- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Pathogenicity Assay of Verticillium nonalfalfae on Hop Plants



(*contributed equally to this work) Published: Vol 7, Iss 6, Mar 20, 2017 DOI: 10.21769/BioProtoc.2171 Views: 10201
Reviewed by: Zhaohui LiuValeria LullaTimo Lehti
Original research article
The authors used this protocol in:

May 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Verticillium nonalfalfae is a soil-borne plant pathogen that infects its hosts through roots. It spreads in the plant’s xylem and causes wilt disease symptoms by secreting different virulence factors. Hop (Humulus lupulus) is a primary host of V. nonalfalfae, so it is used as a model plant for studying this phytopathogenic fungus. Artificial infections of hop plants and disease scoring are prerequisites for studying the pathogen’s virulence/pathogenicity and its interaction with hop plants. In this protocol, we describe the root dipping inoculation method for conducting pathogenicity assay of V. nonalfalfae on hop plants.
Keywords: Verticillium nonalfalfaeBackground
Verticillium spp. infects more than 400 different host plants and every species has its own range of host. The primary host of V. nonalfalfae is hop. However, hop has several disadvantages for use as a test plant for pathogenicity assay; e.g., it is a perennial plant and needs to undergo a dormancy phase. Plants can therefore only be used for pathogenicity assay for a few months in the year, from late spring to late summer. Hop varieties are vegetatively propagated as softwood cuttings in a greenhouse or as dormant cuttings from rootstock. Seeds are obtained by crossing female and male plants and are used only for breeding purposes. The root dipping inoculation method has been widely used for pathogenicity assay of Verticillium spp. on other plant hosts, e.g., tomato (Fradin et al., 2009), N. benthamiana (Klosterman et al., 2011) and Arabidopsis thaliana (Ellendorff et al., 2009).
Materials and Reagents
- Miracloth (EMD Millipore, catalog number: 475855-1R )
- Petri dish (Golias, catalog number: PE01K )
- Host plants (hop Humulus lupulus, susceptible cultivar ‘Celeia’)
- Fungal conidia (Verticillium nonalfalfae; lethal pathotype PV1 [isolate T2]) (Radisek et al., 2006)
- Fertilizer YaraKristalon yellow NPK 13-40-13 + ME [ME - trace elements: B - 0.025%; Cu * - 0.01%; Fe * - 0.07%; Mn * - 0.04%; Mo - 0.004%; Zn * - 0.025%; * - Chelate base] (Yara International ASA)
- Fertilizer YaraKristalon special NPK 18-18-18 + ME [ME - trace elements: B - 0.025%; Cu * - 0.01%; Fe * - 0.07%; Mn * - 0.04%; Mo - 0.004%; Zn * - 0.025%; * - Chelate base] (Yara International ASA)
- Growing medium
- For fungus inoculum preparation: liquid GFM - general fungal medium (Kayser, 1992) (see Recipes)
- For plants: soil substrate (soil substrate for growing plants)
- For fungus re-isolation: potato dextrose agar + antibiotics (streptomycin sulphate, neomycin, chloramphenicol; each 100 mg/ml) = PDA + A plates (see Recipes)
- Sterile distilled water (IDT, catalog number: 231-791-5 )
- 96% ethanol
- Peptone (Sigma-Aldrich, catalog number: 73049-73-7 )
- Yeast extract (AMRESCO, catalog number: J850 )
- Glucose (Kemika, catalog number: 07051 )
- Potassium nitrate (KNO3) (EMD Millipore, catalog number: 105063 )
- Potato dextrose agar (Biolife, catalog number: 4019352 )
- Streptomycin sulphate (Duchefa Biochemie, catalog number: S0148 )
- Neomycin (Duchefa Biochemie, catalog number: M0135 )
- Chloramphenicol (Sigma-Aldrich, catalog number: C0378 )
Equipment
- 500 ml Erlenmeyer flask (BRAND, catalog number: 92824 )
- 2 L plastic cup (BRAND, catalog number: 87822 )
- Plastic pots 0.5 L
- Rotary shaker (Infrost, catalog number: 29313 )
- Growth chamber (Kambič Laboratory Equipment, model: RK-13300 )
- Fluorescent grow lamp (Idealo, model: Osram Fluora L 58 W/77 )
- Thoma counting chamber (BRAND, Wertheim, Germany)
- Wooden sticks for hop’s bine support
- Scalpel and tweezers
- Light microscope (Nikon Instruments)
Procedure
- Preparation of host plants
- For inoculation, use two-month-old, well-rooted plants that have been grown in 0.5 L plastic pots and have been vegetatively produced in a greenhouse from hop mother plants as soft wood cuttings. Plants should be directly produced by a professional grower.
- Grow plants in a greenhouse on mist benches. Water the plants every 2 days and fertilize once per week using foliar fertilizers that contain macro and micro elements (e.g., 0.1% solution of YaraKristalon yellow NPK 13-40-13 + ME).
- To maintain the appropriate health status, spray the plants once per week against pests and foliar diseases using protective plant protection products.
- Use the susceptible cultivar ‘Celeia’.
- For inoculation, use two-month-old, well-rooted plants that have been grown in 0.5 L plastic pots and have been vegetatively produced in a greenhouse from hop mother plants as soft wood cuttings. Plants should be directly produced by a professional grower.
- Inoculum preparation
- Place a small piece of fungal mycelium (of the size of a 2 mm ‘ball’) from stock PDA plates (which should be maintained in a fridge) in each of four 500 ml Erlenmeyer flasks filled with 250 ml of liquid GFM, supplemented with 100 mg/ml streptomycin sulphate and 100 mg/ml chloramphenicol.
- Incubation takes approximately 5 days at 100 rpm on a rotary shaker at room temperature and in the dark (place the shaker in a dark room) (Figure 1).

Figure 1. A picture of fungal culture grown after 5 days. White fungal mycelium in a sphere shape can be seen. The medium with fungal isolates that produce a lot of spores is duller. - Prepare the conidia inoculum (spore size 5 to 7 μm) by filtration of mycelia and the spore through miracloth (typical pore size is 22-25 µm):
- Place a non-autoclaved piece of miracloth (20 x 20 cm) on top of a 2 L plastic cup, hand held, and filter each flask separately, one by one.
- Wash spores and suspend them in sterile distilled water. There is no need to spin down the spores after washing.
- If the concentration is low, all the filtration material is needed.
- Use a Thoma counting chamber to determine the spore concentration. Determine the spore concentration four times for one filtrated suspension and calculate an average.
- Adjust the spore concentration to 5 x 106 conidia/ml with sterile distilled water.
- The final volume of inoculum with a concentration of 5 x 106 conidia/ml for artificial infection should be 1 L. Use a maximum of 12 plants per one volume of inoculum.
- Place a small piece of fungal mycelium (of the size of a 2 mm ‘ball’) from stock PDA plates (which should be maintained in a fridge) in each of four 500 ml Erlenmeyer flasks filled with 250 ml of liquid GFM, supplemented with 100 mg/ml streptomycin sulphate and 100 mg/ml chloramphenicol.
- Artificial infection
- Uproot hop plants that have been grown in 0.5 L plastic pots. Use as much soil substrate as possible from the roots by hand.
- Rinse the roots in sterile water (just gently dip for a few seconds).
- Then dip the roots for 10 min in 1 L of inoculum, poured into a 2 L plastic cup.
- Inoculate a minimum of 10 plants per treatment.
- Dispose of the remaining fungal inoculum after autoclaving.
- Treat control plants similarly but dip their roots in sterile water for 10 min.
- Pot the plants in new pots of size 0.5 L.
- Grow the plants as a single bine in a growth chamber (RK-13300, Kambič) under a 12-h photoperiod of fluorescent light (L 58 W/77; Fluora, Osram) at a temperature of 22 °C and relative humidity of 65% during the light period and 20 °C and 70% during the dark period. During growth in the chamber, water twice a week (every Monday and Friday) and fertilize once per week using foliar fertilizer containing a higher quantity of nitrogen (e.g., 0.2% solution of YaraKristalon special NPK 18-18-18 + ME).
- Uproot hop plants that have been grown in 0.5 L plastic pots. Use as much soil substrate as possible from the roots by hand.
- Symptoms monitoring
- The first disease symptoms occur approximately 20 days post inoculation (dpi). The first symptoms can be seen as pale yellowing of some leaves (Figure 2 – section 1). At that time no wilting is observed.

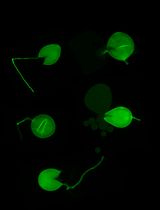
Figure 2. The 0-5 scale used to evaluate foliar symptoms of Verticillium wilt of hop. 0 indicates no leaf symptoms, 1 = 1 to 20% leaf area wilted, 2 = 21 to 40% leaf area wilted, 3 = 41 to 60% leaf area wilted, 4 = 61 to 80% leaf area wilted and 5 = 81 to 100% leaf area wilted. - Subsequently, assess the plants visually for the appearance of foliar symptoms using a 0-5 scale (Radisek et al., 2003) (Figure 2) at 7-day intervals.
- There are 3 to 5 assessment time points. Photograph the plants at the last time point, when the disease symptoms are the most severe. Photographing at earlier time points does not show the biggest difference between fungal inoculated and mock inoculated plants.
- At the end of visual disease assessments, carry out mycological re-isolation of the pathogen to confirm the presence of the pathogen in the inoculated plants.
- Cut the plants at ground level and transfer them to a laminar. Spray the cut stems with 96% ethanol and place very briefly on an open flame in order to sterilise the surface.
- Perform re-isolation by using xylem sections that have been sterilely harvested from the stem of inoculated plants and placed on PDA + A plates.
- The xylem section is actually the whole inner part of the stem, which should be cut into 1-2 cm long pieces. Place three to six pieces from each plant on PDA + A plates.
- Fungal outgrowth can be detected after the plates have been incubated for 3-5 days in the dark at 24 °C. Examine the emerging mycelium (if any) is examined by light microscopy.
- Cut the plants at ground level and transfer them to a laminar. Spray the cut stems with 96% ethanol and place very briefly on an open flame in order to sterilise the surface.
- Only infected plants should be considered and used for further analysis.
- Calculate the disease severity index (DSI) for every time point for each plant and determine the average disease severity index (Equation 1). Also present DSI results graphically (Figure 3).
Equation 1:
Average disease severity index = sum of assessments of infected plants for last assessment time points/number of infected plants (Jakse et al., 2013).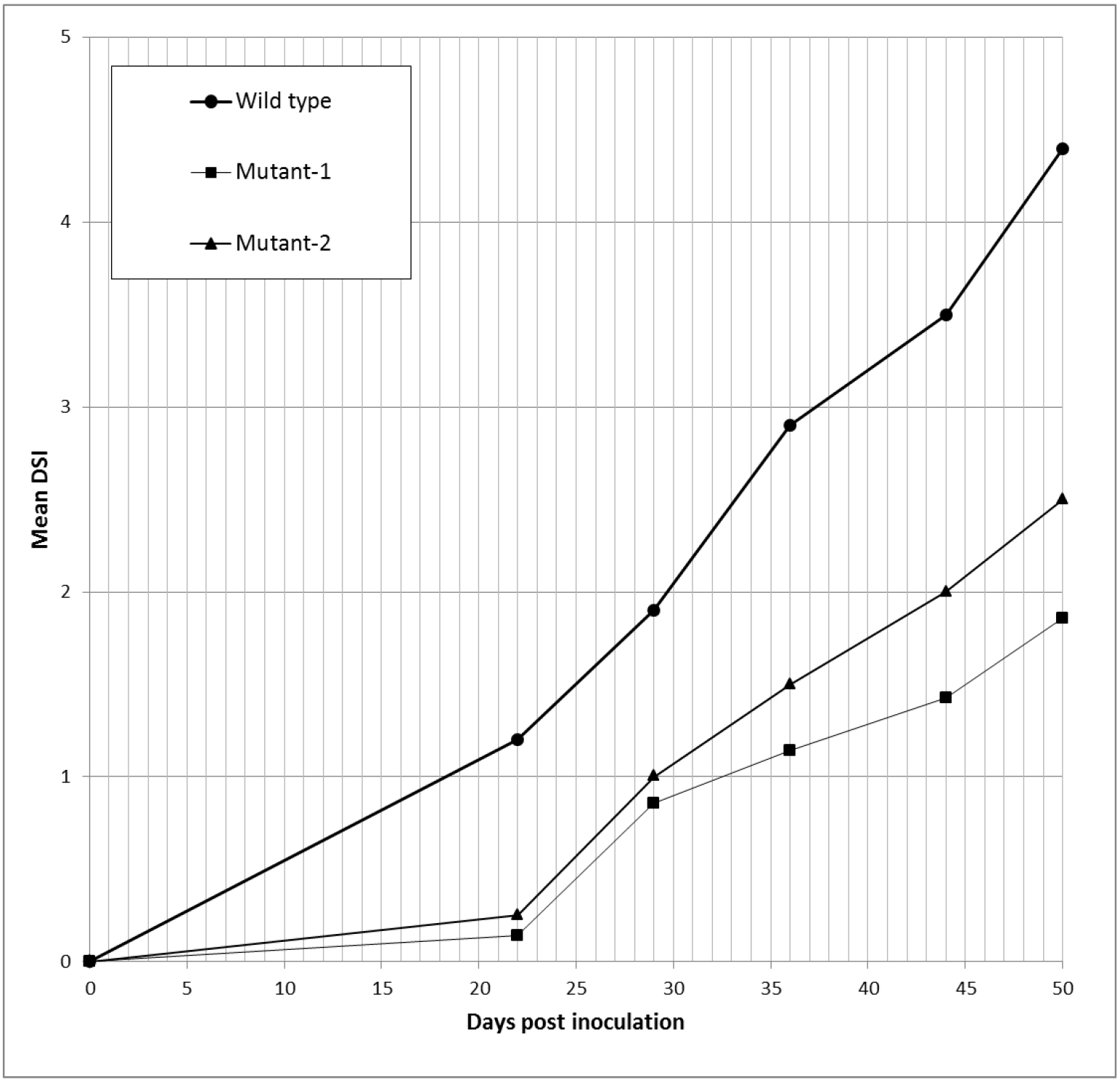
Figure 3. Example of graphic presentation of DSI index. DSI curves of hop plants infected with wild type and two mutants of V. nonalfalfae, which had impaired virulence, are indicated by symptom evaluation of infected plants at five different time points. - The area under the disease progress curve (AUDPC) is calculated as indicated by Campbell and Madden (1990) (Equation 2) and expressed as relative AUDPC - rAUDPC (Simko and Piepho, 2012) (Equation 3).
Equation 2: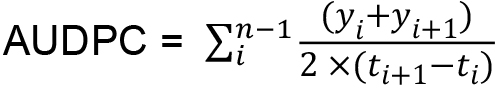
Where,
n = total number of observations,
y = DSI index for each plant,
t = number of days from inoculation.
Equation 3:
rAUDPC = actual AUDPC value/maximum potential AUDPC value
- The first disease symptoms occur approximately 20 days post inoculation (dpi). The first symptoms can be seen as pale yellowing of some leaves (Figure 2 – section 1). At that time no wilting is observed.
Data analysis
Figure 4 shows a representative example of the results of pathogenicity assay.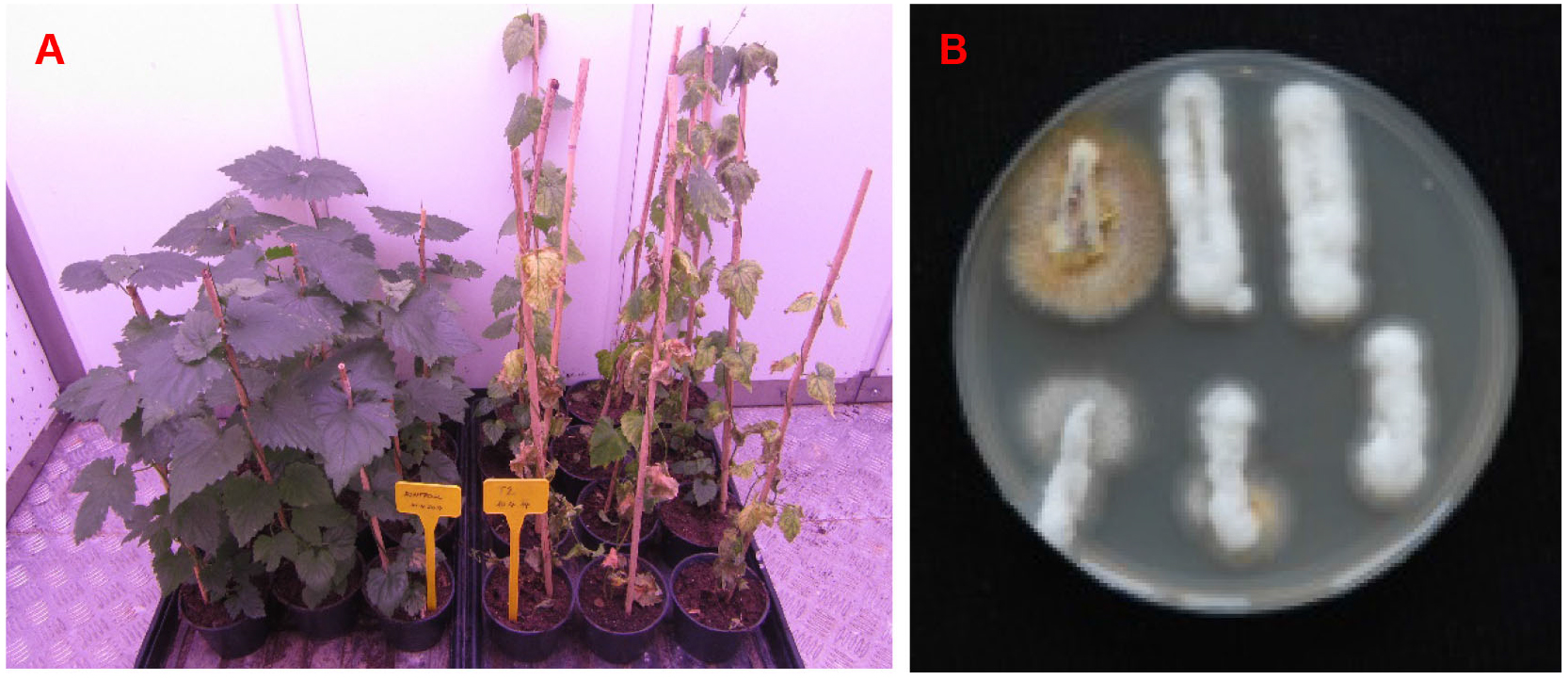
Figure 4. Results of pathogenicity assay. A. Disease symptoms of hop plants; left are mock inoculated asymptomatic plants, right are disease symptoms of hop plants infected with wild type. Plants were imaged 31 days after inoculation. B. Mycological re-isolation of one infected hop plant. Fungal outgrowth is shown 3 days after plating of xylem sections on PDA + A plates at room temperature. White, fluffy mycelium is typical of V. nonalfalfae. However, outgrowth of some other endophytic fungi can also appear, as is clearly seen with the first xylem section in the first row and to a lesser extent with the first and second xylem sections in the second row. Other microorganisms can also arise from xylem sections (see Notes). V. nonalfalfae mycelium prefers to grow on the plant material (xylem sections are completely overgrown by mycelium) and not on the PDA medium.
Statistical analysis
Perform statistical analysis of results of pathogenicity assay on rAUDPC values in the following order:
- Firstly, use Levene’s test to evaluate the difference in variance values.
- Secondly, if there is not a significant difference among variances of rAUDPC values of treatments, subject rAUDPC values calculated for each treatment to standard one-way analysis of variance followed by Dunnett’s test, which compares average rAUDPC values of mutant-infected plants with average rAUDPC values of wild-type-infected plants.
- However, if there is a significant difference among variances of rAUDPC values of treatments, the Kruskal-Wallis test of non-parametrical statistical analysis should be used, which compares the ranks of rAUDPC values.
- If differences among ranks exist, perform Dunn’s test in order to compare the average rank value of mutant-infected plants with the average rank value of wild-type-infected plants.
- Results are presented in a dot graph (Figure 5).
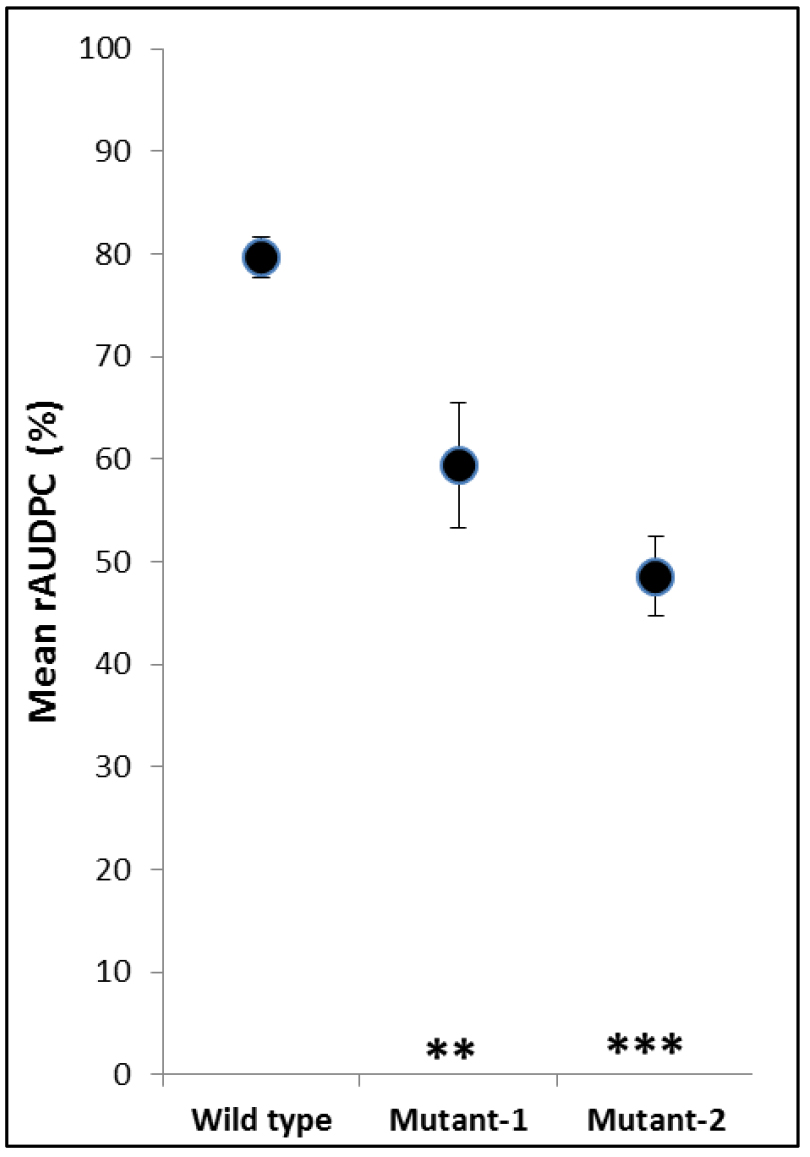
Figure 5. Example of graphic presentation of rAUDPC values. Mean rAUDPC values of wild type and two mutants, which had impaired virulence, are presented. The asterisks show significant differences (***P < 0.001; **P < 0.01). Error bars indicate standard error of the mean.
Notes
- As mentioned in the section ‘Procedure’ (part ‘B. Inoculum preparation’, step 6), a maximum 12 plants per one volume of inoculum (which is 1 L with a concentration of 5 x 106 conidia/ml) should be used. During root dipping of these plants, the inoculum gets diluted. If there are more plants to be inoculated with the same inoculum, the concentration of used inoculum must therefore be recovered to 5 x 106 conidia/ml by adding fresh conidia. It is even better to prepare new inoculum with a concentration of 5 x 106 conidia/ml.
- In relation to disease symptom assessments, we strongly advise that the same person always evaluates disease symptoms. Any mistake of assessment will thus be smaller or, at least equal for all assessments.
- In relation to mycological re-isolation: although the re-isolation medium contains 300 mg/ml antibiotics and re-isolation is done in a sterile environment, many different microorganisms can arise after plating xylem sections on media. Incubation of plates at room temperature should therefore be no longer than 5 days.
- Emerging mycelium on re-isolation plates must be subjected to morphological analysis using light microscopy in order to confirm the presence of V. nonalfalfae. Aerial mycelium of V. nonalfalfae is generally abundant, floccose to pruinose, hyphae are smooth-walled and 1.5-3 µm wide. Conidiophores are erect or slanted, generally determinate, branched or unbranched, formed disjointedly throughout the colonies and hyaline.
Recipes
- GFM
2 g peptone
2 g yeast extract
20 g glucose
1 g KNO3
Mix in 1,000 ml distilled water and autoclave for 20 min at 121 °C - PDA with 300 mg/ml antibiotics
- Add 35 g of potato dextrose agar in 1,000 ml distilled water
- Shake and mix the powder and autoclave it for 20 min
- Cool down the PDA broth to 55 °C
- Add 1 ml of 100 mg/ml streptomycin sulphate, 1 ml of 100 mg/ml neomycin and 1 ml of 100 mg/ml chloramphenicol and mix well
- Pour in 90 x 15 mm Petri dishes
Acknowledgments
This protocol is adapted from previously published papers (Radisek et al., 2006; Jakše et al., 2013; Cregeen et al., 2015; Mandelc et al., 2013; Flajsman et al., 2016). We acknowledge the Slovenian Research Agency, research program P4-0077, for funds.
References
- Campbell, C. L. and Madden, L. V. (1990). Introduction to plant disease epidemiology. John Wiley & Sons.
- Cregeen, S., Radisek, S., Mandelc, S., Turk, B., Stajner, N., Jakse, J. and Javornik, B. (2015). Different gene expressions of resistant and susceptible hop cultivars in response to infection with a highly aggressive strain of Verticillium albo-atrum. Plant Mol Biol Report 33(3): 689-704.
- Ellendorff, U., Fradin, E. F., De Jonge, R. and Thomma, B. P. (2009). RNA silencing is required for Arabidopsis defence against Verticillium wilt disease. J Exp Bot 60(2): 591-602.
- Flajsman, M., Mandelc, S., Radisek, S., Stajner, N., Jakse, J., Kosmelj, K. and Javornik, B. (2016). Identification of novel virulence-associated proteins secreted to xylem by Verticillium nonalfalfae during colonization of hop plants. Mol Plant Microbe Interact 29(5): 362-373.
- Fradin, E. F., Zhang, Z., Juarez Ayala, J. C., Castroverde, C. D., Nazar, R. N., Robb, J., Liu, C. M. and Thomma, B. P. (2009). Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiol 150(1): 320-332.
- Jakše, J., Cerenak, A., Radisek, S., Satovic, Z., Luthar, Z. and Javornik, B. (2013). Identification of quantitative trait loci for resistance to Verticillium wilt and yield parameters in hop (Humulus lupulus L.). Theor Appl Genet 126(6): 1431-1443.
- Kayser, T. (1992). Protoplasten fusion sowie elektrophoretische Chromosomentrennung und Genkartierung bei filamentösen Pilzen: Penicillium janthinellum, Absidia glauca und Cochliobolus heterostrophus. Dissertation.
- Klosterman, S. J., Subbarao, K. V., Kang, S., Veronese, P., Gold, S. E., Thomma, B. P., Chen, Z., Henrissat, B., Lee, Y. H., Park, J., Garcia-Pedrajas, M. D., Barbara, D. J., Anchieta, A., de Jonge, R., Santhanam, P., Maruthachalam, K., Atallah, Z., Amyotte, S. G., Paz, Z., Inderbitzin, P., Hayes, R. J., Heiman, D. I., Young, S., Zeng, Q., Engels, R., Galagan, J., Cuomo, C. A., Dobinson, K. F. and Ma, L. J. (2011). Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog 7(7): e1002137.
- Mandelc, S., Timperman, I., Radisek, S., Devreese, B., Samyn, B. and Javornik, B. (2013). Comparative proteomic profiling in compatible and incompatible interactions between hop roots and Verticillium albo-atrum. Plant Physiol Biochem 68: 23-31.
- Radisek, S., Jakse, J. and Javornik B. (2006). Genetic variability and virulence among Verticillium albo-atrum isolates from hop. Eur J Plant Pathol 116: 301-314.
- Radisek, S., Jakse, J., Simoncic, A. and Javornik, B. (2003). Characterization of Verticillium albo-atrum field isolates using pathogenicity data and AFLP analysis. Plant Dis 87: 633-638.
- Simko, I. and Piepho, H. P. (2012). The area under the disease progress stairs: calculation, advantage, and application. Phytopathology 102(4): 381-389.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Flajšman, M., Radišek, S. and Javornik, B. (2017). Pathogenicity Assay of Verticillium nonalfalfae on Hop Plants. Bio-protocol 7(6): e2171. DOI: 10.21769/BioProtoc.2171.
Category
Plant Science > Plant immunity > Disease bioassay
Microbiology > Microbe-host interactions > Fungus
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.