µl of deionized water. Shake the suspension well, and label as ‘10-1’.
- Before the sample settles, remove 50 µl of the cell suspension with a sterile pipette and transfer it to a 450 µl-deionized water blank. Vortex the sample, and label as ‘10-2’.
- Repeat this dilution step, using 50 µl of the previous suspension and a 450 µl-deionized water blank. Label these sequentially as tubes ‘10-3’.
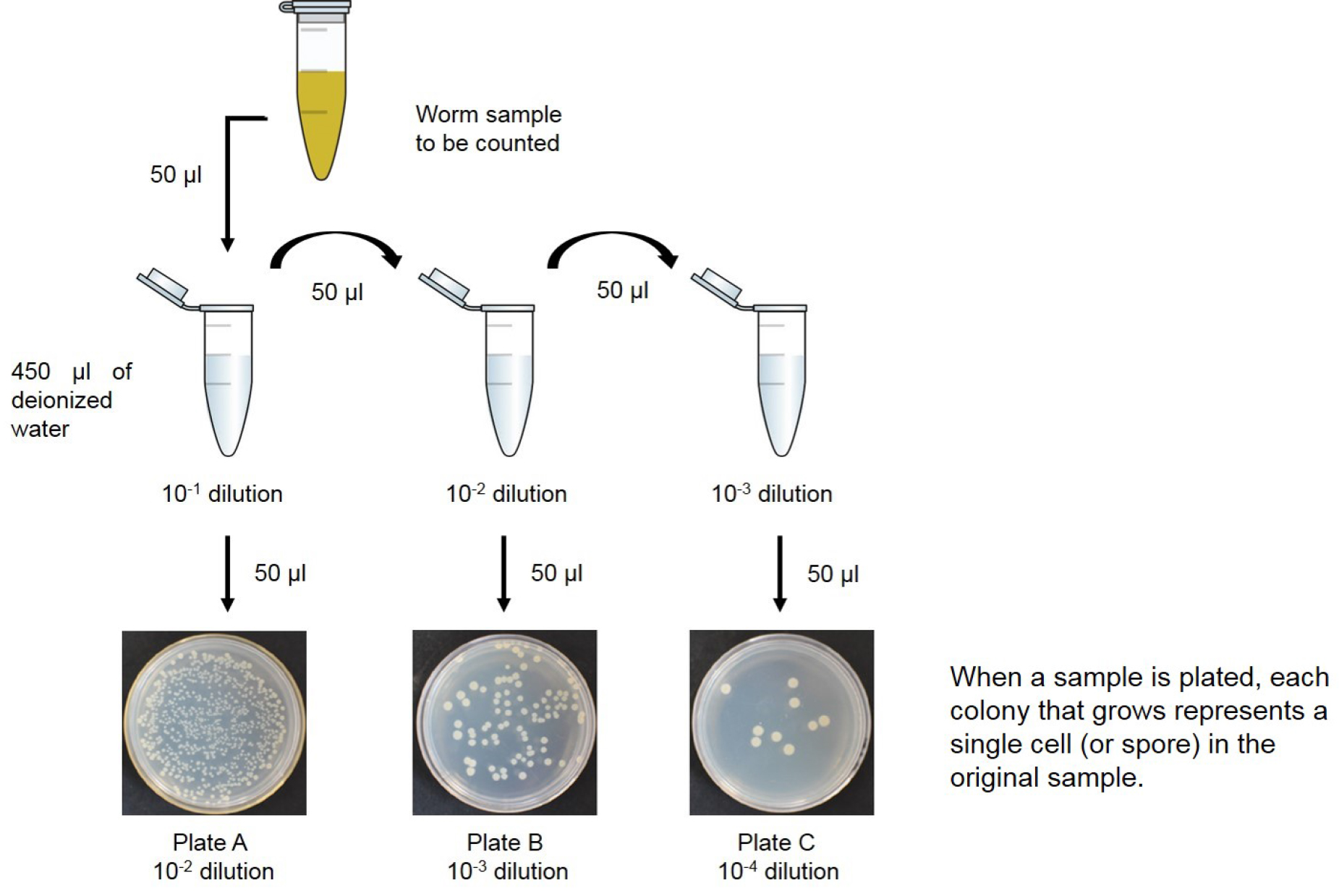
Figure 1. Diagram of how to prepare bacterial dilutions. Serial dilution of an initial culture to obtain solutions that are 1/10th, 1/100th and 1/1,000th the concentration of the initial sample (10-1, 10-2 and 10-3 dilutions, respectively). For each dilution, tubes initially have 450 µl of water in them so that the final volume will be 500 µl after the addition of 50 µl of culture.
- To begin the procedure, take 50 µl of the cell suspensions and add to 450 µl of deionized water. Shake the suspension well, and label as ‘10-1’.
- Making spread plates for bacterial culture
- To get bacterial colonies from worm gut isolated E. coli and vegetative B. subtilis (see Figure 2A), take three LB agar plates (see Note 3) and label them as A, B, and C. Vortex samples 10-1, 10-2, and 10-3, and pipette 50 µl onto each plate. This increases the dilution value further, by a factor of ten (A = 10-2, B = 10-3, C = 10-4) (see Figure 1).
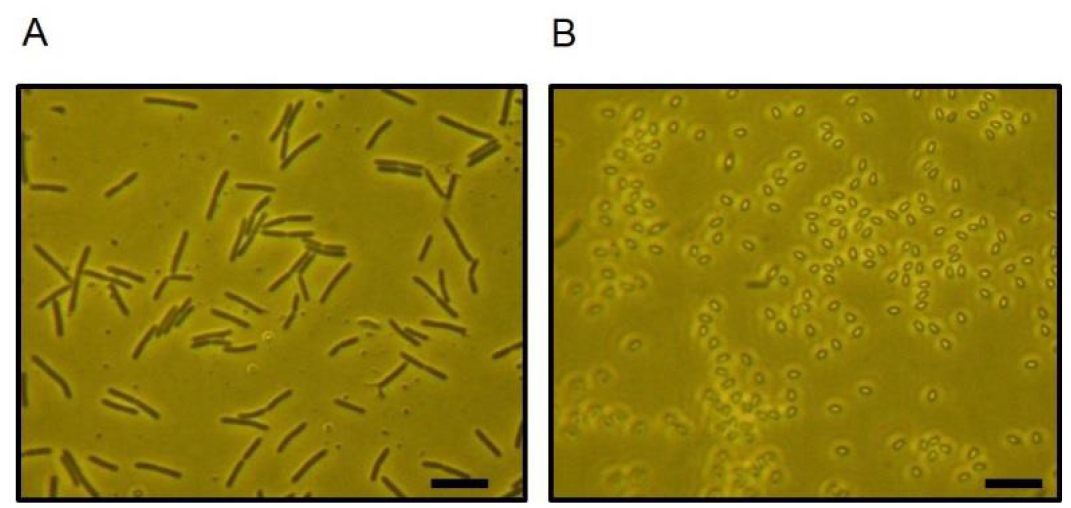
Figure 2. The micrographs show the vegetative cells (A) and spores (B) of Bacillus subtilis. Bar size = 5 µm. - Next, dip a glass spreader into ethanol. Place the spreader in a flame for a few seconds to ignite and burn off the ethanol. This will sterilize the spreader.
- Hold the spreader above the first plate until the flame is extinguished. Open the plate quickly, holding the lid close by. Touch the spreader to the agar away from the inoculum (Inoculum = cells used to begin a culture) to cool, and then spread the drop of inoculum around the surface of the agar until traces of free liquid disappear.
- Re-flame the spreader and repeat the process with the next plate, working quickly so as not to contaminate the agar with airborne organisms.
- Incubate the bacteria plates at 37 °C for 16-18 h. Make sure the plates are inverted during the incubation to prevent drops of moisture from condensation from falling onto the agar surface.
- To get bacterial colonies from worm gut isolated E. coli and vegetative B. subtilis (see Figure 2A), take three LB agar plates (see Note 3) and label them as A, B, and C. Vortex samples 10-1, 10-2, and 10-3, and pipette 50 µl onto each plate. This increases the dilution value further, by a factor of ten (A = 10-2, B = 10-3, C = 10-4) (see Figure 1).
- Making spread plates for spore culture
- Heat-treat the tubes with B. subtilis samples labeled as 10-1, 10-2, and 10-3 for 20 min at 80 °C to kill vegetative cells. Vortex spores samples (see Figure 2B) and pipette 50 µl onto each plate. This increases the dilution value further, by a factor of ten (A = 10-2, B = 10-3, C = 10-4).
- Next, dip a glass spreader into ethanol. Place the spreader in a flame for a few seconds to ignite and burn off the ethanol. This will sterilize the spreader.
- Hold the spreader above the first plate until the flame is extinguished. Open the plate quickly, holding the lid close by. Touch the spreader to the agar away from the inoculum to cool, and then spread the drop of inoculum around the surface of the agar until traces of free liquid disappear.
- Re-flame the spreader and repeat the process with the next plate, working quickly so as not to contaminate the agar with airborne organisms.
- Incubate the bacteria plates at 37 °C for 16-18 h. Make sure the plates are inverted during the incubation to prevent drops of moisture from condensation from falling onto the agar surface.
- Heat-treat the tubes with B. subtilis samples labeled as 10-1, 10-2, and 10-3 for 20 min at 80 °C to kill vegetative cells. Vortex spores samples (see Figure 2B) and pipette 50 µl onto each plate. This increases the dilution value further, by a factor of ten (A = 10-2, B = 10-3, C = 10-4).
- Bacterial counts
- After incubation, examine all of the bacteria plates carefully, and note differences in colony size and shape.
- Count and record the number of bacterial colonies. Only count and use for calculation, plates with 30-300 colonies per plate. The number of colonies per plate bellow 30 is not statistically significant whereas a number higher than 300 colonies per plate is hard to count, and it is possible a colony arise from more than one cell.
- The number of CFU/worm is calculated as follows:
Number of CFU/worm in plate A = (number of colonies x 102)/50
Number of CFU/worm in plate B = (number of colonies x 103)/50
Number of CFU/worm in plate C = (number of colonies x 104)/50
- After incubation, examine all of the bacteria plates carefully, and note differences in colony size and shape.
Data analysis
- For each strain or condition, use at least 50-100 animals to obtain results that are more accurate.
- For each experiment, the same number of nematodes should be examined for each strain and condition.
- Each assay should be run in triplicate and repeated at least three times (Duveau and Felix, 2012).
- Use the Student’s t-test with a significance cut-off level of P < 0.05 for comparisons between two groups.
- Use the one-factor (ANOVA) variance analysis and correct by the post hoc Bonferroni test for multiple comparisons.
Notes
- Take a toothpick and glue an eyebrow hair to the tip of it. Let it dry at room temperature. Then, use this tool to pick nematodes. Before using the eyebrow hair always, sterilize it by using ethanol.
- To obtain pure spores, the heat-treated culture (20 min at 80 °C to kill vegetative cells in a water bath) was treated three times with lysozyme, washed each time with cold water and centrifuged until 100% of the culture consisted of phase-bright spores.
- For the LB plates used to seed Bacillus subtilis, we strongly recommend drying plates at 45 °C for 20 min or 37 °C for 40 min before to drop the dilutions onto each plate to avoid the sliding movement of this bacterium in solid surface with water drops from condensation.
- Samples should be used immediately or store for 1 week at -20 °C.
- To surface sterilize, resuspend 50 worms in 500 µl M9 buffer, add 0.25 mM levamisole (5 µl of 25 mM levamisole stock solution) and 3 % commercial Bleach (stock solution). Mix samples by gentle inversion for 15 min. Then, wash the worms with M9 buffer at least 3 times.
- After the worms are surface sterilized, pour the 200 µl worm suspension in M9 buffer in a 1.5 ml microcentrifuge tube. Add the extraction solution of choice (1% Triton X-100), and the pestle is used to grind the sample 10 times or until a creamy white to yellow suspension is obtained. After centrifugation, cellular debris and bacteria remain firmly in the bottom of the conical tube, and the supernatant is easily pipetted out.
Recipes
- Nematode growth medium (NGM)
Dissolve 3 g NaCl, 2.5 g Bacto peptone and 17 g agar to 1 L of dH2O
Autoclave
Store at room temperature - M9 buffer
Dissolve 5 g NaCl, 6 g Na2HPO4 and 3 g KH2PO4 to 1 L of dH2O
Autoclave
Store at room temperature - 5 mg/ml cholesterol
Dissolve 0.25 g of cholesterol in 50 ml of 100% ethanol
Do not autoclave
Store at room temperature - 1 M MgSO4
Dissolve 6 g MgSO4 heptahydrate in 50 ml of dH2O
Autoclave
Store at room temperature - 1 M CaCl2
Dissolve 5.55 g CaCl2 dihydrate in 50 ml of dH2O
Autoclave
Store at room temperature - Phosphate buffer
Dissolve 10.7 g K2HPO4 and 32.5 g KH2PO4 to 300 ml of dH2O
Adjust pH to 6.0
Autoclave
Store at room temperature - 1 N NaOH
Dissolve 2 g NaOH in 50 ml of dH2O - 25 mM levamisole
Dissolve 0.3 g levamisole in 50 ml of dH2O
Acknowledgments
This work was supported by CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas) and FONCyT (Fondo para la Investigación Científica y Tecnológica) with the aid of the Pew Latin-American Program in Biological Sciences (Philadelphia, PA, USA), the Fulbright Committee (Washington, DC, USA) and former Fundación Antorchas (Buenos Aires, Argentina). We modified the media and NGM plate’s preparation from Stiernagle T. Maintenance of C. elegans. WormBook. 2006 11:1-11. We modified C. elegans synchronization from Montserrat Porta-de-la-Riva et al. (2012). Basic Caenorhabditis elegans Methods: Synchronization and Observation. Journal of Visualized Experiments; 64, e4019, 1-9.
References
- Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77(1): 71-94.
- Bumbarger, D. J., Riebesell, M., Rodelsperger, C. and Sommer, R. J. (2013). System-wide rewiring underlies behavioral differences in predatory and bacterial-feeding nematodes. Cell 152(1-2): 109-119.
- Duveau, F. and Felix, M. A. (2012). Role of pleiotropy in the evolution of a cryptic developmental variation in Caenorhabditis elegans. PLoS Biol 10(1): e1001230.
- Garsin, D. A., Villanueva, J. M., Begun, J., Kim, D. H., Sifri, C. D., Calderwood, S. B., Ruvkun, G. and Ausubel, F. M. (2003). Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300(5627): 1921.
- Porta-de-la-Riva, M., Fontrodona, L., Villanueva, A. and Cerón, J. (2012). Basic Caenorhabditis elegans methods: synchronization and observation. J Vis Exp 64: e4019.
- Portal-Celhay, C. and Blaser, M. J. (2012). Competition and resilience between founder and introduced bacteria in the Caenorhabditis elegans gut. Infect Immun 80(3): 1288-1299.
- Rosenberg, E. and Zilber-Rosenberg, I. (2011). Symbiosis and development: the hologenome concept. Birth Defects Res C Embryo Today 93(1): 56-66.
- Stiernagle, T. (2006). Maintenance of C. elegans. WormBook 11:1-11.
- Wollenberg, A. C., Visvikis, O., Alves, A. F. and Irazoqui, J. E. (2013). Staphylococcus aureus killing assay of Caenorhabditis elegans. Bio-protocol 3(19): e916.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rodriguez Ayala, F., Cogliati, S., Bauman, C., Leñini, C., Bartolini, M., Villalba, J. M., Argañaraz, F. and Grau, R. (2017). Culturing Bacteria from Caenorhabditis elegans Gut to Assess Colonization Proficiency. Bio-protocol 7(12): e2345. DOI: 10.21769/BioProtoc.2345.
Category
Microbiology > Microbe-host interactions > Nematode
Cell Biology > Cell isolation and culture > Cell growth
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.