- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Loading of Extracellular Vesicles with Chemically Stabilized Hydrophobic siRNAs for the Treatment of Disease in the Central Nervous System
Published: Vol 7, Iss 12, Jun 20, 2017 DOI: 10.21769/BioProtoc.2338 Views: 9967
Reviewed by: Longping Victor TseVaibhav B ShahAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Efficient delivery of oligonucleotide therapeutics, i.e., siRNAs, to the central nervous system represents a significant barrier to their clinical advancement for the treatment of neurological disorders. Small, endogenous extracellular vesicles were shown to be able to transport lipids, proteins and RNA between cells, including neurons. This natural trafficking ability gives extracellular vesicles the potential to be used as delivery vehicles for oligonucleotides, i.e., siRNAs. However, robust and scalable methods for loading of extracellular vesicles with oligonucleotide cargo are lacking. We describe a detailed protocol for the loading of hydrophobically modified siRNAs into extracellular vesicles upon simple co-incubation. We detail methods of the workflow from purification of extracellular vesicles to data analysis. This method may advance extracellular vesicles-based therapies for the treatment of a broad range of neurological disorders.
Keywords: RNA interferenceBackground
siRNAs are one type of oligonucleotide therapeutics, a new class of drugs directly targeting messenger RNAs (mRNAs) to prevent the expression of proteins leading to disease phenotypes. The therapeutic application of siRNAs is extremely promising as siRNAs can be designed to target any gene, including genes not ‘druggable’ with small molecules or protein-based therapies. The progress made in the chemistry of oligonucleotide therapeutics enables the design of fully stabilized hydrophobically modified siRNAs (hsiRNAs, modified with 2’-O-Methyl or 2’-Fluoro as well as phosphorothioates and sense strand covalently conjugated to cholesterol), which promote cellular self-internalization of hsiRNAs and maintain an ability to be efficiently loaded into the RNA-induced silencing complex (RISC) (Byrne et al., 2013; Khvorova and Watts, 2017). A cholesterol conjugate, linked to the 3’ end of the passenger strand, is essential for rapid cellular membrane association (Byrne et al., 2013; Alterman et al., 2015). The single-stranded phosphorothioate tail promotes cellular internalization (Geary et al., 2015). We recently demonstrated that hsiRNAs bind cellular membranes within seconds after treatment, enter cells and promote potent gene silencing in vitro (Byrne et al., 2013; Alterman et al., 2015; Ly et al., 2017). However, upon local bolus injection in vivo in mouse brain, hsiRNA spread and efficacy are limited to the region surrounding the site of administration (Alterman et al., 2015). Whereas hsiRNAs remain therapeutically promising due to potent and specific gene silencing, their delivery to the brain hampers their advancement for the treatment of diseases in the central nervous system.
Endogenously produced extracellular vesicles mediate intercellular transfer of lipids, proteins, and RNAs between cells over short and long distances, thus playing a crucial role in health and disease (Distler et al., 2005; Muralidharan-Chari et al., 2010). The ability of extracellular vesicles to carry functional RNAs has attracted considerable interest to their use as novel vehicles to transport and deliver RNA-based therapeutics. The cargo includes siRNAs or other oligonucleotide therapeutics (Tetta et al., 2013). Strategies used to load RNA-based therapeutics into extracellular vesicles include electroporation (Alvarez-Erviti et al., 2011; Ohno et al., 2013) or overexpression of miRNAs in extracellular vesicle-producing cells (Kosaka et al., 2012; Ohno et al., 2013; Mizrak et al., 2013). Though both strategies have been able to promote the transfer of siRNA-loaded extracellular vesicles into target cells and the silencing of the target gene, they cannot be controlled or scaled up for clinical-stage manufacturing (Kooijmans et al., 2013). Moreover, electroporation compromises the integrity of extracellular vesicles (Kooijmans et al., 2013). We demonstrated the efficient loading of extracellular vesicles with hsiRNAs without modifying the vesicle size distribution, concentration and integrity. hsiRNA-loaded extracellular vesicles were shown to induce gene silencing of the target gene, huntingtin mRNA, in vitro in mouse primary neurons, and in vivo in mouse brain (Didiot et al., 2016).
Here, we describe a method exploring the ability of hsiRNAs to bind membranes to promote their loading into extracellular vesicles. The co-incubation of hsiRNAs with extracellular vesicles purified from cell culture conditioned medium, promotes loading into extracellular vesicles. We provide details on our methods for extracellular vesicles purification, loading of extracellular vesicles with hsiRNAs, size, charge and integrity characterization of hsiRNA-loaded extracellular vesicles, as well as in vivo testing of hsiRNAs-loaded extracellular vesicles in mouse brain, and data analysis. This technology may promote the loading of several other classes of oligonucleotide therapeutics (i.e., antisense, splice-switching oligonucleotides, sterically blocking oligonucleotides, aptamers and others) to extracellular vesicles, thus providing a significant leap forward to advance multiple classes of oligonucleotide therapeutics for the treatment of diseases in the brain. Subsequently, exploiting the natural properties of extracellular vesicles to functionally transport small RNAs (Valadi et al., 2007; Pegtel et al., 2010; Wang et al., 2010) offers a strategy for improving the in vivo distribution of and cellular uptake of oligonucleotide therapeutics (Zomer et al., 2010; El Andaloussi et al., 2013; Kooijmans et al., 2012; Lasser, 2012; Lee et al., 2012; Pan et al., 2012; Marcus and Leonard, 2013; Nazarenko et al., 2013; Didiot et al., 2016).
Materials and Reagents
- Tips (from 0.2 µl to 1,000 µl) (VWR)
- Paper towel
- Serological pipettes individually wrapped (from 5 ml to 50 ml) (Olympus)
- 0.22-μm filter-sterilization system (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 567-0020 )
- Tissue culture treated multilayer flask–T500 cm2 triple flask (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 132867 )
- 50 ml conical centrifuge tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339652 )
- Vacuum-connected Pasteur pipette
- 1.7 ml microcentrifuge tubes (Genesee Scientific, catalog number: 22-282 )
- Aluminum foil
- UV-transparent, flat-bottom 96-well plate (Corning, catalog number: 3635 )
- 1 ml syringe (BD, catalog number: 309659 )
- Parafilm (Bemis, Parafilm M®)
- Whatman No. 1 filter paper (GE Healthcare, Whatman, catalog number: 10010155 )
- 30 G ½ needle
- Electron microscopy grids (Electron Microscopy Sciences, catalog number: FCF2010-Ni ) and clean forceps to manipulate the grid
- Grid storage box (Electron Microscopy Sciences, catalog number: 71156 )
- Mouse (FVB/NJ) (THE JACKSON LABORATORY, catalog number: 001800 )
- 200-proof ethanol (Decon Labs, catalog number: 2805M )
- Fetal bovine serum (FBS) (Mediatech, catalog number: 35-010-CV )
- Phosphate-buffered saline (PBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14190250 )
- Protease inhibitor cocktail stock solution (Sigma-Aldrich, catalog number: P8340 )
- Cy3-labeled or biotinylated cholesterol-conjugated hsiRNAs (produced in-house)
- Cy3-OO-PNA strands, fully complementary to the hsiRNA guide strand (20 nucleotides long) (PNA Bio)
- Peptide-nucleic acid (PNA) hybridization assay (developed by Axolabs, Kulmbach, Germany)
- RIPA buffer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 89900 )
- Proteinase K (Thermo Fisher Scientific, InvitrogenTM, catalog number: 25530049 )
- Glycine (Sigma-Aldrich, catalog number: G7126 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153 )
- Saponin (Sigma-Aldrich, catalog number: S1252 )
Note: This product has been discontinued. - Glutaraldehyde (Polysciences, catalog number: 01909-10 )
- Avertin (Sigma-Aldrich, catalog number: T48402 )
- QuantiGene 2.0 Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: QS0011 )
- Quantigene 2.0 Probesets (Varies by gene)
- Dulbecco’s modified Eagle medium (DMEM)
- MSC culture media (ATCC, catalog number: PCS-500-041 )
- Sucrose (Sigma-Aldrich, catalog number: S0389 )
- Tris base (TRIZMA) (Sigma-Aldrich, catalog number: T6066 )
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: H1758 )
- Ethylenediaminetetraacetate acid disodium salt (EDTA) (Sigma-Aldrich, catalog number: E6758 )
- Sodium hydroxide (NaOH) (~50 ml of NaOH) (Sigma-Aldrich, catalog number: 72068 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771 )
- Sodium phosphate monobasic monohydrate (NaH2PO4·H2O)
- Sodium phosphate monobasic (NaH2PO4) (Sigma-Aldrich, catalog number: S3139 )
- Acetonitrile (50% acetonitrile solution, diluted in distilled H2O) (Fisher Scientific, catalog number: A998-4 )
- Sodium perchlorate monohydrate (NaClO4·xH2O) (Fisher Scientific, catalog number: S490-500 )
- Methyl cellulose (Sigma-Aldrich, catalog number: M6385 )
- PFA powder (Sigma-Aldrich, catalog number: P6148 )
- Uranyl acetate (Electron Microscopy Sciences, catalog number: 22400 )
- Oxalic acid
- Tris-HCl (pH 8.5) (Fisher Scientific, catalog number: BP153 )
- Ammonium hydroxide (NH4OH) (Fisher Scientific, catalog number: A669-212 )
- Cell culture medium (see Recipes)
- 1 M sucrose (see Recipes)
- 1 M Tris-HCl (see Recipes)
- 0.5 M EDTA (see Recipes)
- 3 M KCl (see Recipes)
- 10% SDS (see Recipes)
- 0.1 M sodium phosphate buffer (see Recipes)
- HPLC buffer A (see Recipes)
- HPLC buffer B (see Recipes)
- Glutaraldehyde, 1% (v/v) (see Recipes)
- Methyl cellulose, 2% (w/v) (see Recipes)
- Paraformaldehyde (PFA), 2% and 4% (w/v) (see Recipes)
- Uranyl acetate (4% w/v), pH 4 (see Recipes)
- Uranyl-oxalate, pH 7 (see Recipes)
- Methyl cellulose-UA, pH 4 (see Recipes)
Equipment
- Tissue culture hood
- Soft brush
- Ultracentrifuge 70 ml polycarbonate bottles (Beckman Coulter, catalog number: 355655 )
- Fixed-angle Ti45 ultracentrifuge rotor (Beckman Coulter, model: Type 45 Ti , catalog number: 339160)
- Refrigerated ultracentrifuge (Beckman Coulter, model: Optima XE )
- Refrigerated benchtop centrifuge (Beckman Coulter, model: Allegra® X-15R )
- Micropipettes from 0.5 µl to 1 ml (Labnet International, model: BioPetteTM Plus )
- Fixed-angle TLA-110 rotor (Beckman Coulter, model: TLA-110 , catalog number: 366735)
- 1.5 ml microcentrifuge adapters (Beckman Coulter, catalog number: 360951 )
- Refrigerated benchtop ultracentrifuge (Beckman Coulter, model: OptimaTM MAX-TL )
- Orbital thermo-shaker for 1.5 ml microtube (Grant Instruments, model: PHMT series )
- Plate reader spectrophotometer (Tecan Trading, model: Infinite® M1000 Pro )
- Stereotactic frame (KOPF INSTRUMENTS, model: Model 963 )
- HPLC system with fluorescent detector with autosampler (Agilent Technologies, model: HPLC 1100 series )
- Hamilton syringe (Hamilton, model: Gastight #1002 )
- Extracellular vesicles size distribution and concentration reader (Malvern Instruments, model: NanoSight NS300 )
- Extracellular vesicles charge reader (Malvern Instruments, model: Zetasizer Nano NS )
- Glass micro-electrophoresis cuvette Dip Cell kit (Malvern Instruments, catalog number: ZEN1002 )
- Transmission electron microscope (JEOL, model: JEM-1011 )
Note: This product has been discontinued. - ALZET® osmotic pumps (DURECT, ALZET, catalog numbers: 1003D and 1007D )
- Water bath at 37 °C
- Microscissors (Fine Science Tools, catalog numbers: 14060-10 ; 14002-12 )
- Set of two forceps (Fine Science Tools, catalog number: 11251-30 )
- Vibratome (Leica Biosystems, model: Leica VT1000 S)
- Autoplate washer (BioTek Instruments, model: ELx405 )
- Tissue culture incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: HeracellTM 150i )
- Pipet-aid (Drummond Scientific, model: Portable Pipet-Aid® XP )
- Tissue culture phase-contrast inverted microscope (Motic, model: AE2000 )
- Anion exchange column (Thermo Fisher Scientific, Thermo ScientificTM, model: DNAPacTM PA100 )
- Heat block (Fisher Scientific, model: IsotempTM 2050FS )
- µPlate carrier (Beckman Coulter, model: SX4750 )
- -86 °C freezer (Thermo Fisher Scientific, Thermo ScientificTM, model: FormaTM 900 Series )
- Biological safety cabinet connected to vacuum (Thermo Fisher Scientific, Thermo ScientificTM, model: 1300 Series Class II , Type A2)
Software
- Nanoparticles Tracking Analysis (NTA) software
- Microsoft Office Excel (Microsoft Pack Office)
- GraphPad Prism 6 software (GraphPad Software, Inc.)
Procedure
- Extracellular vesicles production and purification
Notes:- The purification of extracellular vesicles is shown in Figure 1.
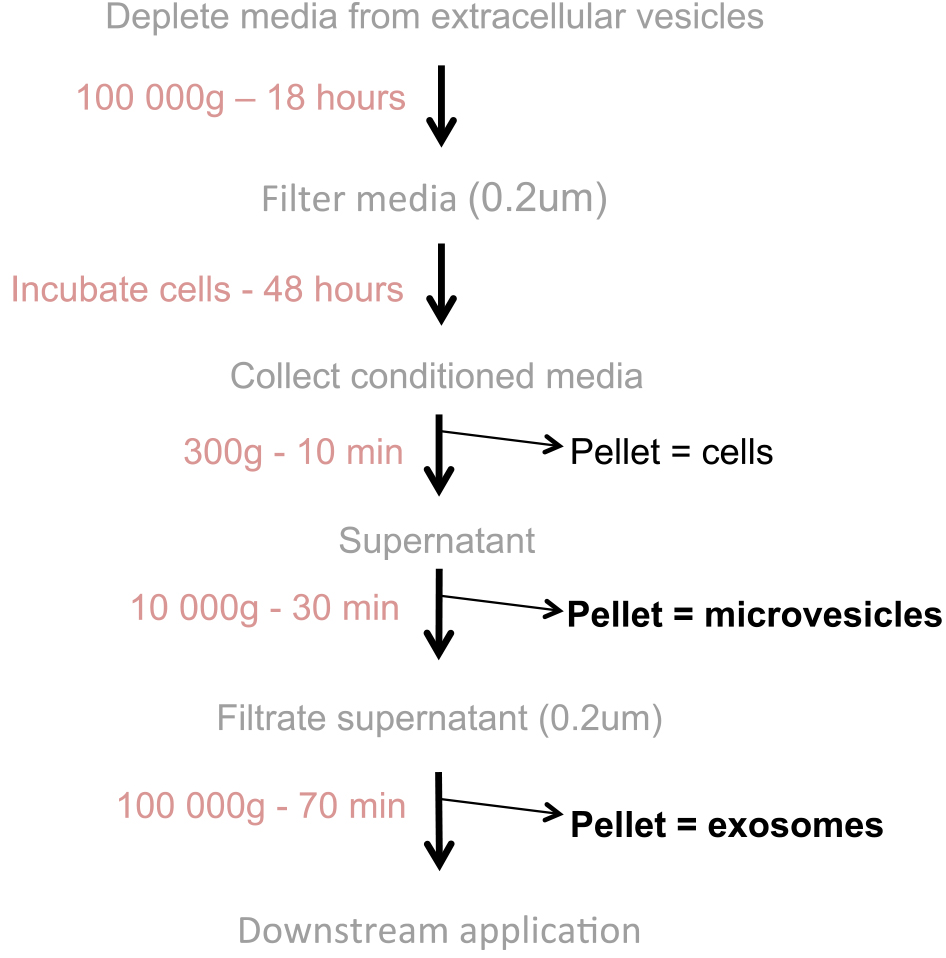
Figure 1. Flowchart of extracellular vesicle production: Extracellular vesicles (microvesicles and exosomes) are purified from conditioned media via differential ultracentrifugation - All steps that involve opening the ultracentrifuge bottles must be performed in a tissue culture hood. To sterilize the bottles, wash the bottles and their lids with soap using a soft brush and rinse carefully with sterile water to remove any trace of soap. In a tissue culture hood, cleaned bottles and their lids are sterilized in a 70% ethanol bath and dried on a paper towel under UV light for a final sterilization. Polycarbonate bottles are sensitive to ethanol and UV light, so avoid leaving the bottles in 70% ethanol for longer than 2 min and UV for longer than 30 min.
- The extracellular vesicle pellets are transparent brown and very small, which make them difficult to detect. Using a permanent marker, label the side of each 70 ml ultracentrifugation bottle to orient the bottles in the rotor and to indicate where to look for the extracellular vesicle pellet. All 70 ml bottles should be full to the bottle shoulder (~65 ml) for high-speed centrifugation. Otherwise the bottle will collapse and damage the rotor. If not filled to the shoulder, complete the volume with PBS.
- Preparing extracellular vesicle-depleted medium and cell culture
Note: Extracellular vesicles are naturally present in body fluid, including the fetal bovine serum (FBS) usually used to supplement cell culture medium. Thus, prior any use, the cell culture medium, from which the extracellular vesicles are purified, is depleted of the serum-extracellular vesicles by overnight ultracentrifugation to minimize the contamination. - For each cell line, prepare the recommended medium supplemented with 40% (v/v) FBS. Depleted medium supplemented with 40% FBS allows preparation of a large volume of extracellular vesicle-free culture medium in a single round of centrifugation.
- Transfer 65 ml of medium in 70 ml bottles using a 50 ml serological pipette.
- Place the bottles, with the label facing up, in a pre-cooled Type 45 Ti rotor (stored at 4 °C) and pre-cool the ultracentrifuge to 4 °C.
- Ultracentrifuge for at least 18 h at 100,000 x g, 4 °C.
- To filter sterilize the supernatant, transfer the contents of each bottle into a vacuum-connected 0.22 μm filter-sterile rapid-flow disposable unit using a 50 ml serological pipette. At the bottom of the bottle, leave 2-3 ml of medium containing higher concentration of brown sediments. The pellet is brown and very sticky and will not detach from the edge of the bottle (see Note 1).
- Store the sterilized extracellular vesicle-depleted medium, diluted or not, up to 4 weeks at 4 °C.
- Prior to cell culture, dilute the extracellular vesicle-depleted medium with additional medium to obtain medium with the recommended concentration of FBS (usually 10% FBS) required for the cell line. If necessary, supplement the medium with any additional nutrients and/or antibiotics.
- Production and purification of extracellular vesicles
Notes:- Extracellular vesicles are purified from conditioned medium by a method adapted from Thery et al. (2006) combining sequential centrifugation and filtration steps. All extracellular vesicles quality controls were performed following the experimental requirements recommended by the International Society of Extracellular Vesicles (Lotvall et al., 2014; Thery et al., 2006).
- In vivo experiments need a significant concentration of extracellular vesicles for efficient silencing. To collect large quantities of extracellular vesicles, we purify extracellular vesicles from conditioned medium from adherent cells in culture. The yield of purified extracellular vesicles correlates with the increase of medium volume. Thus, to produce large volume of conditioned medium and to scale up the concentration of extracellular vesicles, we use T500 cm2 triple flasks.
- In multilayer flasks, grow adherent cells of interest in the regular recommended medium until they reach 70% to 80% confluency. We observed that 60 ml is the minimal volume of medium that can be used in T500 cm2 triple flask.
- Pour the culture medium in a liquid trash, as serological pipette cannot be used to remove liquid from multilayer flasks. Using a 50 ml serological pipette, replace the medium with a similar volume of recommended EV-depleted medium.
- Incubate the cells for 48 h at 37 °C, 5% CO2 and 100% humidity.
- Collect the medium and proceed to extracellular vesicle purification as followed. If the cells are not 100% confluent, replace the collected medium with fresh extracellular vesicle-depleted medium, incubate for 48 h and repeat the extracellular vesicle purification as followed.
- Harvest the culture medium containing extracellular vesicles in 50 ml centrifuge conical tubes.
- Centrifuge at 300 x g for 10 min at room temperature to pellet and eliminate the floating cells and large debris.
- Transfer the supernatant into new 50 ml centrifuge conical tubes using 50 ml serological pipette. Leave 2-3 ml of medium in the bottom of the tube to avoid disturbing the pellet. Discard the pellet.
- Centrifuge the supernatant at 10,000 x g, 4 °C for 30 min to isolate large vesicles or microvesicles.
- Pour and filter sterilize the supernatant through a vacuum-connected sterile 0.22 μm filter unit to further eliminate larger vesicles.
Note: The 10,000 g pellet, containing microvesicles (300 nm to 1 µm), is white and sticky and can be used for additional experiments. If microvesicles are needed, completely remove the remaining medium forming a drop at the bottom of the bottle using a vacuum-connected Pasteur pipette. Using a micropipette, suspend the pellet in 1 ml of PBS. The pellet should detach easily with the flow of PBS. Centrifuge the collected microvesicles at 10,000 x g, 4 °C for 30 min, as an additional wash step, and suspend the pellet in 100 µl of PBS. - Using a 50 ml serological pipette, transfer the filtered medium in 70 ml polycarbonate bottles.
Note: Mark the bottles with a permanent marker and place the bottles in the centrifuge with the mark facing away from the center. This will help the localization of the pellet later. - Ultracentrifuge for at least 70 min at 100,000 x g, 4 °C in a pre-cooled Type 45 Ti rotor.
- Aspirate completely the supernatant from all bottles using a vacuum-connected Pasteur pipette.
Note: Remaining medium will fall down the wall of the bottle and form a final drop of supernatant that has to be aspirated by vacuum-connected Pasteur pipette. - Detect the extracellular vesicles pellet on the bottom of the bottle. The pellet is brownish transparent in color and approximately 5 mm in diameter.
Note: The pellet will be localized to the marked side of the bottle. - To concentrate the extracellular vesicles, suspend all the pellets in the same 1 ml PBS using a micropipette. With the tip, scrape the bottle in and around the pellet to recover a maximum of extracellular vesicles.
- Transfer the suspended pellet into 1.5 ml microcentrifuge tubes. Carefully recover the remaining drops of extracellular vesicle-containing PBS forming at the bottom of each bottle.
- Place the 1.5 ml tubes in the tube holders in a pre-cooled TLA-110 rotor with the tube-cap orientated to find the pellet.
- Centrifuge for at least 70 min at 100,000 x g, 4 °C in a tabletop ultracentrifuge.
- Resuspend the pellet in 100 µl PBS, 1x protease inhibitor cocktail and 10 mM sucrose for further experiments. Store at 4 °C for a short-term use and -80 °C for long term storage. Avoid repeated freezing and thawing (see Notes 2 and 3).
Note: For proteomic or lipidomic analysis, store the pellet as is at -80 °C.
- Extracellular vesicles are purified from conditioned medium by a method adapted from Thery et al. (2006) combining sequential centrifugation and filtration steps. All extracellular vesicles quality controls were performed following the experimental requirements recommended by the International Society of Extracellular Vesicles (Lotvall et al., 2014; Thery et al., 2006).
- The purification of extracellular vesicles is shown in Figure 1.
- Preparation and characterization of hsiRNA-loaded extracellular vesicles
- Loading of extracellular vesicles with hsiRNAs.
The loading of extracellular vesicles with hsiRNA is performed as shown in Figure 2.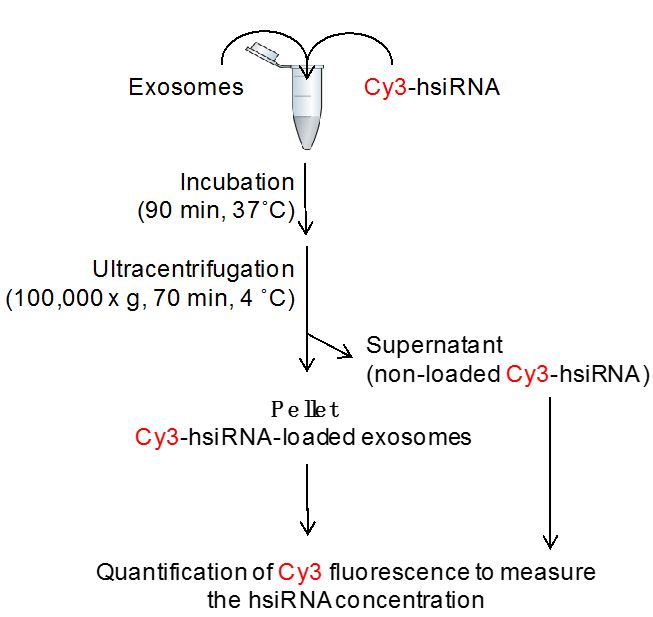
Figure 2. Flowchart of exosome loading procedure: co-incubation of Cy3-hsiRNA and extracellular vesicles results in Cy3-hsiRNA-loaded extracellular vesicles that pellet by ultracentrifugation - Into a 1.5 ml microcentrifuge tube, mix required concentration of hsiRNAs (3,000-5,000 molecules of hsiRNA per vesicle) and extracellular vesicles resuspended in 100 µl PBS by gently pipetting up and down with micropipette (see Notes 4 and 5).
Note: It is advised to determine the concentration of the extracellular vesicles first (see step B4). If the concentration of hsiRNA is above 100 µM, the volume of hsiRNA solution needed to reach the 3,000-5,000 hsiRNA per vesicle ratio will be negligible (less than 10 µl). - Incubate the mix in a thermo-shaker for at least 60 min at 37 °C and shake at 500 rpm. If the oligonucleotide is fluorescently labeled, protect the tube from light using aluminum foil.
- Place the 1.5 ml tubes in the tube holder in a pre-cooled TLA-110 rotor with the tube-cap oriented to find the pellet.
- Centrifuge for at least 70 min at 100,000 x g, 4 °C in a tabletop ultracentrifuge. Unloaded hsiRNAs remain in the supernatant and hsiRNA-loaded extracellular vesicles form a pellet.
- Remove and save the supernatant into a 1.5 ml centrifugation for further measurement. The quantification of hsiRNA remaining in the supernatant can be used to estimate the efficiency of loading (see steps B2 and B3).
- Resuspend the pellet in required volume of medium or artificial CSF for cell based assays, or for in vivo brain injection, respectively. Resuspended hsiRNA-loaded extracellular vesicles are immediately used for further experiment without being stored. Alternatively extracellular vesicles can be stored at -80 °C in 0.1 M sucrose. We observed that storage did not affect the number or activity of vesicles for up to 6 months.
- Save an aliquot of hsiRNA-loaded extracellular vesicles for quantification of hsiRNAs and extracellular vesicles concentration.
- Determination of hsiRNA concentration by direct quantification of fluorescently labeled hsiRNAs.
Two methods are used to estimate the loading efficiency of hsiRNAs in extracellular vesicles (Figure 1) (see Notes 6 and 7). First, directly measuring the Cy3 fluorescence in the hsiRNA-extracellular vesicles pellet. We observed that extracellular vesicles pellets may display autofluorescent signal, thus may confound measurement of Cy3 fluorescence coming from loaded hsiRNA. Therefore we recommend a second strategy: calculating the difference between the total amount of Cy3-hsiRNAs originally added to the sample and the amount of Cy3-hsiRNAs remaining in the supernatant (unloaded) after incubation with extracellular vesicles and ultracentrifugation. To determine an accurate concentration of hsiRNAs, prepare negative controls (PBS only with no hsiRNA) and a hsiRNAs calibration curve into the well of a UV-transparent, flat-bottom 96-well plate. The minimal volume of sample usually used in 96-well plate is 50 µl. For the calibration curve make a 1:2 dilution series of hsiRNA, spanning amounts comparable to what was loaded into the extracellular vesicles. - For the negative control, dispense 50 µl of PBS (blank) in the first well of the plate (well #A1).
- In the following well (well #A2), prepare a Cy3 fluorescent positive control stock solution. Dilute a known concentration of Cy3 labeled hsiRNAs in 100 µl PBS. Use twice the amount of hsiRNA than what was used in the hsiRNA-extracellular vesicles sample. Dilute 1:2 in PBS to get an 8-points calibration curve.
- Dispense 50 µl of PBS into three following wells (wells #A3, A4 and A5). To make a series dilution, transfer 50 µl of the stock solution (well #A2) into the next well (well #A3) containing 50 µl of PBS and mix gently with a micropipette.
- Using a fresh micropipette tip, transfer 50 µl (from well #A3) into the next well (well #A4) and proceed until the last well of the calibration curve.
- Transfer 50 µl of supernatant (containing unloaded hsiRNAs) and pellet (containing hsiRNA-extracellular vesicles) of each sample into the plate.
- Measure the Cy3 fluorescence of each sample using a spectrophotometer plate-reader at 547 nm excitation and 570 nm emission wavelengths.
- Using the calibration curve, calculate the Cy3 fluorescence in both the pellet and supernatant samples.
- The Cy3 fluorescence of the pellet sample gives a direct estimate of hsiRNAs concentration in the hsiRNA-extracellular vesicles sample.
- As a second method to estimate the concentration of hsiRNAs in the pellet, calculate the difference between the total amount Cy3-hsiRNAs originally added to the sample and the amount of Cy3 fluorescence remaining in the supernatant.
- Determination of hsiRNA concentration by peptide-nucleic acid hybridization assay
To confirm the concentration of hsiRNAs obtained by measuring the Cy3 fluorescence using a spectrophotometer, we determine the hsiRNAs guide strand concentration (not Cy3-labeled) in samples using a peptide-nucleic acid (PNA) hybridization assay (developed by Axolabs, Kulmbach, Germany) (Roehl et al., 2011). PNAs are single strand analogues of DNA with the sugar-phosphate backbone replaced by a polyamide backbone. Therefore, free of any charge, PNAs bind to their complementary strand with a very high affinity, forming double-stranded DNA-RNA structures. PNA assay is a more accurate but also more laborious measurement of hsiRNA concentration in the pellet than spectrophotometric fluorescence measurement. Furthermore, the samples used for PNA assay is lysed, and cannot be used in downstream experiments.- Dilute hsiRNA-extracellular vesicles sample in 100 µl of RIPA buffer containing 2 mg/ml Proteinase K in a 1.5 ml microcentrifuge tube.
- Sonicate for 15 min to lyse the vesicles at room temperature.
- To prepare a calibration curve, add a known amount of hsiRNA duplex into lysed extracellular vesicles sample.
- Add 20 µl of 3 M KCl and mix gently with a micropipette to precipitate SDS (component of RIPA buffer).
- Centrifuge for 15 min at 5,000 x g at room temperature to pellet insoluable membrane matieral and precipitated SDS.
- Transfer the cleared supernatant containing the hsiRNAs guide strands in a new 1.5 ml microcentrifuge tube.
- Dilute the supernatant to obtain a concentration below 5 pmol hsiRNA/100 µl sample.
- Add 5 pmol (or an amount exceeding the amount of hsiRNA loaded into extracellular vesicles) of Cy3-labeled PNA strands fully complementary to guide strand of hsiRNA and mix gently with the micropipette.
- To anneal the PNA strands to the hsiRNA guide strands, incubate for 15 min at 95 °C (to melt hsiRNA duplexes) followed by an incubation at 50 °C (to anneal PNA to hsiRNA guide strands, replacing sense strands) for 15 min and cool to room temperature.
- Transfer the samples in a 96-well plate or in vials compatible with your HPLC system.
- Inject the samples containing PNA-hsiRNA guide strand hybrids into HPLC anion exchange column, automatically using an autosampler or manually using a Hamilton syringe. The buffer A and buffer B used for the mobile phase are described in the Recipes section. Optimize the speed of gradient between 10% buffer B to 100% buffer B according to your HPLC system and anion exchange column. An example of protocol is described in Table 1. The elution time will be determined by the negative charge of the hsiRNA guide strands and not influenced by the neutral PNA.
- Read the Cy3 fluorescence at 550 nm excitation and 570 nm emission wavelengths. The signal is coming from the Cy3 label on the PNA. Integrate the peak corresponding to Cy3-PNA-hsiRNA guide strand hybrid.
- Using the calibration curve, calculate the hsiRNA guide strand concentration.
Table 1. Table describing the HPLC standard operating procedure for Cy3-PNA-hsiRNA hybrid concentration estimation
- Dilute hsiRNA-extracellular vesicles sample in 100 µl of RIPA buffer containing 2 mg/ml Proteinase K in a 1.5 ml microcentrifuge tube.
- Characterization of extracellular vesicles size distribution and concentration
For each sample, the concentration and size of vesicles are determined by recording and analyzing the Brownian motion of particles in PBS and using a NanoSight NS300 system and a Nanoparticles Tracking Analysis (NTA) software (see Note 8).- Dilute 1 to 5 µl of sample in 1 ml PBS (dilution ratio 1:200 to 1:1,000) at room temperature in a 1.5 ml microtube and mix gently using 1 ml micropipette.
- Inject the diluted sample in the NanoSight NS300 system using a 1 ml syringe. Use a new, clean syringe for each sample. Before injecting a new sample, flush out the previous sample to empty the tubing from vesicles.
- Adjust camera shutter and gain manually. To accurately compare the concentration of different samples, use similar camera shutter and gain values.
- Monitor the sample in duplicate for 30 to 60 sec.
- Process the recorded videos with NTA software to determine the particle size and concentration.
- Calculate the number of hsiRNAs loaded per extracellular vesicle by dividing the total amount of hsiRNAs in the sample by the number of extracellular vesicles detected by NTA, assuming normal distribution.
- Dilute 1 to 5 µl of sample in 1 ml PBS (dilution ratio 1:200 to 1:1,000) at room temperature in a 1.5 ml microtube and mix gently using 1 ml micropipette.
- Characterization of extracellular vesicles surface charge
The hsiRNA-EV surface charges are determined by Laser Doppler Micro-electrophoresis (DLM) using a Zetasizer Nano. The particles charge is obtained by applying an electric field on the sample and measuring the velocity of the particles present in the sample. The surface charge of the particles is directly correlated to their velocity.- Dilute 1 to 5 µl of sample in 1 ml distilled H2O (dilution ratio 1:200 to 1:1,000) at room temperature in a 1.5 ml microtube and mix gently using 1 ml micropipette. We noticed that diluting the sample in PBS provoked the burn of the solution that induces false measurements.
- Transfer the diluted sample in a universal glass micro-electrophoresis cuvette. A minimum volume of 700 µl of sample is required to completely immerse the electrodes. However do not overfill the cuvette as this will produce a thermal gradient within the sample and reduce the accuracy of the measurement.
- Slowly insert the Dip cell avoiding the formation of any bubble between the sample and the electrodes. For more detail, follow the procedure recommended by the manufacturer (Zetasizer Nano accessories guide). Clean the Dip cell electrodes thoroughly with distilled water between each measurement.
- Create a standard operating procedure following the manufacturer’s instructions (Zetasizer Nano User Manual).
- Monitor the sample charges in duplicate.
- The instrument gives an absolute value of the particles charge. We observed that hsiRNA-loaded extracellular vesicles have more negative charges than non-loaded extracellular vesicles.
- Dilute 1 to 5 µl of sample in 1 ml distilled H2O (dilution ratio 1:200 to 1:1,000) at room temperature in a 1.5 ml microtube and mix gently using 1 ml micropipette. We noticed that diluting the sample in PBS provoked the burn of the solution that induces false measurements.
- Analysis of extracellular vesicles and hsiRNA-loaded extracellular vesicles integrity by electron microscopy.
To detect hsiRNAs bound to extracellular vesicles by electron microscopy, we use biotinylated hsiRNA and streptavidin immune-gold particles. Samples and grids used for electron microscopy are prepared at room temperature unless otherwise specified.- To fix the sample, add an equal volume of 4% paraformaldehyde to the hsiRNA-loaded extracellular vesicle sample and incubate for 2 h. For example, add 50 µl of 4% paraformaldehyde to 50 µl of hsiRNA-extracellular vesicle sample.
- Drop 3 µl of fixed hsiRNA-EV aliquots onto EM grids
- Incubate for 20 min in a dry environment to let the membrane adsorb.
- To wash the grid, drop 100 µl of PBS on a sheet of Parafilm and transfer the grids upside down to the drop of PBS.
Note: Subsequent steps are performed similarly, transferring the grid upside down from drop to drop of reagents on a sheet of Parafilm. - Transfer the grid to a 100 µl drop of 50 mM glycine/PBS and incubate for 5 min.
- Transfer the grid to a 100 µl drop of 5% BSA/PBS for 10 min to block the sample.
Note: The addition of 0.1% saponin to the reagent will permeabilize the extracellular vesicles to enable the detection of any intraluminal hsiRNA molecule. - Wash the grid twice in 100 µl PBS drop.
- Incubate the grid with 50 µl drop of 6-nm or 10-nm streptavidin immune-gold particles diluted 1:10 to 1:20 in 0.5% BSA/PBS for 1 h in presence or absence of 0.1% saponin.
- Wash the grid three times in 100 µl PBS drop.
- Incubate the grid in 1% glutaraldehyde for 5 min.
- Wash the grid eight times in water for 2 min each wash.
- Transfer the grid in uranyl oxalate pH 7 for 5 min to contrast the sample.
- Contrast and embed the samples in a 9:1 solution of respectively 1% methyl cellulose and 4% uranyl acetate for 10 min on ice.
- Remove the excess liquid with a Whatman No. 1 filter paper and let the grids air-dry for 5 to 10 min.
- Observe the grid in a transmission electron microscope at 60-80 kV.
- Store the grid in grid storage boxes at room temperature. Grids can be stored indefinitely.
- To fix the sample, add an equal volume of 4% paraformaldehyde to the hsiRNA-loaded extracellular vesicle sample and incubate for 2 h. For example, add 50 µl of 4% paraformaldehyde to 50 µl of hsiRNA-extracellular vesicle sample.
- Loading of extracellular vesicles with hsiRNAs.
- Treatment of mouse central nervous system with hsiRNA-loaded extracellular vesicles
Note: All animal procedures are performed according to your facility Institutional Animal Care and Use Committee regulations (IACUC). For the validation of hsiRNATarget-extracellular vesicles dependent silencing of target mRNA in vivo, control groups include mice treated with artificial CSF, extracellular vesicles alone, hsiRNANTC-extracellular vesicles and hsiRNATarget alone. (NTC: non-targeting control) (see Note 9).- Prior to any treatment in the brain, animals are deeply anesthetized by intraperitoneal injection of 1.2% Avertin.
- Place the anesthetized mouse in the stereotactic frame (see Note 10).
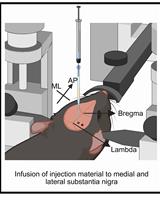
- For direct injection, mice receive microinjections by stereotactic placement into the right striata. The coordinates relative to bregma are 1.0 mm anterior, 2.0 mm lateral, and 3.0 mm ventral.
- For brain infusion, we use ALZET® osmotic pumps that infuse the sample for 3 days (flow rate of 1 µl/h) or for 1 week (flow rate of 0.5 µl/h). Using a 30 G ½ needle attached to a 1 ml syringe, prefill the ALZET® pumps with the required volume of sample: 100 µl for both 3 days and 1 week pumps, following the manufacturer’s instructions. Incubate the pump overnight at 37 °C in a water bath. The next day, perform the mouse surgery to implant the ALZET® pumps in mouse brain.
- Upon 3 days or 7 days of treatment, mice are euthanized by intraperitoneal injection of 1 ml Avertin.
- Lay the mouse on its back and clean the abdominal surface with 70% ethanol to sterilize the area.
- Using microscissors, make a vertical incision in the abdominal muscles to open the abdominal cavity of the mouse.
- In the mouse heart, still beating, place a needle connected to the peristaltic pump to infuse room temperature PBS for 5 min to remove the blood from the brain.
- Using microscissors, carefully cut the skull of the mouse in half from the neck to the forehead. Carefully remove the two sides of the skull.
- Using forceps beneath the brain, carefully lift up and remove the brain from the skull.
- Process the brains accordingly to further experiments.
- mRNAs are unstable and degraded rapidly after the mouse anesthesia. Thus, for an accurate mRNA quantification and evaluation of in vivo silencing efficiency, the brains are processed immediately after harvest (Alterman et al., 2015; Coles et al., 2016; Didiot et al., 2016). Brains are cut in 300 µM coronal sections in PBS at -1 to 0.5 °C using a vibratome according to the manufacturer’s instructions. The temperature of the PBS should be stable around 0 ± 0.2 °C). If the temperature of the PBS increases, the brains soften and become more difficult to cut.
- Depending on the size of the brain region treated, collect a minimum of 3 biopsies, representing technical replicates, per animal. The 4th section from the front of the brain (olfactory bulbs) corresponds to the beginning of the striatum, which is also visually confirmed. The next 3 consecutive coronal sections (5th, 6th and 7th) are selected to collect the 2 mm biopsies from the striatal region easily identified in the mouse brain. The position of the biopsy are strictly fixed and the samples are always collected from the same region of the brain for all animals.
- Brain samples are processed for mRNA quantification following the method from Alterman et al., 2015; Coles et al., 2016; Didiot et al., 2016.
- The target and control mRNAs are quantified using the QuantiGene® 2.0 assay as recommended by the manufacturer protocol (Affymetrix). The amount of target mRNA is always normalized to the amount of the control housekeeping mRNA (Coles et al., 2016) (see Note 11).
- Prior to any treatment in the brain, animals are deeply anesthetized by intraperitoneal injection of 1.2% Avertin.
Data analysis
Data are processed using Microsoft Office Excel and analyzed using GraphPad Prism 6 software.
The levels of targeted and housekeeping genes in the biopsies are independently measured using the QuantiGene® bDNA assay. The average of the 3 technical replicates represents the mRNA expression value per animal (see Notes 12 and 13).
- For each plate and each individual probe set, average the values obtained in the DLM (blank) wells and subtract this average from all sample wells.
- Divide the target gene luminescence value by the housekeeping gene luminescence value to normalize the target gene expression.
- Average the normalized untreated cell values.
- Divide each individual normalized well by the average of the untreated wells and multiply by 100 to calculate the percent of target gene expression relative to untreated cells.
- If compounds are tested in replicate, average the replicates and calculate the standard deviation.
- For dose response analysis, graph concentration-dependent IC50 curves using a non-lineal regression curve fit, log(inhibitor) vs. response–variable slope (four parameters). If necessary, set the lower limit of the curve at zero, and the upper limit of the curve at 100.
- The statistical analysis for the in vivo silencing experiments is performed using standard package in GraphPad Prism software. Compare efficacy of target sequences to the non-targeting control (NTC) to determine statistical significance. Differences in all comparisons are considered significant at P-values less than 0.05 compared with the NTC group.
- When the injections or infusions are performed unilaterally, the analysis is performed comparing ipsilateral or contralateral values groups (artificial CSF, extracellular vesicles only, hsiRNAtarget only and hsiRNAtarget-extracellular vesicles) to the hsiRNANTC-extracellular vesicles group.
Notes
- After ultracentrifugation performed to make extracellular vesicle-depleted medium or purify extracellular vesicles or hsiRNA-loaded extracellular vesicles, it is important to remove the supernatant quickly to avoid the pellet to dissolve back in the supernatant.
- We find that it is best to prepare fresh extracellular vesicles for each functional in vitro and in vivo assay.
- Purified extracellular vesicles are kept at 4 °C for a maximum of 48 h prior any functional assay.
- Particular attention should be given to the hsiRNAs modification pattern. Functionally efficient hsiRNA, loaded or not in extracellular vesicles, requires extensive chemical stabilization. Chemical modification of the ribose with 2’-O-Methyl and 2’-Fluoro result in a significant increase of the hsiRNA-resistance to nuclease degradation, providing higher stability in vitro and in vivo (Byrne et al., 2013). Unmodified or partially modified hsiRNAs will undergo nucleases degradation affecting hsiRNA-extracellular vesicles functional efficacy. Nucleases can potentially be present in the extracellular vesicles sample purified from cell culture medium.
- We noticed that extracellular vesicles purified from different cell sources (e.g., glioblastoma-, mesenchymal stem cell- or umbilical-derived extracellular vesicles) show similar ability to be loaded with hsiRNAs.
- The loading of extracellular vesicles with hsiRNA is saturatable with maximum of ~3,000-5,000 hsiRNA loaded per individual vesicle. However, incubation of extracellular vesicles with higher concentration of hsiRNAs slightly increase the number of hsiRNAs per extracellular vesicles. With increase in the hsiRNA/vesicle loading ratio, there is a slight increase in surface negative charge indicative that of the hsiRNAs association with the vesicle membrane. Thus, it is possible that too much loading might interfere with native extracellular vesicles cellular-uptake mechanism.
- The co-incubation of cholesterol-conjugated hsiRNAs with extracellular vesicles provides a robust, efficient and highly reproducible method for loading extracellular vesicles with chemically synthesized oligonucleotides. We observed that the cholesterol is essential for loading as non-cholesterol conjugated hsiRNAs did not associate with extracellular vesicles (Coles et al., 2016). We used the same procedure to load extracellular vesicles with siRNAs conjugated to various hydrophobic moiety (e.g., DHA). We observed that the more hydrophobic is the conjugate, the more efficient is the loading (data not published).
- The measurement of protein concentration of purified extracellular vesicles by Bradford assay represents an additional reproducible approach to quantify extracellular vesicles.
- The hsiRNAHTT, targeting the Huntingtin gene, and hsiRNANTC (NTC: non-targeting control) used to develop this method were designed and synthesized in our laboratory (Alterman et al., 2015). hsiRNAHTT silencing efficiency was confirmed without or with loading into extracellular vesicles in mouse brain (Coles et al., 2016).
- Bollus injection and pumps loaded with hsiRNA-extracellular vesicles samples are performed by stereotaxic surgery. Stereotaxic surgery allows an optimized delivery of oligonucleotide therapeutics in the mouse brain with excellent spatiotemporal control; essentially any brain region of choice can be targeted to silence any gene at any postnatal developmental stage up to adulthood. This clean and highly reproducible procedure, makes it an attractive approach for delivery of a therapeutic agent in the brain. The entire procedure can be completed in 2 h for 2 mice.
- hsiRNA-loaded extracellular vesicles are stable and loaded hsiRNAs are resistant to nucleases. However, only a fraction of the loaded hsiRNA is internalized into the vesicle lumen, and another fraction associates to the outer membrane. At this point, we cannot define which of the extracellular vesicles-associated oligonucleotides (surface bound or internalized) are functionally active. Further studies are essential to determine the optimal loading, long-term stability, and in vivo behavior of the hsiRNA-loaded extracellular vesicles.
- Based on preliminary data, the level of target mRNA expression variation from animal to animal has ~15% error, which theoretically requires n = 8 animals to be able to detect 20-30% gene modulation with 80% confidence. For in vivo validation of hsiRNA-extracellular vesicles sample efficacy using pump-based infusion, the mice groups are powered with n = 8 to n = 10 mice.
- The pump implantation requires extensive surgery that can result in uncontrolled animal death during the procedure and account for the difference in number of animals for different groups.
Recipes
Note: For all recipes and at any step of the procedure use Milli-Q-purified water.
- Cell culture medium
Note: Recommended culture medium supplemented with FBS and antibiotics.
- 1 M sucrose
Dissolve 34.23 g of sucrose in 100 ml of distilled H2O
Filter sterilize with 0.22 µm syringe filter and store at 4 °C - 1 M Tris-HCl
- Dissolve 121 g of Tris base (TRIZMA) in 700 ml of distilled H2O
- Adjust the pH to 8.5 with HCl
- Fill up to volume 1 L with distilled H2O
- Sterilize by autoclaving and store at room temperature
- 0.5 M EDTA
- Add 186.1 g of EDTA in 800 ml of distilled H2O
- Adjust the pH to 8 with NaOH (~50 ml of NaOH)
- Bring the volume to 1 L with distilled H2O
- Mix on a magnetic stirrer
- Sterilize by autoclaving and store at room temperature
- 3 M KCl
Dissolve 22.368 g of KCl in 100 ml of distilled H2O and mix thoroughly - 10% SDS
Dissolve 10 g of SDS in 80 ml of distilled H2O. Adjust the volume to 100 ml with distilled water - 0.1 M sodium phosphate buffer
Dissolve 3.1 g of NaH2PO4·H2O and 10.9 g of Na2HPO4 (anhydrous) to distilled H2O to make a volume of 1 L. This buffer can be stored for up to 1 month at 4 °C - HPLC buffer A
50 v/v % acetonitrile, 50 v/v % water, containing 25 mM Tris-HCl (pH 8.5) and 1 mM EDTA in water - HPLC buffer B
Buffer A containing 800 mM NaClO4 - Glutaraldehyde, 1% (v/v)
Dilute EM-grade glutaraldehyde fixatives (commercially available as 8%, 25% or 70% aqueous solutions; Sigma-Aldrich) in 0.1 M sodium phosphate buffer, pH 7.4, to the appropriate dilution. Store up to 6 months at -20 °C or up to 1 week at 4 °C after thawing
Note: The buffers used to prepare fixatives need to have a good buffering capacity to maintain a pH of about 7.4 during fixation. - Methyl cellulose, 2% (w/v)
- Heat 196 ml of distilled water to 90 °C and add 4 g methyl cellulose with stirring
- Rapidly cool on ice with stirring, until the solution has reached 10 °C. Continue slow stirring overnight at 4 °C
- Stop stirring and allow the solution ‘ripen’ for 3 days at 4 °C. Bring to a final volume of 200 ml with water
- Centrifuge using polycarbonate centrifuge bottles with cap assemblies 95 min at 100,000 x g, 4 °C
- Collect the supernatant and store up to 3 months at 4 °C
- Paraformaldehyde (PFA), 2% and 4% (w/v)
- Dissolve 4 g of PFA powder in 90 ml of 0.1 M sodium phosphate buffer and heat to 65 °C while stirring. If needed, add drops of 1 N NaOH until the solution becomes clear
- Bring to 100 ml with 0.1 M sodium phosphate buffer. Cool and filter. Aliquot and store at -20 °C. Use thawed aliquots immediately and do not refreeze
- To make 2% paraformaldehyde, dilute 4% paraformaldehyde with 0.1 M sodium phosphate buffer
- Uranyl acetate (4% w/v), pH 4
- Weigh 2 g of uranyl acetate and dissolve in 50 ml distilled water. Store up to 4 months at 4 °C protected from light
- Just before use, filter the amount needed of uranyl solution with a 0.22-μm syringe filter
CAUTION: Consult with the institutional Radiation Safety Office for guidelines concerning proper handling and disposal of radioactive material. The uranyl acetate crystals are difficult to dissolve and it may be necessary to use a rotating wheel for mixing.
- Uranyl-oxalate, pH 7
- Mix 4% uranyl acetate, pH 4 with 0.15 M solution of oxalic acid (0.945 g in 50 ml distilled water), in a 1:1 ratio
- Adjust the pH to 7 by adding 25% (w/v) NH4OH in drops to prevent formation of insoluble precipitates
- Store in the dark up to 1 month at 4 °C
- Methyl cellulose-UA, pH 4
9 parts 2% methyl cellulose and 1 part 4% uranyl acetate mixed just before use
Acknowledgments
We thank the members of the Khvorova and Aronin Laboratories, NIH Extracellular RNA Consortium and CHDI Foundation Inc. for helpful discussions. This work is supported in part by grants from the NIH UH2-TR000888 and UH3-4UH3TR000888-03, NIH NS38194 and CHDI Foundation (Research Agreement A-6119). The authors declare no conflict of interest.
References
- Alterman, J. F., Hall, L. M., Coles, A. H., Hassler, M. R., Didiot, M. C., Chase, K., Abraham, J., Sottosanti, E., Johnson, E., Sapp, E., Osborn, M. F., Difiglia, M., Aronin, N. and Khvorova, A. (2015). Hydrophobically modified siRNAs silence huntingtin mRNA in primary neurons and mouse brain. Mol Ther Nucleic Acids 4: e266.
- Alvarez-Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S. and Wood, M. J. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 29(4): 341-345.
- Byrne, M., Tzekov, R., Wang, Y., Rodgers, A., Cardia, J., Ford, G., Holton, K., Pandarinathan, L., Lapierre, J., Stanney, W., Bulock, K., Shaw, S., Libertine, L., Fettes, K., Khvorova, A., Kaushal, S. and Pavco, P. (2013). Novel hydrophobically modified asymmetric RNAi compounds (sd-rxRNA) demonstrate robust efficacy in the eye. J Ocul Pharmacol Ther 29(10): 855-864.
- Coles, A. H., Osborn, M. F., Alterman, J. F., Turanov, A. A., Godinho, B. M., Kennington, L., Chase, K., Aronin, N. and Khvorova, A. (2016). A high-throughput method for direct detection of therapeutic oligonucleotide-induced gene silencing in vivo. Nucleic Acid Ther 26(2): 86-92.
- Didiot, M. C., Hall, L. M., Coles, A. H., Haraszti, R. A., Godinho, B. M., Chase, K., Sapp, E., Ly, S., Alterman, J. F., Hassler, M. R., Echeverria, D., Raj, L., Morrissey, D. V., DiFiglia, M., Aronin, N. and Khvorova, A. (2016). Exosome-mediated delivery of hydrophobically modified siRNA for huntingtin mRNA silencing. Mol Ther 24(10): 1836-1847.
- Distler, J. H., Huber, L. C., Hueber, A. J., Reich, C. F., 3rd, Gay, S., Distler, O. and Pisetsky, D. S. (2005). The release of microparticles by apoptotic cells and their effects on macrophages. Apoptosis 10(4): 731-741.
- El Andaloussi, S., Lakhal, S., Mager, I. and Wood, M. J. (2013). Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev 65(3): 391-397.
- Geary, R. S., Norris, D., Yu, R. and Bennett, C. F. (2015). Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev 87: 46-51.
- Khvorova, A. and Watts, J. K. (2017). Oligonucleotide therapeutics: from chemistry advances to clinical utility. Nat Biotechnol. In Press.
- Kooijmans, S. A., Stremersch, S., Braeckmans, K., de Smedt, S. C., Hendrix, A., Wood, M. J., Schiffelers, R. M., Raemdonck, K. and Vader, P. (2013). Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release 172(1): 229-238.
- Kooijmans, S. A., Vader, P., van Dommelen, S. M., van Solinge, W. W. and Schiffelers, R. M. (2012). Exosome mimetics: a novel class of drug delivery systems. Int J Nanomedicine 7: 1525-1541.
- Kosaka, N., Iguchi, H., Yoshioka, Y., Hagiwara, K., Takeshita, F. and Ochiya, T. (2012). Competitive interactions of cancer cells and normal cells via secretory microRNAs. J Biol Chem 287(2): 1397-1405.
- Lasser, C. (2012). Exosomal RNA as biomarkers and the therapeutic potential of exosome vectors. Expert Opin Biol Ther 12 Suppl 1: S189-197.
- Lee, Y., El Andaloussi, S. and Wood, M. J. (2012). Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet 21(R1): R125-134.
- Lotvall, J., Hill, A. F., Hochberg, F., Buzas, E. I., Di Vizio, D., Gardiner, C., Gho, Y. S., Kurochkin, I. V., Mathivanan, S., Quesenberry, P., Sahoo, S., Tahara, H., Wauben, M. H., Witwer, K. W. and Thery, C. (2014). Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 3: 26913.
- Ly, S., Navaroli, D. M., Didiot, M. C., Cardia, J., Pandarinathan, L., Alterman, J. F., Fogarty, K., Standley, C., Lifshitz, L. M., Bellve, K. D., Prot, M., Echeverria, D., Corvera, S. and Khvorova, A. (2017). Visualization of self-delivering hydrophobically modified siRNA cellular internalization. Nucleic Acids Res 45(1): 15-25.
- Marcus, M. E. and Leonard, J. N. (2013). FedExosomes: engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals (Basel) 6(5): 659-680.
- Mizrak, A., Bolukbasi, M. F., Ozdener, G. B., Brenner, G. J., Madlener, S., Erkan, E. P., Strobel, T., Breakefield, X. O. and Saydam, O. (2013). Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol Ther 21(1): 101-108.
- Muralidharan-Chari, V., Clancy, J. W., Sedgwick, A. and D'Souza-Schorey, C. (2010). Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 123(Pt 10): 1603-1611.
- Nazarenko, I., Rupp, A. K. and Altevogt, P. (2013). Exosomes as a potential tool for a specific delivery of functional molecules. Methods Mol Biol 1049: 495-511.
- Ohno, S. I., Takanashi, M., Sudo, K., Ueda, S., Ishikawa, A., Matsuyama, N., Fujita, K., Mizutani, T., Ohgi, T., Ochiya, T., Gotoh, N. and Kuroda, M. (2013). Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther 21(1): 185-191.
- Pan, Q., Ramakrishnaiah, V., Henry, S., Fouraschen, S., de Ruiter, P. E., Kwekkeboom, J., Tilanus, H. W., Janssen, H. L. and van der Laan, L. J. (2012). Hepatic cell-to-cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut 61(9): 1330-1339.
- Pegtel, D. M., Cosmopoulos, K., Thorley-Lawson, D. A., van Eijndhoven, M. A., Hopmans, E. S., Lindenberg, J. L., de Gruijl, T. D., Wurdinger, T. and Middeldorp, J. M. (2010). Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 107(14): 6328-6333.
- Roehl, I., Schuster, M. and Seiffert, S. (2011). Oligonucleotide detection method. Application.
- Tetta, C., Ghigo, E., Silengo, L., Deregibus, M. C. and Camussi, G. (2013). Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine 44(1): 11-19.
- Thery, C., Amigorena, S., Raposo, G. and Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3: Unit 3 22.
- Valadi, H., Ekstrom, K., Bossios, A., Sjostrand, M., Lee, J. J. and Lotvall, J. O. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9(6): 654-659.
- Wang, K., Zhang, S., Weber, J., Baxter, D. and Galas, D. J. (2010). Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res 38(20): 7248-7259.
- Zomer, A., Vendrig, T., Hopmans, E. S., van Eijndhoven, M., Middeldorp, J. M. and Pegtel, D. M. (2010). Exosomes: Fit to deliver small RNA. Commun Integr Biol 3(5): 447-450.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Haraszti, R. A., Coles, A., Aronin, N., Khvorova, A. and Didiot, M. (2017). Loading of Extracellular Vesicles with Chemically Stabilized Hydrophobic siRNAs for the Treatment of Disease in the Central Nervous System. Bio-protocol 7(12): e2338. DOI: 10.21769/BioProtoc.2338.
Category
Microbiology > Microbial biochemistry > Lipid
Neuroscience > Nervous system disorders > Animal model
Biochemistry > Lipid > Extracellular lipids
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.