- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Reversible Cryo-arrests of Living Cells to Pause Molecular Movements for High-resolution Imaging


(*contributed equally to this work) Published: Vol 7, Iss 8, Apr 20, 2017 DOI: 10.21769/BioProtoc.2236 Views: 8594
Reviewed by: Vivien Jane Coulson-ThomasFederica PisanoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Fluorescence live-cell imaging by single molecule localization microscopy (SMLM) or fluorescence lifetime imaging microscopy (FLIM) in principle allows for the spatio-temporal observation of molecular patterns in individual, living cells. However, the dynamics of molecules within cells hamper their precise observation. We present here a detailed protocol for consecutive cycles of reversible cryo-arrest of living cells on a microscope that allows for a precise determination of the evolution of molecular patterns within individual living cells. The usefulness of this approach has been demonstrated by observing ligand-induced clustering of receptor tyrosine kinases as well as their activity patterns by SMLM and FLIM (Masip et al., 2016).
Keywords: Cryo-arrestBackground
Understanding molecular processes in cells, e.g., the ligand-induced response of receptor-tyrosine kinases (RTKs), requires the precise spatio-temporal observation of molecular patterns. Due to variance in cellular states, this response needs to be monitored in individual cells rather than in cell populations (Snijder and Pelkmans, 2011). Using SMLM, individual molecules can be localized with high precision (Betzig et al., 2006). This allows, for instance, extracting information about the clustering of RTKs in the plasma membrane. Complementarily, confocal FLIM can unveil how molecules react as an ensemble within a diffraction-limited volume element. This can reveal interaction patterns of RTKs with downstream molecules, phosphorylation patterns as well as activity patterns by the use of conformational sensors (Offterdinger et al., 2004; Sabet et al., 2015). However, the acquisition time in FLIM and SMLM is in the order of minutes. Because many molecular arrangements in living cells–including those of RTKs–evolve on a much faster time scale, confocal FLIM images get blurred and spatial resolution is reduced severely (Masip et al., 2016). In SMLM, molecules are localized successively in consecutive frames. Performing these measurements on dynamic molecules in living cells will lead to a falsified image of localizations since molecules diffuse to different positions in the course of data acquisition. For instance, the epidermal growth factor receptor (EGFR) moves on average with a diffusion constant of 2-5 10-2 μm2/sec in the 2D environment of the membrane (Orr et al., 2005; Xiao et al., 2008). To allow for long acquisition times, irreversible chemical fixation with aldehydes is typically used. Yet, the use of chemical fixation agents is inefficient in immobilizing membranes and can lead to protein extraction or artificial clustering (Saffarian et al., 2007; Tanaka et al., 2010; Schnell et al., 2012). Further, the lethal fixation disrupts the out-of-equilibrium physiological state and prohibits the observation of the evolution of molecular patterns within an individual cell. We have therefore developed a reversible cryo-arrest that halts molecular movements for high-resolution imaging, yet maintains cells in a viable state. Upon cooling, the concentration of cryoprotective dimethyl sulfoxide (DMSO) is increased stepwise (Masip et al., 2016). This precludes the formation of ice crystals that can be lethal to cells while scattering light and altering cellular structure by displacing organic material (Dubochet et al., 1988 and 2012; Huebinger et al., 2016). At the same time, adding DMSO at low temperatures greatly reduces its toxicity (Farrant, 1965). A further advantage of the cryo-approach is that fluorophores become less reactive in the excited state, resulting in the emission of more photons before bleaching as well as a lowered production of cytotoxic radicals (Kaufmann et al., 2014), which can severely damage cells during acquisition at physiological temperatures (Wäldchen et al., 2015).
Materials and Reagents
- 10 μm thick double-sided adhesive tape (D80 19 x 50 mm) (Modulor, catalog number: 0332054 )
- Cover slides 21 x 26 mm (No.1) (Thermo Fisher scientific, Thermo scientificTM, catalog number: BBAD02100260#A* )
- 6-well plates for cell culture (SARSTEDT, catalog number: 83.3920 )
- 15-ml reaction tubes (e.g., SARSTEDT, catalog number: 62.554.502 )
- 200-μl pipette tips (e.g., Greiner Bio One International, catalog number: 739290 )
- Silicon tube
- Flexible silicon tubes (inner diameter [i.d.] 2 mm; outer diameter [o.d.] 4 mm and i.d. 1.5 mm; o.d. 3 mm) (e.g., VWR, catalog numbers: 228-0704 and 228-0702 )
- Cell line of interest and suitable cell culture medium
Note: The protocol has so far been tested for the adherently growing cell lines HeLa (ATCC, catalog number: CCL-2 ), MDCK (ATCC, catalog number: CCL-34 ), COS-7 (ATCC, catalog number: CRL-1651 ), MCF7 cells (ATCC, catalog number: HTB-22 ) and HCT116 (ATCC, catalog number: CCL-2 47). Other cell lines may be used after proper testing for the reversibility of the cryo-arrest. - Ethanol, absolute (e.g., Fisher Scientific, catalog number: 10342652 )
- Sterile water (sterilized by filtration)
- DMSO (min 99%) (e.g., Serva Electrophoresis, catalog number: 20385 )
- Liquid nitrogen
- Immersion oil (e.g., Olympus, catalog number: IMMOIL-F30CC )
- Transfection reagents and plasmid expressing fluorescent proteins of interest or cell lines stably expressing a fluorescent protein of interest. The protocol has been successfully used for confocal FLIM with a conformational activity sensor for EphrinA2 and EGFR tagged to mCitrine in combination with a phosphotyrosine binding domain tagged to mCherry. Single molecule localization has been performed with EGFR-mEos2 and with Vinculin tagged to a SNAP-Tag and labelled with SNAP-Cell TMR-star (New England Biolabs, catalog number: S9105S ). Requests about these plasmids can be addressed to the corresponding author
- HEPES or phosphate buffered imaging medium without phenol red (e.g., DMEM, PAN-Biotech, catalog number: P04-01163 )
- 10% fetal bovine serum (FBS) (PAN-Biotech, catalog number: P30-1505 )
- Streptomycin/penicillin (PAN-Biotech, catalog number: P06-07100 )
- L-glutamine (200 mM) (PAN-Biotech, catalog number: P04-80100 )
- 1% nonessential amino acids (PAN-Biotech, catalog number: P08-32100 )
- Different DMSO solutions (see Recipes)
- Cell culture medium (see Recipes)
Equipment
- Scalpel
- Laminar flow hood
- Incubator (e.g., Nuaire, model: NU-5510/E )
- Fine forceps (e.g., Electron Microscopy Sciences, catalog number: 0203-7-PO )
- Anodized (black) aluminum flow-through chamber (custom-built; see Figure 1)
Note: Anodizing the aluminum reduces its corrosion and thereby solvation of aluminum ions, which might otherwise influence cellular reactions. Anodizing with black color reduces reflection of light, which might interfere with the microscopic imaging.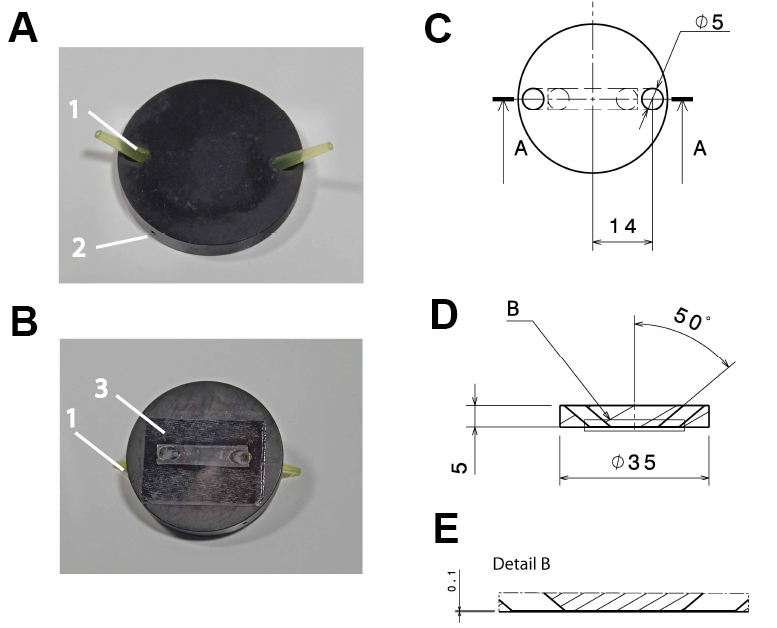
Figure 1. Design of the flow-through chamber. Photographic representation (A, B) and technical drawings (C-E) of the self-built flow-through chamber made out of aluminum. A. Top view on the flow through chamber with pipette tips inserted into the in- and outlet (1) and a drill hole (2; depth: 15 mm; diameter: 1 mm) for the insertion of a thermocouple. B. Bottom view with the cover slide glued to the flow-through chamber (3); C. Top view of the flow-through chamber; D. Side view section through the flow-through chamber (section a:a in C); E. Detail of the side view section (detail b in D). All dimensions in c-e are in mm. - Polyvinyl chloride (PVC) insert for the microscope table (custom-built; see Figure 2), with 2 threaded holes to fix the stage using a metal clamp (see Figure 3B)
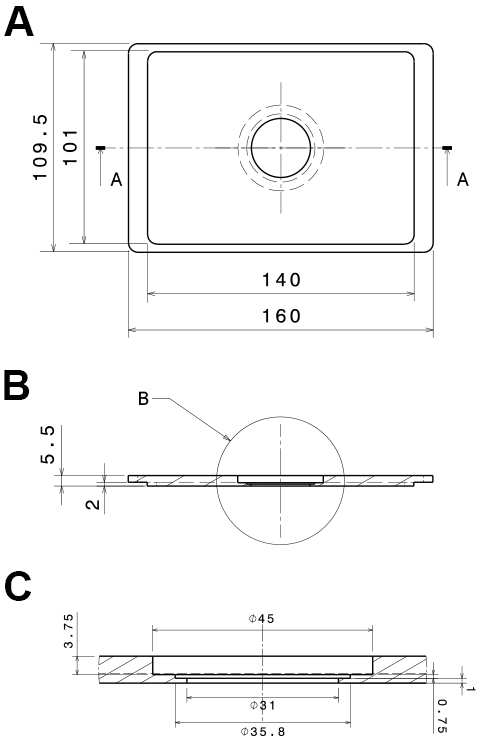
Figure 2. Technical, drawings of the PVC-insert for the microscope stage. The technical drawings show a mounting that fits into Scan IM stages (Märzhäuser Wetzlar GmbH & Co. KG, Wetzlar, Germany). A. Bottom view; B. Side view section (a:a in A); C. Side view detail of the central part (b in B). All dimensions are in mm. - Low-pressure syringe pump with a computer interface (Cetoni, model: neMESYS low pressure syringe pump )
- Cryo-stage with temperature control (Linkam Scientific, model: MDS600 , other models with similar cooling heads may also work)
- Thermocouple (e.g., PTFE-insulated type T with 0.08 mm wires; Omega Engineering, Deckenpfronn, Germany) with a data acquisition device connected to a computer for continuous temperature recording (e.g., OMEGA Engineering, model: OMB-DAQ-2408-2AO )
- For confocal FLIM: Confocal laser scanning microscope equipped with a time correlated single photon counting unit and a ps-pulsed laser (e.g., PicoQuant, model: MicroTime 200 )
- For SMLM: Microscope equipped with sensitive cameras (EMCCD or sCMOS) as well as lasers with adequate wavelength and power to image, switch on and bleach fluorophores
- Inverted microscope (Olympus, model: IX-81 )

Figure 3. Cryo-stage mounted on a microscope. Photographic representation of the cryo-stage mounted on an inverted microscope ( IX-81 ; Olympus GmbH, Hamburg, Germany). A. Overview of the cryo-stage with peripheral instruments on the microscope. 1: Liquid nitrogen pump with control unit (Note: This should normally be disconnected from the optical table, since it may cause vibrations of the sample); 2: 15-ml tubes with medium of different DMSO-concentrations, inlet tube of the low-pressure syringe pump is inserted via a lid with a hole; 3: Low-pressure syringe pump; 4: Data acquisition device for thermocouple; 5: Central part of the cryo-stage as detailed in (B); 6: Liquid nitrogen reservoir. B. Detailed image of the central part of the microscope. 1: Tube connected to nitrogen pump; 2: Metal clamp to fix the stage to the PVC-insert; 3: Thermocouple to measure the temperature inside the silver block; 4: Medium inlet connected to the low pressure syringe pump; 5: Aluminum flow-through chamber (compare Figure 1); 6: PVC-insert for microscope stage (compare Figure 2); 7: Tube connected to the nitrogen reservoir; 8: Temperature controlled silver block with electrical counter heater; 9: Medium outlet tube; 10: Thermocouple to measure temperature of the aluminum block.
Software
- ThunderSTORM Plugin (Ovesny, Bioinformatics, 2014)
- ImageJ (Rasband, ImageJ, National Institutes of Health, Bethesda, Maryland, USA)
Procedure
- Culturing of cells
Note: The following steps should be performed under a laminar flow hood. All instruments should be sterilized before. The adhesive tape should be taken out of its packing only under a laminar flow hood.- Cut double-sided adhesive tape into 26-mm long pieces using a scalpel (Figure 4A).
Note: Use one layer of adhesive tape with release liner as support. Then, add a second layer on top and cut this into size. This facilitates working with the adhesive tape, since it does not stick to the release liner.
Figure 4. Preparation of culture dish with cover slides. A. A layer of double-sided adhesive tape (1) is glued on a smooth surface (e.g., an anodized aluminum plate). A second layer of double-sided adhesive tape is glued on top (2) and cut to length of a cover slide (left image). Afterwards, a rectangular of app. 21 x 5 mm is cut out in the center and removed (right image). B. The piece of double-sided adhesive tape is glued on a cover slide (4; left image). Mild pressing ensures tight contact and prevents leakage (right image). C. Photographic representation of a 6-well cell culture dish with cover slides with double sided sticky tape (arrowheads) in cell culture medium with phenol red. The double-sided sticky tapes have cut-outs fitting the cavity of the flow through chamber (arrows; compare Figure 1B). - Cut out a 21 x 5 mm rectangular in the middle of each piece of adhesive tape using a scalpel and remove the inner rectangular (Figure 4A).
- Glue the adhesive tape on a 21 x 26 mm cover slide. Mildly press to ensure a tight contact without breaking the cover slide (Figure 4B).
- Sterilize the cover slides by dipping them in ethanol and wash them with sterile, distilled water.
- Place the cover slides in the 6-well-plate (Figure 4C).
- Detach cultured cells and plate them in the 6-well dish in regular cell culture medium (see Figure 4). The adhesive tape is not affected by the cell culture medium and remains sticky.
Note: Let the cover slides dry or wash with cell culture medium before adding the cells, since distilled water is harmful to the cells. - Incubate at 37 °C/5% CO2 at least until they are firmly attached. Cells can be cultured under this condition for days and transfected with the respective constructs of interest.
- Cut double-sided adhesive tape into 26-mm long pieces using a scalpel (Figure 4A).
- Assembly of the cryo-stage on the microscope
- Prepare 10%, 20%, 30% and 50% (vol/vol) solutions of DMSO in imaging medium in 15-ml reaction tubes.
Note: The inlet tube of the low temperature syringe pump can be inserted into the 15-ml reaction tubes through a hole in the lid (Figure 3A). - Take the silver cooling head out of the casing of the cryo-stage.
- Connect the nitrogen pump, the liquid nitrogen reservoir using flexible silicon tube (i.d. 2 mm; o.d. 4 mm) and the electronic temperature control to the silver cooling head (see Figure 3B).
- Set the temperature of the silver block to 37 °C, using the control unit for the liquid nitrogen pump (Figure 3A-1).
- Attach a piece of flexible silicon tube (i.d. 1.5 mm; o.d. 3.5 mm) to the outlet of the low-pressure syringe pump.
Note: The volume of the tube connected to the syringe pump should not exceed 200 μl. Otherwise, this would increase the amount of medium that needs to be exchanged during every step of the cryo-arrest. - Fill the syringe and the tube with imaging medium without DMSO using the software of the automated syringe. Avoid air bubbles.
Note: For the first filling, small amounts of medium have to be filled in the syringe and pressed through its outlet to remove air bubbles in the syringe and tubes before filling the syringe. - Assemble the PVC-insert into the microscope table and center the objective below the opening for the flow through chamber (Figure 3B).
- Press 200-μl pipette tips through the in- and outlet of the flow-through chamber from the bottom. Remove the parts sticking out of the bottom with a scalpel. Also, cut of a few millimeters of the tapered end of the pipette tip with scissors (compare Figures 1A and 1B).
- Prepare 10%, 20%, 30% and 50% (vol/vol) solutions of DMSO in imaging medium in 15-ml reaction tubes.
- Mounting of cells to the cryo-stage
Note: Steps C1-C4 have to be performed quickly to protect the cells from drying out.- Take the cover slide with adhesive tape and cells grown on it out of the 6-well plate
- Remove the release liner with fine forceps.
- Glue the cover slide to the flow-through chamber so that the cut-out in the middle fits to the cavity of the flow through chamber (Figure 1B).
- Fill the flow-through chamber with regular cell culture medium by pipetting through one of the pipet tips attached to the flow-through chamber. Fill the chamber and both pipet tips completely. Avoid air bubbles.
- Place the flow through chamber in the cavity of the PVC-insert (Figures 3B-5 and 3B-6).
- Connect the silicon tube from the syringe pump to one of the pipette tips of the flow through chamber (Figure 3B-4).
- Connect a piece of silicon tube (i.d. 1.5 mm; o.d. 3.5 mm; length ≤ 100 mm) to the other pipet tip and place an empty petri dish below the open end of the silicon tube (Figure 3B-9).
- Put a small drop of oil (e.g., immersion oil) on top of the aluminum chamber and mount the cooling head on it using the metal clamp (Figures 3B-2 and 3B-9)
- Connect the thermocouple into the drilling hole of the aluminum stage. Start the temperature measurement. The measured temperature should be above 36 °C, if the thermocouple is in reasonably good contact with the aluminum chamber.
- Take the cover slide with adhesive tape and cells grown on it out of the 6-well plate
- Reversible cryo-arrest
- Focus and image cells at 37 °C.
Note: During this step it might be useful to supply the cells with fresh medium with a constant flow of 1 μl sec-1 using the automated syringe. - For cryo-arrest: empty the syringe pump and load it with 10% DMSO solution, using the software control of the syringe pump.
- Set the temperature in the cryo-stage to 4 °C, using the control unit for the liquid nitrogen pump (Figure 3A-1).
- When the measured temperature in the aluminum chamber is stable, exchange the medium by applying a flow of 3 μl sec-1 for a total of 300 μl using the syringe pump.
- Empty the syringe and load it with 20% DMSO solution.
- Apply a flow of 3 μl sec-1 for a total of 300 μl using the syringe pump.
- Set temperature to -5 °C.
- Empty the syringe and load it with 30% DMSO solution.
- When the measured temperature in the aluminum chamber is stable, apply a flow of 3 μl sec-1 for a total of 300 μl using the syringe pump.
- Set temperature to -10 °C.
- Empty the syringe and load it with 50% DMSO solution.
- When the measured temperature in the aluminum chamber is stable, apply a flow of 3 μl sec-1 for a total of 600 μl using the syringe pump.
- Set temperature to -45 °C.
- After the temperature in the aluminum chamber is stable, cells can be imaged in the arrested stage.
- To go back to physiological temperature: Set temperature to -10 °C first.
- Empty the syringe and load it with 30% DMSO solution.
- When the measured temperature in the aluminum chamber is stable, apply a flow of 3 μl sec-1 for a total of 300 μl using the syringe pump.
- Set temperature to -5 °C.
- Empty the syringe and load it with 20% DMSO solution.
- When the measured temperature in the aluminum chamber is stable, apply a flow of 3 μl sec-1 for a total of 300 μl using the syringe pump.
- Set temperature to 4 °C.
- Empty the syringe and load it with 10% DMSO solution.
- Apply a flow of 3 μl sec-1 for a total of 300 μl using the syringe pump.
- Empty the syringe and load it with 0% DMSO solution.
- Apply a flow of 3 μl sec-1 for a total of 300 μl using the syringe pump.
- Set temperature to 37 °C.
- At 37 °C cells can be stimulated, e.g., with ligands for RTKs, light, etc.
- To arrest cells at a specific time after stimulation, start again with the cryo-arrest cycle. An example of EGFR localized by photoactivated localization microscopy in HeLa cells before and after stimulation with EGF is shown in Figure 5. More examples are shown in the original publication of the method (Masip et al., 2016).
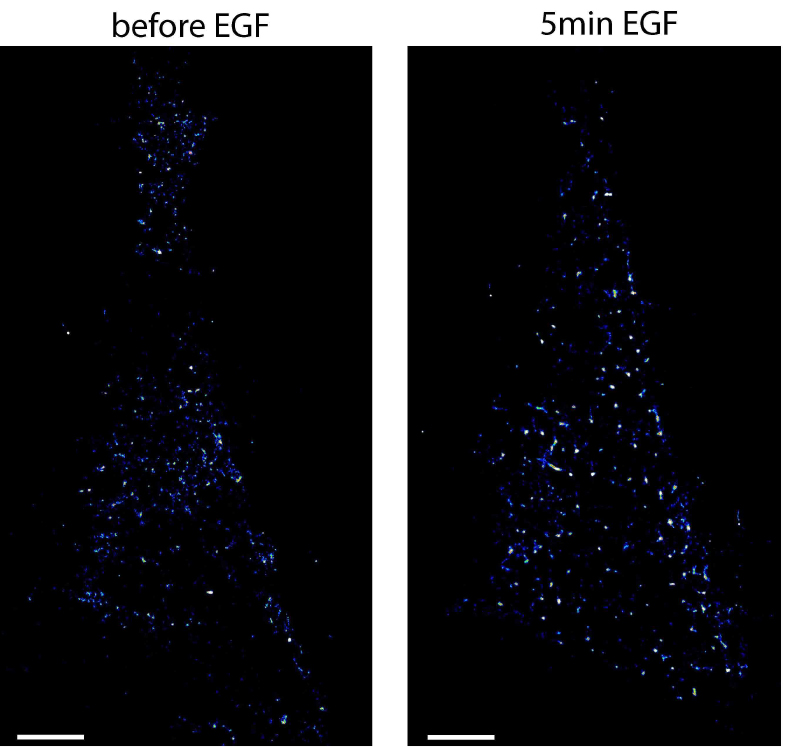
Figure 5. Photoactivated localization microscopy of EGFR in the same HeLa cell before and after stimulation. Shown are localization maps of a HeLa cell transfected with EGFR-mEos2 that was cryo-arrested before (left image) and 5 min after (right image) stimulation with 200 ng/ml EGF. Under reversible cryo-arrest EGFR molecules were localized by photo-activated localization microscopy with a precision of 21 ± 6 nm (mean ± SD). Scale bars = 5 μm
- Focus and image cells at 37 °C.
Data analysis
Reversible cryo-arrest is compatible with almost every fluorescence microscopy technique. Therefore, the data analysis will depend on the microscopy performed under cryo-arrest. So far reversible cryo-arrest has been applied with SMLM and FRET-FLIM. Details about the analysis of this can be found in the freely available authors' version of Masip et al., 2016 (Europe PubMed Central plus, Manuscript #69708). In brief, SMLM images may be reconstructed using the ThunderSTORM Plugin (Ovesny, Bioinformatics, 2014) for ImageJ (Rasband, ImageJ, National Institutes of Health, Bethesda, Maryland, USA). For FRET-FLIM data it is recommended to quantify the fraction of fluorescent molecules that undergo FRET in each pixel on the image by global analysis (Grecco et al., 2009).
Notes
- In this protocol, we focus on the application of the cryo-arrest to study RTK-activation in adherently growing mammalian cells. In the tested cells, general morphology was preserved. They did not induce expression of stress response genes and growth factor induced signaling proceeded normally after the cryo-arrest (Masip et al., 2016). The protocol is likely adaptable to various types of biological samples and processes. However, when reversible cryo-arrest is established in a new laboratory or a different cellular system is used, it is recommended that controls be performed to ensure that the cryo-arrest does not influence the produced data. At least three different experiments with fluorescently labeled cells (e.g., by transfecting a fluorescent protein such as EGFP) should be performed to judge their morphology. After the cryo-arrest, cells should be labeled with a dye that stains dead cells (e.g., propidium iodide). Dead cells can be quantified by dividing the number of fluorescently labeled cells that are positive for the dead cell marker by the total number of fluorescently labeled cells. Additionally, the expression of cold and heat stress proteins should be tested and the reaction under investigation should be quantified with and without cryo-arrest to exclude influences of the cryo-arrest procedure on the process under investigation (Masip et al., 2016).
Recipes
- Different DMSO solutions (10%, 20%, 30% and 50% [vol/vol])
Different DMSO solutions were prepared by pipetting DMSO and HEPES-buffered medium without phenol red in the corresponding ratios into 15-ml reaction tubes and inverting the tube several times - Cell culture medium (for the cell line indicated in the equipment section)
Dulbecco’s modified Eagle medium (DMEM) with phenol red supplemented with 10% fetal bovine serum (FBS), 100 μg ml-1 streptomycin plus 100 U ml-1 penicillin, 1% L-glutamine (200 mM) and 1% nonessential amino acids
Acknowledgments
The authors would like to thank Michael Reichl and Petra Glitz for excellent technical support, the workshop of the Max-Planck Institute of Molecular Physiology for construction of custom parts, Astrid Krämer for critical proofreading of the manuscript and Sven Müller for photographic images of the setup. This study was funded by the Fraunhofer Society and the Max-Planck Society for the Promotion of Science in the joint initiative ‘CryoSystems’ and the European Research Council (ERC AdG 322637). This protocol is based on a previously published protocol (Masip et al., 2016).
References
- Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., Davidson, M. W., Lippincott-Schwartz, J. and Hess, H. F. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313(5793): 1642-1645.
- Dubochet, J. (2012). Cryo-EM-the first thirty years. J microsc 245(3): 221-224.
- Dubochet, J., Adrian, M., Chang, J. J., Homo, J. C., Lepault, J., McDowall, A. W. and Schultz, P. (1988). Cryo-electron microscopy of vitrified specimens. Q Rev Biophys 21(2): 129-228.
- Endres, N. F., Das, R., Smith, A. W., Arkhipov, A., Kovacs, E., Huang, Y., Pelton, J. G., Shan, Y., Shaw, D. E., Wemmer, D. E., Groves, J. T. and Kuriyan, J. (2013). Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152(3): 543-556.
- Farrant, J. (1965). Mechanism of cell damage during freezing and thawing and its prevention. Nature 205(4978): 1284-1287.
- Grecco, H. E., Roda-Navarro, P. and Verveer, P. J. (2009). Global analysis of time-correlated single photon counting FRET-FLIM data. Opt Express 17(8): 6493-6508.
- Huebinger, J., Han, H. M., Hofnagel, O., Vetter, I. R., Bastiaens, P. I. and Grabenbauer, M. (2016). Direct measurement of water states in cryopreserved cells reveals tolerance toward Ice crystallization. Biophys J 110(4): 840-849.
- Kaufmann, R., Hagen, C. and Grünewald, K. (2014). Fluorescence cryo-microscopy: current challenges and prospects. Curr Opin Chem Biol 20: 86-91.
- Masip, M. E., Huebinger, J., Christmann, J., Sabet, O., Wehner, F., Konitsiotis, A., Fuhr, G. R. and Bastiaens, P. I. (2016). Reversible cryo-arrest for imaging molecules in living cells at high spatial resolution. Nat Methods 13(8): 665-672.
- Offterdinger, M., Georget, V., Girod, A. and Bastiaens, P. I. (2004). Imaging phosphorylation dynamics of the epidermal growth factor receptor. J Biol Chem 279(35): 36972-36981.
- Orr, G., Hu, D., Ozcelik, S., Opresko, L. K., Wiley, H. S. and Colson, S. D. (2005). Cholesterol dictates the freedom of EGF receptors and HER2 in the plane of the membrane. Biophys J 89(2): 1362-1373.
- Sabet, O., Stockert, R., Xouri, G., Bruggemann, Y., Stanoev, A. and Bastiaens, P. I. (2015). Ubiquitination switches EphA2 vesicular traffic from a continuous safeguard to a finite signalling mode. Nat Commun 6: 8047.
- Saffarian, S., Li, Y., Elson, E. L. and Pike, L. J. (2007). Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys J 93(3): 1021-1031.
- Schnell, U., Dijk, F., Sjollema, K. A. and Giepmans, B. N. (2012). Immunolabeling artifacts and the need for live-cell imaging. Nat Methods 9(2): 152-158.
- Snijder, B. and Pelkmans, L. (2011). Origins of regulated cell-to-cell variability. Nat Rev Mol Cell Biol 12(2): 119-125.
- Tanaka, K. A., Suzuki, K. G., Shirai, Y. M., Shibutani, S. T., Miyahara, M. S., Tsuboi, H., Yahara, M., Yoshimura, A., Mayor, S., Fujiwara, T. K. and Kusumi, A. (2010). Membrane molecules mobile even after chemical fixation. Nat Methods 7(11): 865-866.
- Verveer, P. J., Wouters, F. S., Reynolds, A. R. and Bastiaens, P. I. (2000). Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science 290(5496): 1567-1570.
- Wäldchen, S., Lehmann, J., Klein, T., van de Linde, S. and Sauer, M. (2015). Light-induced cell damage in live-cell super-resolution microscopy. Sci Rep 5: 15348.
- Xiao, Z., Zhang, W., Yang, Y., Xu, L. and Fang, X. (2008). Single-molecule diffusion study of activated EGFR implicates its endocytic pathway. Biochem Biophys Res Commun 369(2): 730-734.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Huebinger, J., Masip, M. E., Christmann, J., Wehner, F. and Bastiaens, P. I. H. (2017). Reversible Cryo-arrests of Living Cells to Pause Molecular Movements for High-resolution Imaging. Bio-protocol 7(8): e2236. DOI: 10.21769/BioProtoc.2236.
Category
Cell Biology > Cell imaging > Live-cell imaging
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.