- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR-PCS Protocol for Chromosome Splitting and Splitting Event Detection in Saccharomyces cerevisiae


Published: Vol 7, Iss 10, May 20, 2017 DOI: 10.21769/BioProtoc.2306 Views: 10967
Reviewed by: Renate WeizbauerJudd F HultquistAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Chromosome engineering is an important technology with applications in basic biology and biotechnology. Chromosome splitting technology called PCS (PCR-mediated Chromosome Splitting) has already been developed as a fundamental chromosome engineering technology in the budding yeast. However, the splitting efficiency of PCS technology is not high enough to achieve multiple splitting at a time. This protocol describes a procedure for achieving simultaneous and multiple chromosome splits in the budding yeast Saccharomyces cerevisiae by a new technology called CRISPR-PCS. At least four independent sites in the genome can be split by one transformation. Total time and labor for obtaining a multiple split yeast strain is drastically reduced when compared with conventional PCS technology.
Keywords: Saccharomyces cerevisiaeBackground
Chromosome engineering technologies that enable rapid and efficient manipulation of multiple genetic loci or chromosomal regions have become increasingly important. Such technologies offer a powerful means for elucidating chromosome and genome function. Additionally, it can be used for breeding useful strains through the creation of a wide array of genetic variants. A chromosome splitting technology called PCS (PCR-mediated Chromosome Splitting) technology has been developed in the budding yeast Saccharomyces cerevisiae. This technology allows the splitting of yeast chromosomes at any desired site by introducing centromeres and telomere seed sequences based on the homologous recombination mechanism. The resulting chromosomes possess one centromere and telomeres at both ends, thus function as normal chromosomes (Sugiyama et al., 2005). However, low splitting efficiency is a drawback in PCS, therefore simultaneous and multiple splitting of chromosomes has been impossible. In this situation, we developed a novel chromosome splitting technology called CRISPR-PCS. It is well known that double strand break (DSB) markedly increases homologous recombination activity around the DSB site in yeast (Agmon et al., 2009). The CRISPR/Cas9 system is a genome editing technology that can induce targeted DSBs. By utilizing CRISPR/Cas9 system, we can induce DSB at any genomic locus and thus activate homologous recombination activity. CRISPR-PCS is a technology that combines CRISPR/Cas9 system with PCS, thus allowing the increase of splitting efficiency by approximately 200 fold. This drastically increased efficiency enables simultaneous and multiple chromosome spitting. Overview of the CRISPR-PCS technology is illustrated in Figure 1.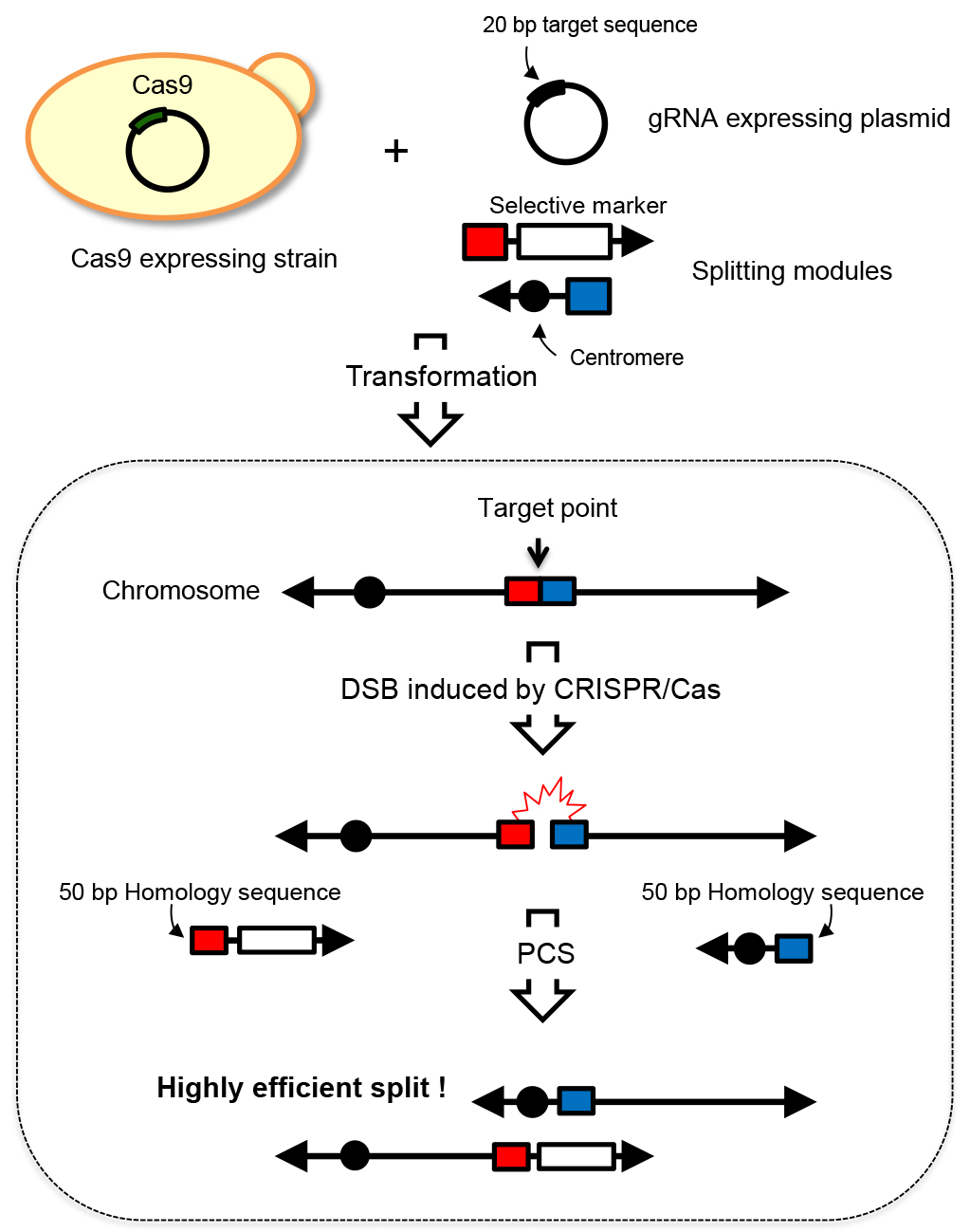
Figure 1. Overview of CRISPR-PCS. In CRISPR-PCS, one gRNA expressing plasmid for the specific targeting site and two splitting modules are required to split yeast chromosome at a specific site. These DNA molecules are introduced into the Cas9 expressing strain, i.e., the strain carrying p414-TEF1p-Cas9-CYC1t plasmid. Transformants where the expected split event occurred are selected by auxotrophic marker selection. Closed black circles represent the centromere. Red and blue boxes represent the homology sequences for recombination. Arrows represent the telomere sequence.
Materials and Reagents
- 10-100 μl pipette tips (e.g., Greiner Bio One International, catalog number: 685280 )
- 100-1,000 μl pipette tips (e.g., Greiner Bio One International, catalog number: 686290 )
- PCR tube (e.g., Greiner Bio One International, catalog number: 683201 )
- p426-SNR52p-gRNA.CAN1.Y-SUP4t (Addgene, catalog number: 43803 )
- p414-TEF1p-Cas9-CYC1t (Addgene, catalog number: 43802 )
- Escherichia coli DH5α competent cell (NIPPON GENE, catalog number: 316-06233 )
- DNA, MB-grade from fish sperm (Roche Diagnostics, catalog number: 11467140001 )
- KOD plus neo (TOYOBO, catalog number: KOD-401 )
- 2 mM dNTP solution (Attached in the KOD plus neo)
- 25 mM magnesium sulfate (MgSO4) (Attached in the KOD plus neo)
- Oligonucleotide primer 1 for construction of a gRNA expressing plasmid (5’-N20GTTTTAGAGCTAGAAATAGCAAG-3’) (synthesized in Sigma-Aldrich Japan)
- Oligonucleotide primer 2 for construction of gRNA expressing plasmid (5’-cN20GATCATTTATCTTTCACTGCGGA-3’) (synthesized in Sigma-Aldrich Japan)
- DpnI (Takara Bio, catalog number: 1235A )
- QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28704 )
- 10x NEB buffer 2 (New England Biolabs, catalog number: B7002S )
- BSA solution (attached in the T4 DNA polymerase) (New England Biolabs, catalog number: M0203 )
- 10x T4 DNA ligase buffer (New England Biolabs, catalog number: M0202 )
- T4 DNA polymerase (New England Biolabs, catalog number: M0203 )
- 2.5 mM each dNTP mix (Takara Bio, catalog number: 4030 )
- Ampicillin (Wako Pure Chemical Industries, catalog number: 015-10382 )
- Oligonucleotide primer 1 for construction of a splitting module (5’-N50GGCCGCCAGCTGAAGCTTCG-3’) (synthesized in Sigma-Aldrich Japan)
- Oligonucleotide primer 2 for construction of a splitting module (5’-CCCCAACCCCAACCCCAACCCCAACCCCAACCCCAAAGGCCACTAGTGGATCTGAT-3’) (synthesized in Sigma-Aldrich Japan)
- Oligonucleotide primer for sequencing (5’-ACGCCAAGCGCGCAATTAAC-3’) (synthesized in Sigma-Aldrich Japan)
- Lithium acetate dihydrate (Wako Pure Chemical Industries, catalog number: 120-01535 )
- Polyethylene glycol 4,000 (Wako Pure Chemical Industries, catalog number: 162-09115 )
- ECL Direct Nucleic Acid Labelling and Detection System (GE Healthcare, catalog number: RPN3000 )
- Ethidium bromide solution (10 mg/ml) (Nacalai Tesque, catalog number: 14631-94 )
- LB broth (Sigma-Aldrich, catalog number: L3022-1KG )
- Agar (Wako Pure Chemical Industries, catalog number: 010-08725 )
- Glucose (Wako Pure Chemical Industries, catalog number: 043-31163 )
- Yeast nitrogen base without amino acids (e.g., BD, Difco, catalog number: 291940 )
- Sodium hydroxide (NaOH) (e.g., Wako Pure Chemical Industries, catalog number: 192-15985 )
- Peptone (e.g., BD, BactoTM, catalog number: 211677 )
- Yeast extract (e.g., BD, BactoTM, catalog number: 288620 )
- Hydrochloric acid (HCl) (e.g., Wako Pure Chemical Industries, catalog number: 087-10361 )
- LB plate (see Recipes)
- Yeast minimum medium (SD medium) (see Recipes)
- YPD medium (see Recipes)
Equipment
- Pipettes
- Thermal cycler (e.g., Takara Bio, model: Dice® Touch, catalog number: TP350 ) (Use at Procedure B and Procedure C)
- Incubator (e.g., Panasonic Healthcare, model: MIR-H163 ) (Use at Procedure B and Procedure D)
- Air shaker (e.g., TAITEC, model: BR-21UM MR ) (Use at Procedure B and Procedure D)
- Heat block (e.g., TAITEC, model: DTU-1BN ) (Use at Procedure B, Procedure D and Procedure E)
- DNA sequencer (e.g., Applied Biosystems, model: ABI PRISM® 3100 Genetic Analyzer ) (Use at Procedure B)
- CHEF-DR® III pulsed field gel electrophoresis system (Bio-Rad Laboratories, model: CHEF-DR III Chiller System , catalog number: 1703700)
Procedure
- Designing of gRNA target site
Once you decide a genomic locus to be split, search PAM sequence for Cas9 cleavage (5’-NGG-3’ or 5’-CCN-3’ for opposite strand) in the vicinity of the target locus. Decide the most appropriate 20 bp guide RNA (gRNA) target sequence so that the gRNA target sequence is not present in the DNA splitting module (see Procedure C) and the gRNA target sequence is not predicted to induce any off-target cleavages. CRISPRdirect (https://crispr.dbcls.jp/) may be helpful for designing target sequences. Paste your target sequence into the box and choose S. cerevisiae S288C genome for the specificity check. Then you can easily know the most appropriate guide RNA target sequence. It is known that the cleavage point by the CRISRP/Cas9 system is 3 bp upstream of PAM sequence (Jinek et al., 2012). It is desirable that the point to be split is designed at the same point cleaved by CRISPR/Cas9. However, this is not absolute requirement unless splitting modules don’t contain gRNA target sites. - Construction of a plasmid expressing gRNA with designed 20 bp target sequence
We used p426-SNR52p-gRNA.CAN1.Y-SUP4t as a template plasmid to construct a desired gRNA expressing plasmid by PCR. You can use any plasmids to express your desired gRNA. The following protocol is a version when p426-SNR52p-gRNA.CAN1.Y-SUP4t is used as a template plasmid. The principle for plasmid construction is based on SLIC technology (Li and Elledge, 2012).- Perform PCR reaction

- After PCR reaction, add 1 μl of DpnI directly to the PCR tube to destroy methylated template plasmid. Incubate at 37 °C for 1 h.
- Perform gel electrophoresis and purify the PCR product by QIAquick Gel Extraction Kit.
- Purified fragment is dissolved into 50 μl of elution buffer.
- T4 DNA polymerase reaction to generate 5’ overhangs by 3’ exonuclease activity.

Incubate at 22 °C for 30 min. Then add 2 μl of dNTP mix (2.5 mM each) to terminate the reaction. Keep on ice. - Annealing reaction
To 9 μl solution from step B5, add 1 μl of 10x T4 ligase buffer (NEB). Incubate at 37 °C for 30 min. - Transformation of Escherichia coli
Transform E. coli DH5α strain by introducing 2 μl of solution from step B6. After incubating at 37 °C for 1 h, spread onto LB plate containing ampicillin. - Confirmation of plasmid construction by sequencing analysis
Note: Although most of the E. coli transformants harbor the desired gRNA plasmid, it is better to check by sequencing analysis using the primer 5’-ACGCCAAGCGCGCAATTAAC-3’.
- Perform PCR reaction
- Construction of a splitting module with 50 bp homology sequence
- After deciding a point to be split at Procedure A, design and purchase oligonucleotide primers to amplify any genetic marker gene flanked with 50 bp homology sequence corresponding to the upstream and downstream of the target splitting point at both primers (see Figure 2). For PCR templates we used a pUG6 based plasmid set (Guldener et al., 1996) for PCR amplification but with different genetic marker genes (Candida glabrata LEU2 gene, Candida glabrata HIS3 gene, URA3, and KanMX) or centromere (CEN4) (Sugiyama et al., 2005). Select appropriate plasmid as a template for PCR so that newly generated chromosomes contain one centromere. These plasmids are available at NBRP (http://yeast.lab.nig.ac.jp/nig.v2.1/index.html).
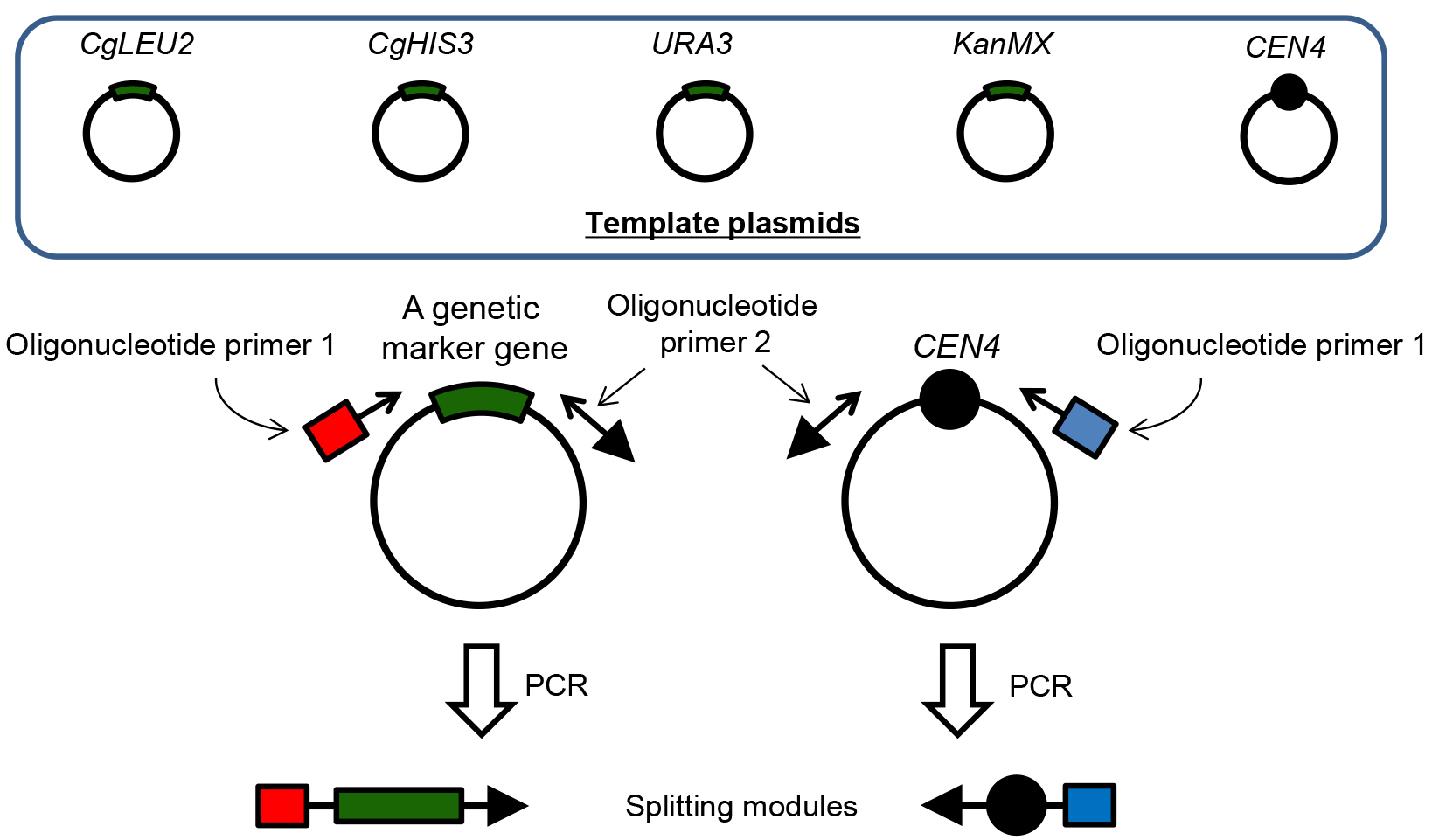
Figure 2. Detailed illustration for construction of splitting modules. There are five template plasmids to construct splitting modules. CEN4 is a centromere sequence of the chromosome IV in S. cerevisiae. All sequences of the plasmids are identical except for those of genetic marker gene, because all template plasmids have the same plasmid backbone (pUG6 plasmid). Therefore, primer sequences required for annealing to the template plasmid is common when you use any plasmids as a template. In oligonucleotide primer 1 and 2, in addition to the sequence for annealing, 50 bp sequence that is identical to the upstream and downstream sequence of the target splitting point in the chromosome should be added, respectively. Closed black circles represent the centromere. Red and blue boxes represent a 50 bp sequence upstream and downstream from the target splitting point in the chromosome, respectively. Arrows represent the telomere sequence. - PCR reaction

After PCR reaction, purify the PCR product by gel extraction by QIAquick Gel Extraction Kit.
Dissolve the purified splitting module DNA into 50 μl of elution buffer.
- After deciding a point to be split at Procedure A, design and purchase oligonucleotide primers to amplify any genetic marker gene flanked with 50 bp homology sequence corresponding to the upstream and downstream of the target splitting point at both primers (see Figure 2). For PCR templates we used a pUG6 based plasmid set (Guldener et al., 1996) for PCR amplification but with different genetic marker genes (Candida glabrata LEU2 gene, Candida glabrata HIS3 gene, URA3, and KanMX) or centromere (CEN4) (Sugiyama et al., 2005). Select appropriate plasmid as a template for PCR so that newly generated chromosomes contain one centromere. These plasmids are available at NBRP (http://yeast.lab.nig.ac.jp/nig.v2.1/index.html).
- Transformation
- Prepare a yeast strain in advance which constitutively express codon optimized Cas9 for yeast expression by introduction of p414-TEF1p-Cas9-CYC1t.
Note: Yeast strains which can perform many auxotrophic marker selections are desirable. We use FY834 strain (MATα ura3-52 his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63). FY834 strain is transformed by introduction of p414-TEF1p-Cas9-CYC1t plasmid by LiAc/PEG method. Because p414-TEF1p-Cas9-CYC1t plasmid has a TRP1 marker, transformants that can grow in the medium without tryptophan were selected. - Cultivate the strain overnight in liquid yeast minimum medium (SD medium supplemented by necessary amino acids and nucleic acid base).
- Inoculate fresh 50 ml YPD medium with the yeast cell pre-culture to obtain an initial OD600 of approximately 0.2-0.3. Then incubate with shaking speed 140 rpm at 30 °C until OD600 reaches 0.8-1.0 (about 4 to 6 h).
- Mix the splitting DNA module(s) (1-5 μg each) and gRNA expressing plasmid(s) (7.5 μg each) and perform transformation by the conventional LiAc/PEG method (Gietz and Schiestl, 2007). LiAc/PEG method uses DNA MB-grade from salmon sperm, lithium acetate and polyethylene glycol 4000 (listed in the Materials and Reagents section).
Note: In our protocol, the maximum transformation efficiency is achieved when the amount of gRNA expressing plasmid is 7.5 μg. However, multiple split transformants can be obtained even when using less gRNA expressing plasmid (for example, 1 μg). - After transformation, cells are suspended in 200 μl of sterilized water and spread onto an appropriate selection plate and incubate at 30 °C for 2 or 3 days.
Note: Selection of transformants by a genetic marker gene on a gRNA expressing plasmid is not necessary and may decrease transformation efficiency. It usually causes decreased number of transformants probably due to continual targeted Cas9-endonuclease activity.
- Prepare a yeast strain in advance which constitutively express codon optimized Cas9 for yeast expression by introduction of p414-TEF1p-Cas9-CYC1t.
- Confirmation of the splitting event by Pulsed Field Gel Electrophoresis and subsequent Southern blotting
- Prepare plugs in accordance with the kit protocol (e.g., CHEF Genomic DNA Plug Kits Instruction Manual, Bio-Rad Laboratories).
- Perform Pulsed Field Gel Electrophoresis (PFGE) with an appropriate apparatus (e.g., CHEF system in Bio-Rad Laboratories). A typical setting for PFGE is described in Sugiyama et al., 2005.
- After electrophoresis, stain the gel by ethidium bromide solution (1 μg/ml) to check the karyotype of the transformants.
- Perform membrane blotting by the conventional blotting method (Southern, 2006).
- Prepare probes for detection of a specific region by an appropriate DNA labeling kit (e.g., ECL Direct Nucleic Acid Labelling and Detection System) and perform probe hybridization.
Note: A probe is a labeled single strand DNA having a specific sequence to detect the presence of DNA molecule having the same sequence with the probe. Typically, the size of a probe is from 500 bp to 1,000 bp. Although any region in the target chromosome can be used as a probe to check the splitting event, you may choose a region extending over the splitting point. When you use such a probe, two bands will appear corresponding to two chromosomes yielded by the splitting event. On the other hand if a splitting event did not occur, only one band corresponding to the intact chromosome will appear. A typical result is illustrated in Figure 3. - Detect the labeled probe in accordance with the DNA labeling kit manual you use.
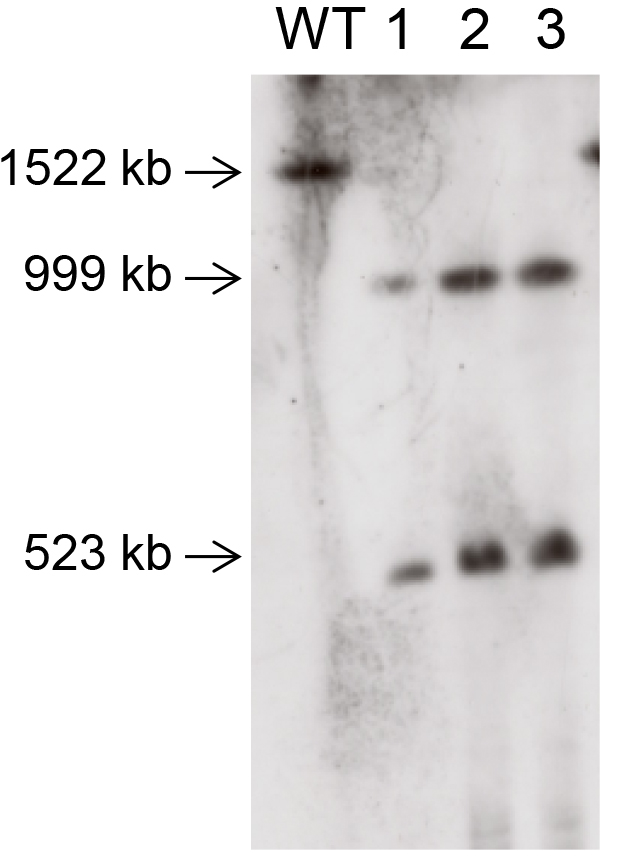
Figure 3. A typical result of chromosome splitting by CRISPR-PCS. Chr. IV was split at the position 999,122.5 by CRISPR-PCS. The probe was designed to extend over the splitting point (from 998,717 to 999,302). The wild type strain (designated as WT) shows single band corresponding to intact Chr. IV, while transformants (designated as 1, 2, and 3) show two bands demonstrating that Chr. IV was split at the targeted point.
- Prepare plugs in accordance with the kit protocol (e.g., CHEF Genomic DNA Plug Kits Instruction Manual, Bio-Rad Laboratories).
Data analysis
The number of transformants varies in every experiment. Generally, when targeting only one genomic site, several hundred or more transformants can be obtained and almost all the transformants harbor expected splitting. Even when targeting multiple sites, you can obtain several hundred transformants, although the frequency of transformants with expected splitting is decreased. Examination of transformants should yield at least one correctly split transformant out of 10 transformants.
Recipes
- LB plate
2% LB broth (Sigma-Aldrich)
1.5% agar - Yeast minimum medium (SD medium)
2% glucose
0.67% yeast nitrogen base without amino acids (e.g., BD Difco)
Note: If required, drop out mix containing all amino acids lacking appropriate amino acids used for auxotrophic selection can be used.
Adjust to pH = 6.0 with NaOH
For plate media, 2% agar is added - YPD medium
2% glucose
2% peptone (e.g., BD BactoTM)
1% yeast extract (e.g., BD BactoTM)
Adjust to pH = 6.0 with HCl
Acknowledgments
This work was supported by Grant-in-Aid for Scientific Research (B) [15H04475] (to S. H.), Grant-in-Aid for Challenging Exploratory Research [26660066] (to S. H.), and Grant-in-Aid for Young Scientists (B) [15K18672] (to Y. S.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
References
- Agmon, N., Pur, S., Liefshitz, B. and Kupiec, M. (2009). Analysis of repair mechanism choice during homologous recombination. Nucleic Acids Res 37(15): 5081-5092.
- Gietz, R. D. and Schiestl, R. H. (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2(1): 31-34.
- Guldener, U., Heck, S., Fielder, T., Beinhauer, J. and Hegemann, J. H. (1996). A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24(13): 2519-2524.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Li, M. Z. and Elledge, S. J. (2012). SLIC: a method for sequence- and ligation-independent cloning. Methods Mol Biol 852: 51-59.
- Southern, E. (2006). Southern blotting. Nat Protoc 1: 518-525.
- Sugiyama, M., Ikushima, S., Nakazawa, T., Kaneko, Y. and Harashima, S. (2005). PCR-mediated repeated chromosome splitting in Saccharomyces cerevisiae. Biotechniques 38(6): 909-914.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sasano, Y. and Harashima, S. (2017). CRISPR-PCS Protocol for Chromosome Splitting and Splitting Event Detection in Saccharomyces cerevisiae. Bio-protocol 7(10): e2306. DOI: 10.21769/BioProtoc.2306.
Category
Molecular Biology > DNA > Chromosome engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.