- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Bioinformatic Analysis for Profiling Drug-induced Chromatin Modification Landscapes in Mouse Brain Using ChlP-seq Data
Published: Vol 7, Iss 3, Feb 5, 2017 DOI: 10.21769/BioProtoc.2123 Views: 12786
Reviewed by: Oneil G. BhalalaSalma HasanSabine Le Saux
Original research article
The authors used this protocol in:
Dec 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Chromatin immunoprecipitation followed by massively parallel sequencing (ChIP-seq) is a powerful technology to profile genome-wide chromatin modification patterns and is increasingly being used to study the molecular mechanisms of brain diseases such as drug addiction. This protocol discusses the typical procedures involved in ChIP-seq data generation, bioinformatic analysis, and interpretation of results, using a chronic cocaine treatment study as a template. We describe an experimental design that induces significant chromatin modifications in mouse brain, and the use of ChIP-seq to derive novel information about the chromatin regulatory mechanisms involved. We describe the bioinformatic methods used to preprocess the sequencing data, generate global enrichment profiles for specific histone modifications, identify enriched genomic loci, find differential modification sites, and perform functional analyses. These ChIP-seq analyses provide many details into the chromatin changes that are induced in brain by chronic exposure to cocaine, and generates an invaluable source of information to understand the molecular mechanisms underlying drug addiction. Our protocol provides a standardized procedure for data analysis and can serve as a starting point for any other ChIP-seq projects.
Keywords: Chromatin immunoprecipitation (ChIP)Background
Chromatin modification has been implicated in the molecular mechanisms of drug addiction and may hold the key to understanding multiple aspects of addictive behaviors (Robison and Nestler, 2011). Chromatin Immuno-Precipitation (ChIP) followed by massively parallel sequencing (ChIP-seq) is the current state of the art technology to profile the chromatin landscape. The typical procedure of ChIP-seq involves: 1) using the antibody against a protein of interest to pull down the binding DNA, which has been fixed to the protein and broken into smaller fragments, 2) the immunoprecipitated DNA is then purified and constructed into a library for high throughput sequencing of short reads (usually 50-100 bp) from the ends of insert DNA fragments, 3) the short reads are aligned to the genome and put through data analysis. Compared with its predecessor – ChIP-chip, ChIP-seq has unparalleled advantages such as unbiased coverage of the entire genome, single base resolution, and significantly improved signal-to-noise ratio (Park, 2009). It has proven to be an invaluable tool to understand numerous types of chromatin modifications.
Brief overview of ChIP-seq experiment: Please see original research articles for greater experimental details (Renthal et al., 2009; Lee et al., 2006). Briefly, adult mice received a standard regimen of repeated cocaine (7 daily IP injections of cocaine [20 mg/kg] or saline) and were used 24 h after the last injection (Robison and Nestler, 2011). The nucleus accumbens, a major brain reward region, was obtained by punch dissection and used for ChIP-seq. Chromatin IP was performed as described previously (Renthal et al., 2009; Lee et al. 2006; http://jura.wi.mit.edu/young_public/hES_PRC/ChIP.html) with minor modifications, using two antibodies, anti-H3K4me3 (tri-methylation of Lys4 in histone H4) (Abcam #ab8580) and anti-H3K9me3 (Abcam #ab8898). Sequencing libraries for each experimental condition were generated in triplicate, and were then sequenced on an Illumina sequencer.
Bioinformatic analysis: The sequencing data generated from libraries under treatment and corresponding control conditions will be used to identify the specific genomic regions that have undergone chromatin changes. We can then associate these regions with biological functions and select the regions of interest for further study. The basic procedure for this kind of bioinformatic analysis can be laid out as a pipeline of eight steps (Figure 1):
1. Perform quality control analyses on the sequencing data files;
2. Align the sequences to the genome;
3. Remove PCR duplicates;
4. Perform cross-correlation quality analysis;
5. Generate coverage files to be loaded into a genome browser;
6. Generate global coverage plots;
7. Perform peak detection and annotation;
8. Perform differential analysis and functional association.
This protocol shall explain each of these steps in more detail, including some of the principles and rationale underlying the bioinformatics analyses, and provide the necessary commands for execution in a Unix-like command-line environment. 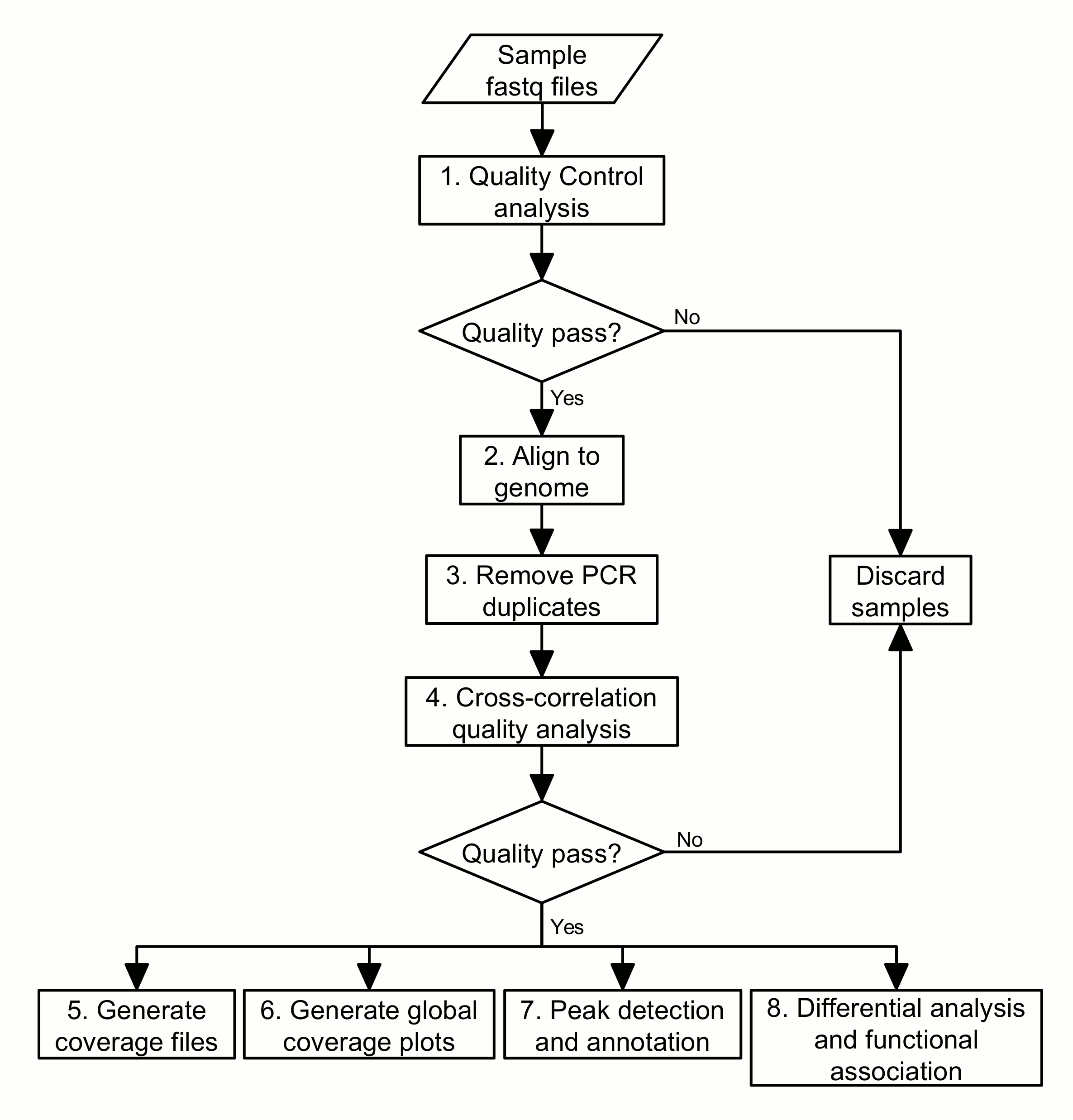
Figure 1. Flowchart of the ChIP-seq analysis pipeline
Equipment
- Personal computer or high performance computing cluster. All software mentioned here can be run under a Unix-like workstation, such as Linux and Mac. For Windows users, a terminal emulator, such as Cygwin (http://cygwin.com/), can be used.
Software
- FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
- Bowtie2 (Langmead and Salzberg, 2012; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
- Samtools (Li et al., 2009; http://samtools.sourceforge.net/)
- phantompeakqualtools (http://code.google.com/archive/p/phantompeakqualtools/)
- IGV and IGVtools (Robinson et al., 2011; http://software.broadinstitute.org/software/igv/)
- ngs.plot (Shen et al., 2014; http://github.com/shenlab-sinai/ngsplot)
- MACS (Zhang et al., 2008; http://github.com/taoliu/MACS)
- diffReps (Shen et al., 2013; http://github.com/shenlab-sinai/diffreps)
- bedtools (Quinlan and Hall, 2010; http://bedtools.readthedocs.io/en/latest/)
- region-analysis (Shao et al., 2016; http://github.com/shenlab-sinai/region_analysis)
- ChIPseqRUs (Loh et al., 2016; https://github.com/shenlab-sinai/chip-seq_preprocess)
- David (Dennis et al., 2003; http://david.ncifcrf.gov/)
- IPA (http://www.ingenuity.com/)
- GREAT (McLean et al., 2010; http://bejerano.stanford.edu/great/public/html/)
Procedure
- Perform quality control analyses on the sequencing data files
We strongly recommend checking the quality of the ChIP-seq sequencing data before moving on to the next steps. FastQC is an excellent program we use which performs a series of quality control steps for a next generation sequencing (NGS) dataset. Some of the more important quality reports to take note of in the evaluation of sequencing quality include the ‘Per base sequence quality’, which should show consistently high quality values across the reads; the ‘Per base sequence content’, which should show non-random distribution of the nucleotides at each base; and ‘Sequence duplication levels’, which should not be excessive. The FastQC documentation in the web link above provides some examples of reports of good and bad sequencing data may look like.
fastqc -o path_to_output_directory -t number_of_threads_to_usesequence_file. fastq
Note: In addition to command line execution, FastQC can also be run as an interactive graphical application. Also, definitions for command line parameters (e.g., ‘-o’, ‘-t’) for FastQC and all other programs used in this protocol can be found in their respective weblinks as provided in the ‘Software’ section above. - Alignment of the sequences to the genome
- For each sample, we performed the alignment of the raw fastq sequence reads to the reference mouse genome using the Bowtie2 program. Bowtie2 generates alignment results in the Sequence Alignment/Map (SAM) format (Li et al., 2009), which contains alignment information including the short read ID, chromosome name, start position, strand, mismatch information, and the raw nucleotides of the read, among others.
bowtie2 -p number_of_compute_cores_to_use -x path_to_mm9_genome_index -1 sequence_file_forward_reads.fastq -2 sequence_file_reverse_reads.fastq -S alignment_file.sam
Note: In cases of single-end sequencing alignments, remove ‘-2 sequence_file_reverse_reads.fastq’ from the above command. - From the SAM file generated, we need to extract only the reads that mapped uniquely to the genome. As multi-mapped reads are represented by Bowtie2 in the SAM file only by way of an ‘XS:i:value’ tag:value pair, we will use the Linux ‘grep’ command to extract all result lines that do not include ‘XS:i:’ in them.
grep -v “XS:i:” alignment_file.sam > alignment_unique.sam
- The SAM format files are text files which typically end up very large and unwieldy for the tens of millions of reads aligned per sample. A companion to SAM, the Binary Alignment/Map (BAM) format (Li et al., 2009), contains the exact same information as in SAM, but enables efficient compression and storing of the alignment data, and also allows for efficient retrieval of the aligned reads. The BAM format is now widely accepted by most programs processing alignment data. Using the samtools program, we shall convert and sort the SAM file into the BAM format for use in subsequent steps.
samtools view -Sb alignment_unique.sam > alignment_unique.bam
samtools sort alignment_unique.bam alignment_sorted
- For each sample, we performed the alignment of the raw fastq sequence reads to the reference mouse genome using the Bowtie2 program. Bowtie2 generates alignment results in the Sequence Alignment/Map (SAM) format (Li et al., 2009), which contains alignment information including the short read ID, chromosome name, start position, strand, mismatch information, and the raw nucleotides of the read, among others.
- Removing PCR duplicates
- The presence of potentially redundant short reads, caused by PCR amplification, represents an intrinsic limitation of second generation sequencing technology. Because PCR preferentially amplifies DNA fragments with certain nucleotide compositions, it makes some genomic locations overrepresented in a ChIP-seq library. While it may be argued that this overrepresentation bias applies equally to treatment and control conditions and can therefore be normalized, it actually perturbs the distribution of the underlying data and may inflate statistical significance. Take a hypothetical example (Table 1), where the actual number of fragments in library A and B are 1 and 3, respectively. If we simply multiply this number by ten then we will have 10 fragments in library A and 30 in library B. Both cases give us the same fold change of 3.0 but dramatically different p-values if assessed by a Chi-square test.
Table 1. A hypothetical example to demonstrate the danger of inflating statistical significance without removing redundancy. The P-values are generated from the Chi-square test.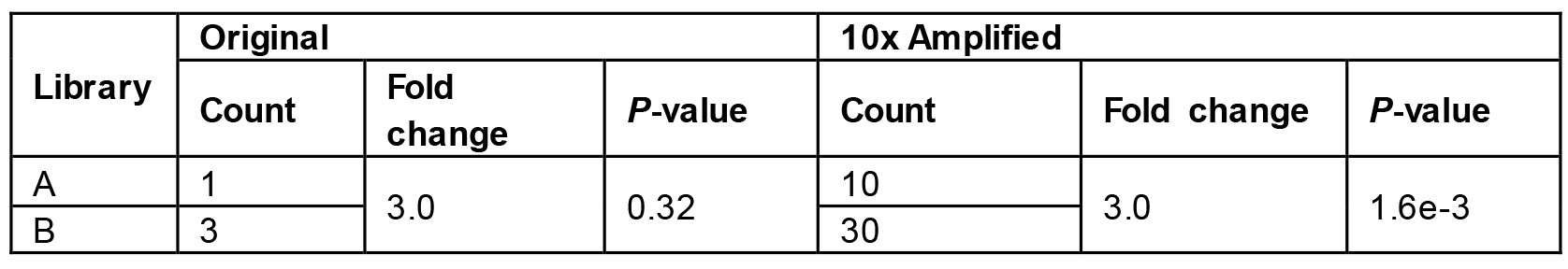
- To remove the PCR duplicates, we utilize the fact that, during the preparation of a ChIP-seq library, a sonicator breaks the whole genome almost uniformly so that most fragments come from unique positions (because a mammalian genome is ~2-3 billion bp long). This process may remove ‘innocent’ fragments that truly come from the same position and strand. But this happens less commonly in comparison with PCR duplication, and, as a general rule in statistical inference, it is always better to be conservative than to make false claims. Therefore, we have made removal of PCR duplicates a part of our ChIP-seq processing pipeline (Loh et al., 2016).samtools rmdup -s alignment_sorted.bam alignment_rmdup.bamNote: In cases of paired-end alignments, use the -S option in place of the -s option.
Special note: When micrococcal nuclease is used in place of sonication during fragmentation step in ChIP preparation, the redundancy removing procedure should not be applied. This is because the strong preference for micrococcal nuclease to cut a DNA sequence at particular positions (with specific nucleotide compositions) can lead to a lot of genuine duplicated fragments. We are not aware of any available approach to specifically remove PCR-based duplication in such a library.
- The presence of potentially redundant short reads, caused by PCR amplification, represents an intrinsic limitation of second generation sequencing technology. Because PCR preferentially amplifies DNA fragments with certain nucleotide compositions, it makes some genomic locations overrepresented in a ChIP-seq library. While it may be argued that this overrepresentation bias applies equally to treatment and control conditions and can therefore be normalized, it actually perturbs the distribution of the underlying data and may inflate statistical significance. Take a hypothetical example (Table 1), where the actual number of fragments in library A and B are 1 and 3, respectively. If we simply multiply this number by ten then we will have 10 fragments in library A and 30 in library B. Both cases give us the same fold change of 3.0 but dramatically different p-values if assessed by a Chi-square test.
- Perform cross-correlation quality analysis
The analysis of strand cross-correlation can be used as a ChIP-seq quality metric (Kharchenko et al., 2008; Landt et al., 2012). The principle behind this analysis is that high-quality ChIP-seq experiments are expected to generate clusters of mapped reads on the forward and reverse strands, with the ChIP binding site centered between them. By increasingly shifting the reads in the direction of the strand they map to and then calculating the between-strand Pearson correlation of read depth at all positions, the plot of cross-correlation coefficient against strand-shift usually yields two peaks, one corresponding to the read length (the so called ‘phantom’ peak) and the other to the average fragment length of the library. The absolute and relative heights of these peaks are then used to calculate two quality metrics, the normalized strand coefficient (NSC) and the relative strand correlation (RSC). The Encode Consortium ChIP-seq guidelines state that high quality ChIP-seq datasets typically have NSC > 1.1 and RSC > 1, and recommend the generation of additional replicates when NSC < 1.05 and RSC < 0.8 (Landt et al., 2012). Here, we use the run_SPP_nodups.R script of the phantompeakqualtools package to calculate the NSC and RSC values. An example of the NSC and RSC metrics and cross-correlation plot generated by the script is shown in Figure 2.
run_spp_nodups.R -savp -c=alignment_rmdup.bam -out=phantompeak_output.txt
Notes:- We recommend assessing the NSC and RSC values with some caution. They tend to be more informative for chromatin marks and transcription factors with punctuated peaks than broad peaks. If your sequencing target is a chromatin mark with broad pattern peaks, such as the H3K9me2, the NSC and RSC values may fall below the Encode suggested criteria. On the other hand, we have found some input control samples to have very high NSC and RSC values.
- The ‘phantom’ peak is a phenomenon caused by biased mappability of the genome. When a genomic region has good mappability, so does its reverse complement. Therefore, both strands of the genomic region will get an equal amount of aligned reads. Because the read depth is calculated based on the 5’ ends of the aligned reads, a strand-shift of the read length will yield a local maximum – the ‘phantom’ peak.
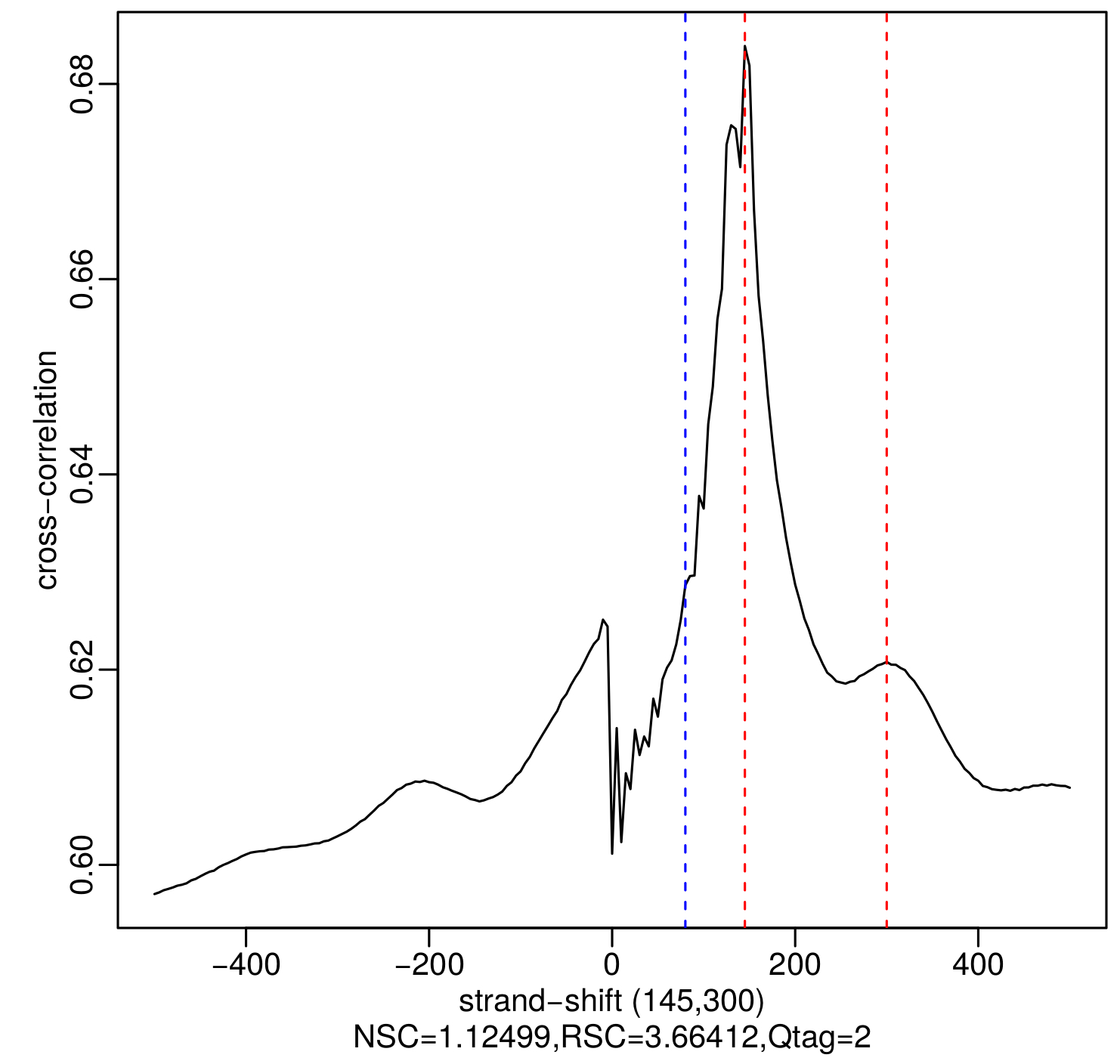
Figure 2. Plot of cross-correlation coefficient as a function of strand-shift for an example H3K27ac ChIP-seq sample, generated by the run_SPP_nodups.R script. The blue dotted line shows the location of the phantom peak (i.e., read length) and the red dotted lines show the best and the second-best guesses (the two numbers in parentheses) of the fragment length. The fragment length estimates are identified as local maxima.
- We recommend assessing the NSC and RSC values with some caution. They tend to be more informative for chromatin marks and transcription factors with punctuated peaks than broad peaks. If your sequencing target is a chromatin mark with broad pattern peaks, such as the H3K9me2, the NSC and RSC values may fall below the Encode suggested criteria. On the other hand, we have found some input control samples to have very high NSC and RSC values.
- Generation of coverage files to be loaded into a genome browser
The effective visualization of a ChIP-seq sample provides valuable information to a bench biologist (Figure 3). Through the use of a genome browser, an investigator can easily see which genomic regions are enriched by ChIP. In Figure 3, the y-axis gives the number of short reads aligned to that location, which can also be normalized by library size for comparing two or more ChIP-seq samples. This can be done by generating a so-called coverage file for each ChIP-seq sample. Many options are available in choosing a genome browser. Most genome browsers support its own file format that best suits its implementation, but often also supports some other common formats. The UCSC genome browser (Kent et al., 2002) is one of the earliest genome browsers and probably still the most comprehensive one. It is a web-based application and usually requires a user to upload the coverage files to a remote server. As sequencing technology advances rapidly, so does the size of coverage files generated from ChIP-seq samples. Therefore, it has become very inconvenient to upload those large files to a remote server. We therefore recommend using a genome browser that can work on a local machine. A very good choice is the IGV genome browser which is a Java-based cross-platform application. It supports a binary format called TDF and provides a utility program (igvtools) for users to generate a TDF file from a BAM format alignment file or several other commonly used formats.
igvtools count alignment_rmdup.bam alignment_rmdup.tdf mm9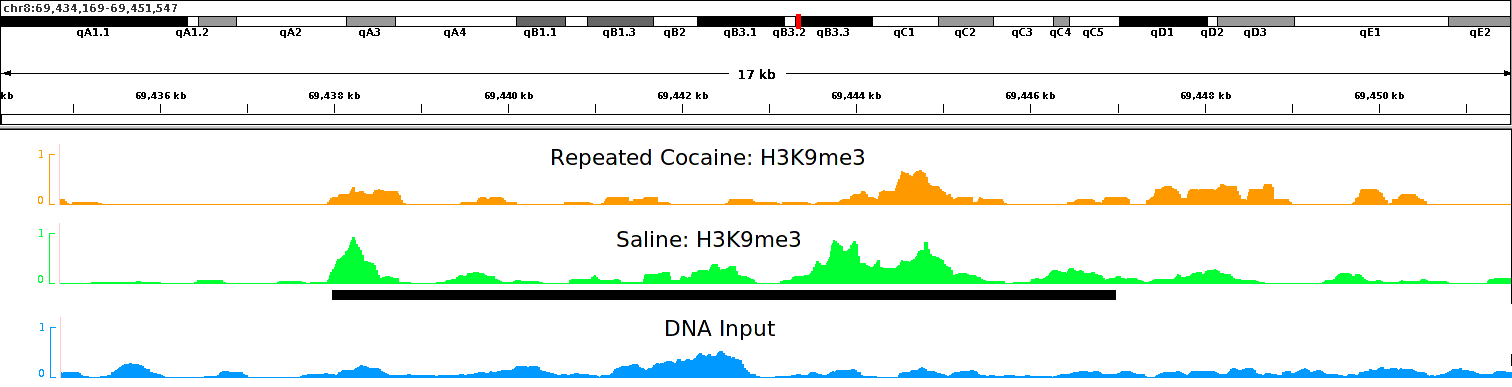
Figure 3. Coverage plot of a histone mark, H3K9me3, after repeated cocaine or saline conditions shown in the IGV genome browser (Robinson et al., 2011). Y-axis represents the normalized coverage of DNA fragments enriched by the antibody against H3K9me3. This is a peak of ~9 Kb long located on chromosome 8, which shows reduced binding after chronic cocaine treatment in mouse nucleus accumbens (NAc) (Maze et al., 2011). - Generation of global coverage plots
A global coverage plot that shows the averaged coverage over all instances of annotated genomic features, such as transcription start sites (TSS), is often extremely helpful in determining enrichment, assessing IP efficiency, or correlating with gene groups. An example is shown in Figure 4, which demonstrates that the enrichment of H3K4me3 correlates positively with gene expression levels. The generation of such a plot is straightforward. The basic principle is first to calculate the genomic coverage vector for a ChIP-seq sample. One then retrieves the genomic coordinates for the selected genomic feature from an annotation database. With the genomic coordinates, the coverage values are then extracted and averaged. We have developed an open source software package called ngs.plot, based on the statistical package R, that would generate these global coverage plots. The command shown below executes a basic ngs.plot analysis with the four mandatory arguments required. Additional customizations of the analysis and plotting can be performed with additional arguments (https://github.com/shenlab-sinai/ngsplot/wiki/weblink). ngs.plot is also available as an add-on to Galaxy (Goecks et al., 2010), a popular web-based genomic analysis framework.
ngs.plot.r -G mm9 -R tss -C alignment_rmdup.bam –O outputfile_prefix
Note: All the above-mentioned steps 1-6 can be considered as pre-processing steps that would be needed to be applied to all samples. While the individual command lines to run each step have been provided here, we have also developed an open source automated pipeline (Loh et al., 2016) (Available at http://github.com/shenlab-sinai/chip-seq_preprocess/) that would automatically run all these steps on all samples. The pipeline also features parallel processing and automatic resume of interrupted jobs. Please refer to the URL above for instructions on how to install and use the pipeline, which would not be covered here.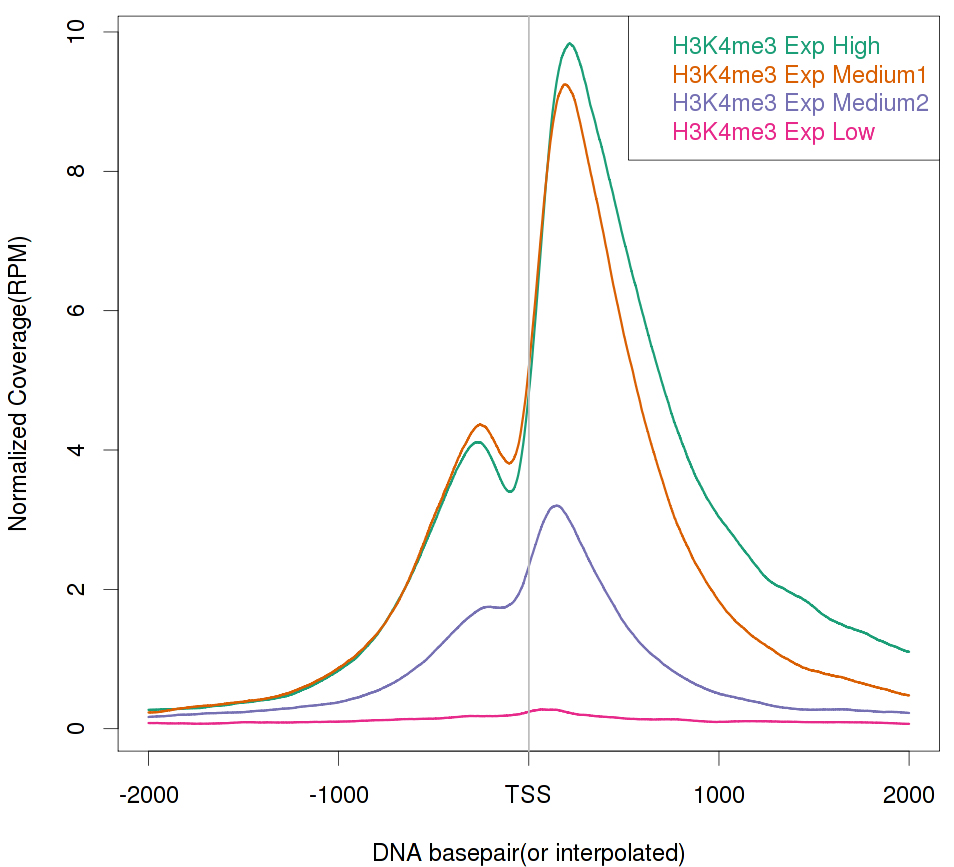
Figure 4. Global TSS plot for a histone mark, H3K4me3, in mouse NAc under saline condition correlating with gene expression levels. H3K4me3 is known as a transcriptional activation mark and shows a strong positive correlation with gene expression levels. Here, four gene groups – High, Medium 1, Medium 2, and Low are defined by normalized RNA-seq read counts. The four gene groups are ranked by their expression levels from high to low and each group contains 1,000 genes randomly selected from the genome. - Peak detection and annotation
Determining binding enrichments for a ChIP-seq library with rigorous statistical evaluation (peak calling) is often extremely useful. Global statistics can be obtained on the peaks to determine their distribution (Figure 5). One can also select top ranked peaks for follow up, or correlate the peak list with functional annotations or gene expression. Peak calling for ChIP-seq has been a heavily researched area in the past few years, with dozens of software tools developed to date (for reviews see [Szalkowski and Schmid, 2010; Pepke et al., 2009; Wilbanks and Facciotti, 2010]). The basic principle of any peak calling program can be summarized as follows: 1) the whole genome is binned into small intervals of fixed size, such as 200 bp; 2) the number of short reads in each bin is used to build a null distribution to describe non-binding regions, typically using Poisson or Negative Binomial (NB) distributions; 3) a sliding window is used to scan the whole genome and identify regions that exceed pre-determined statistical cutoffs. As we examine this procedure, we can see that the most important part of peak calling is in the building of the null distribution that describes a ChIP-seq background. This is a non-trivial task because the short read counts of a ChIP-seq library do not follow a unified random process. The complexity is caused by many factors, but some of the most important ones are: uneven packaging of the chromatin into nucleosomes, PCR bias, and sequencing imperfection (Chen et al., 2012). Modeling these effects by DNA sequences alone is an extremely complicated task, if it is even possible. Therefore, it is always recommended to couple several IP libraries with an input library with the same preparation procedure. One can then easily compare the normalized read counts from the IP and input libraries and determine the enrichment of IP. A very popular peak calling program is MACS, which can be used with or without inputs. MACS utilizes a local Poisson distribution to model the background of a ChIP-seq sample. When working without input, it uses regional counts from IP as background. This is prone to inaccuracy because there is no way to separate the short reads coming from IP and non-specific binding. When an input is available, MACS compares IP with input to identify enrichment with better sensitivity and specificity than IP alone.
macs2 callpeak -t alignment_rmdup.bam -c input.bam -f BAM -g mm -n outputfile_prefix --bw 150 -q 0.05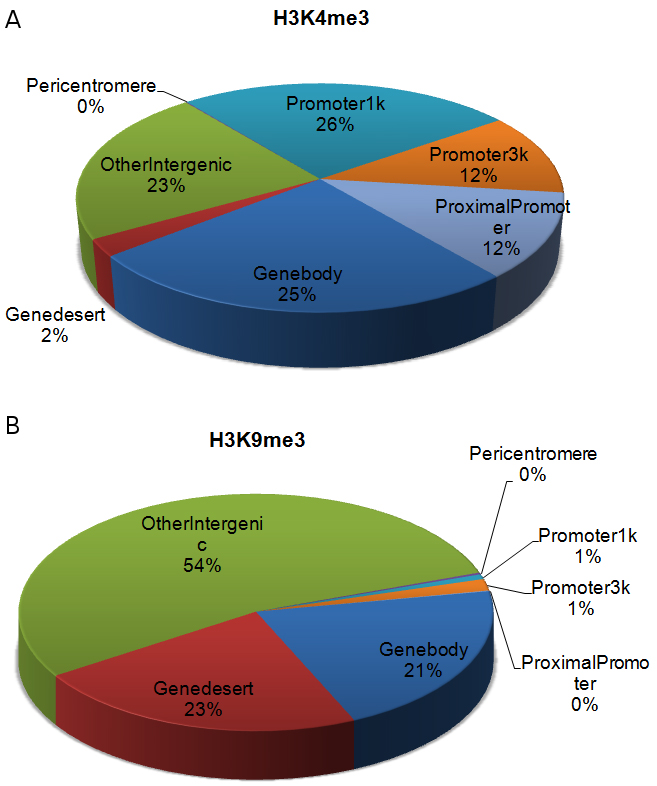
Figure 5. Peak distribution of two histone marks, (A) H3K4me3 and (B) H3K9me3, across the whole genome using existing annotation of gene features. H3K4me3 is a ‘genic’ mark with most of its peaks located around TSSs or on gene bodies. In contrast, H3K9me3 is a ‘non-genic’ mark with most of its peaks located in intergenic regions (Maze et al., 2011). - Differential analysis and functional association
- As we are interested in the characterization of drug-induced chromatin modifications, it is of high interest to compare the two IP libraries of treatment and control and identify differences in binding, or in other words, differential sites. This can be done by comparing the two peak lists from treatment and control and making presence/absence calls. However, we have found this approach to be ineffective for our data because the drug-induced chromatin changes in brain are often relatively small, presumably due to the heterogeneity of the tissue compared, for example, to cell culture systems. Typically, one may observe a peak to be present in both drug-treated and control conditions, but some of the peaks do show significant differences in magnitude (Figure 6A). In addition, there are situations where a peak is not significantly more enriched in one condition than in the comparison condition but still classified as different because an arbitrary cutoff is chosen (Figure 6B). A refinement of this approach is to identify peaks in both conditions and make quantitative assessment on their binding enrichments (Liang and Keles, 2012). This is a significant improvement to the first approach and can usually generate much more reliable differential sites than the former. The approach should work well for transcription factor-like histone marks. However, for some histone marks, this can still be suboptimal. For example, marks like H3K9me3 typically produce peaks as long as several Kb, while differences can be observed well within a peak (Figure 6C). Comparing two peaks can be ineffective in identifying this kind of changes. To deal with the above challenges, we have developed our own program – diffReps to detect differential chromatin modification regions between two conditions with high definition. diffReps employs a sliding window approach to identify all regions that show significant changes. With a tunable window size, it allows one to look for either large blocks or small regions of chromatin modifications. diffReps also takes into account the biological variations within each condition and provides a few statistical tests to choose from for assessing the differences. When biological replicates are available, we recommend the use of NB test which models the over-dispersion (large variation) of discrete counting data within a group. NB test is usually much more sensitive than t-test in detecting differential sites for ChIP-seq data. It also avoids the problem of making false positives on small count data. When there is only one sample in each group, one can choose either Pearson Chi-square or G-test for differential analysis. The following example shows a diffReps run to detect differential sites by comparing two experimental conditions where each condition contains three BED files. Input samples for each condition are also available and used. We use a window size of 200 bp to obtain a high-resolution profiling of the chromatin modification landscape and NB test to assess statistical significance.
diffReps.pl --tr T1.bed T2.bed T3.bed --co C1.bed C2.bed C3.bed --btr T_input.bed --bco C_input.bed --re diffsite.txt --gname mm9 --me nb --wi 200 --st 20
Note: diffReps requires input files to be in the BED format (https://genome.ucsc.edu/FAQ/FAQformat#format1), which can be converted from the BAM format using the bedtools package (Quinlan and Hall, 2010) using the command:
bedtools bamtobed -i inputfile.bam > outputfile.bed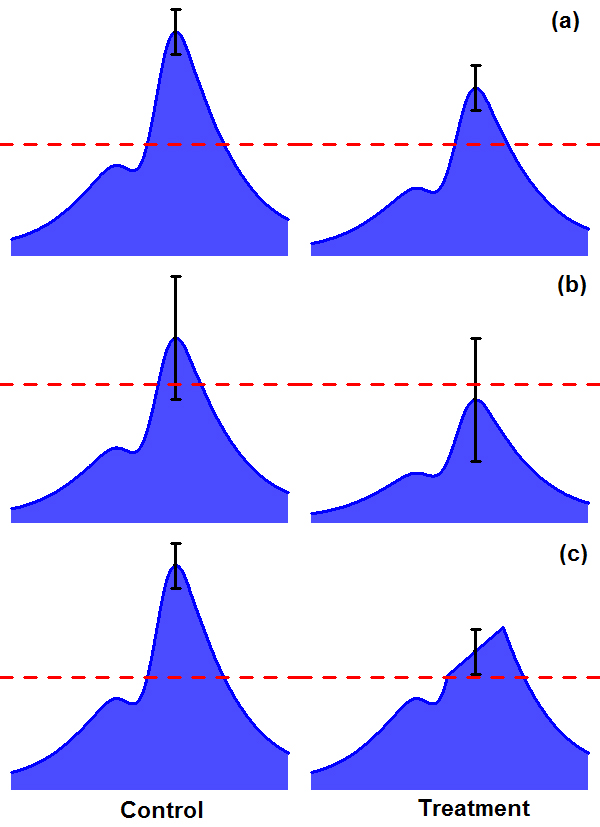
Figure 6. Challenges in detecting differential sites from ChIP-seq data by comparing two conditions. A. Two peaks are both above significance cutoff but show differences in binding. B. One peak is above cutoff and the other peak is below cutoff but they are not significantly different. C. There is a region with significant difference within a peak but may not be detected when the whole peak is considered. - The final step in our analysis is the functional annotation for a peak list or differential site list. This has traditionally been done by first annotating each genomic region to the closest gene and then testing the enrichment of functional terms among this Goecks gene group. To annotate each called peak or differential site according to the type of genomic feature on which they are located, the region_analysis program is used. The region_analysis program is able to annotate any text file where the first three columns are chromosome, start and end positions respectively.
region_analysis.py -i input_file -g mm9 -d refseq –r - Gene enrichment analysis of the gene lists annotated to peaks or differential sites can be accomplished with various open source or commercially available tools. A very popular open source tool is DAVID, while a commercially available alternative is Ingenuity Pathway Analysis (IPA). Additionally, another development in functional interpretation of peaks is to overlap them with gene regulatory regions which can either be proximal regions at TSS or distal regions as far as 1 Mb away. This approach is useful because, with the advent of ChIP-seq, we are now able to profile genomic regions that are not restricted to promoters. Potential genomic regions that may contain regulatory information include distant enhancers as well as gene bodies. This functionality greatly enhances our ability to reveal the previously unknown functions of a histone mark or a DNA binding protein. A representative tool in this category is called ‘Genomic Regions Enrichment of Annotations Tool’ (GREAT). GREAT simply requires the input of a BED file which can be easily obtained from peak calling or diffReps outputs. These analysis tools are mainly web-based and interested readers are referred to their websites for more information.
- As we are interested in the characterization of drug-induced chromatin modifications, it is of high interest to compare the two IP libraries of treatment and control and identify differences in binding, or in other words, differential sites. This can be done by comparing the two peak lists from treatment and control and making presence/absence calls. However, we have found this approach to be ineffective for our data because the drug-induced chromatin changes in brain are often relatively small, presumably due to the heterogeneity of the tissue compared, for example, to cell culture systems. Typically, one may observe a peak to be present in both drug-treated and control conditions, but some of the peaks do show significant differences in magnitude (Figure 6A). In addition, there are situations where a peak is not significantly more enriched in one condition than in the comparison condition but still classified as different because an arbitrary cutoff is chosen (Figure 6B). A refinement of this approach is to identify peaks in both conditions and make quantitative assessment on their binding enrichments (Liang and Keles, 2012). This is a significant improvement to the first approach and can usually generate much more reliable differential sites than the former. The approach should work well for transcription factor-like histone marks. However, for some histone marks, this can still be suboptimal. For example, marks like H3K9me3 typically produce peaks as long as several Kb, while differences can be observed well within a peak (Figure 6C). Comparing two peaks can be ineffective in identifying this kind of changes. To deal with the above challenges, we have developed our own program – diffReps to detect differential chromatin modification regions between two conditions with high definition. diffReps employs a sliding window approach to identify all regions that show significant changes. With a tunable window size, it allows one to look for either large blocks or small regions of chromatin modifications. diffReps also takes into account the biological variations within each condition and provides a few statistical tests to choose from for assessing the differences. When biological replicates are available, we recommend the use of NB test which models the over-dispersion (large variation) of discrete counting data within a group. NB test is usually much more sensitive than t-test in detecting differential sites for ChIP-seq data. It also avoids the problem of making false positives on small count data. When there is only one sample in each group, one can choose either Pearson Chi-square or G-test for differential analysis. The following example shows a diffReps run to detect differential sites by comparing two experimental conditions where each condition contains three BED files. Input samples for each condition are also available and used. We use a window size of 200 bp to obtain a high-resolution profiling of the chromatin modification landscape and NB test to assess statistical significance.
- Anticipated results
The ChIP-seq protocol outlined here is an extremely powerful and comprehensive technique for characterizing chromatin modification landscapes for histone marks in brain. The ChIP-seq data not only allows us to look at traditional regulatory regions – promoters, but also enables us to investigate remote regulatory sites, such as enhancers and exons. With this enhanced capability, we have now entered a new era to study disease-related chromatin modification changes with unprecedented detail.
Data analysis
For some concrete examples of using the above analytic procedures on a dataset of seven histone modification marks and RNA-seq to study cocaine-induced epigenomic and transcriptomic changes in mouse brain, readers are referred to our previous study (Feng et al., 2014).
Acknowledgments
This work is supported by grants: P50MH096890 and P01DA008227.
References
- Chen, Y., Negre, N., Li, Q., Mieczkowska, J. O., Slattery, M., Liu, T., Zhang, Y., Kim, T. K., He, H. H., Zieba, J., Ruan, Y., Bickel, P. J., Myers, R. M., Wold, B. J., White, K. P., Lieb, J. D. and Liu, X. S. (2012). Systematic evaluation of factors influencing ChIP-seq fidelity. Nat Methods 9(6): 609-614.
- Dennis, G., Jr., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C. and Lempicki, R. A. (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4(5): P3.
- Feng, J., Wilkinson, M., Liu, X., Purushothaman, I., Ferguson, D., Vialou, V., Maze, I., Shao, N., Kennedy, P., Koo, J., Dias, C., Laitman, B., Stockman, V., LaPlant, Q., Cahill, M.E., Nestler, E.J., and Shen, L. (2014) Chronic cocaine-regulated epigenomic changes in mouse nucleus accumbens. Genome Biol 15(4):R65.
- Goecks, J., Nekrutenko, A., Taylor, J. and Galaxy, T. (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11(8): R86.
- Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M. and Haussler, D. (2002). The human genome browser at UCSC. Genome Res 12(6): 996-1006.
- Kharchenko, P. V., Tolstorukov, M. Y. and Park, P. J. (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26(12): 1351-1359.
- Landt, S. G., Marinov, G. K., Kundaje, A., Kheradpour, P., Pauli, F., Batzoglou, S., Bernstein, B. E., Bickel, P., Brown, J. B., Cayting, P., Chen, Y., DeSalvo, G., Epstein, C., Fisher-Aylor, K. I., Euskirchen, G., Gerstein, M., Gertz, J., Hartemink, A. J., Hoffman, M. M., Iyer, V. R., Jung, Y. L., Karmakar, S., Kellis, M., Kharchenko, P. V., Li, Q., Liu, T., Liu, X. S., Ma, L., Milosavljevic, A., Myers, R. M., Park, P. J., Pazin, M. J., Perry, M. D., Raha, D., Reddy, T. E., Rozowsky, J., Shoresh, N., Sidow, A., Slattery, M., Stamatoyannopoulos, J. A., Tolstorukov, M. Y., White, K. P., Xi, S., Farnham, P. J., Lieb, J. D., Wold, B. J. and Snyder, M. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22(9): 1813-1831.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Lee, T. I., Jenner, R. G., Boyer, L. A., Guenther, M. G., Levine, S. S., Kumar, R. M., Chevalier, B., Johnstone, S. E., Cole, M. F., Isono, K., Koseki, H., Fuchikami, T., Abe, K., Murray, H. L., Zucker, J. P., Yuan, B., Bell, G. W., Herbolsheimer, E., Hannett, N. M., Sun, K., Odom, D. T., Otte, A. P., Volkert, T. L., Bartel, D. P., Melton, D. A., Gifford, D. K., Jaenisch, R. and Young, R. A. (2006). Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125(2): 301-313.
- Liang, K. and Keles, S. (2012). Detecting differential binding of transcription factors with ChIP-seq. Bioinformatics 28(1): 121-122.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and 1000 Genome Project Data Processing Subgroup. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Loh, Y. H., Shao, N. Y. and Shen, L. (2016). ChIPseqRUs: a pipeline for ChIP-seq preprocessing. Github repository. Zenodo.
- Maze, I., Feng, J., Wilkinson, M. B., Sun, H., Shen, L. and Nestler, E. J. (2011). Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci U S A 108(7): 3035-3040.
- McLean, C. Y., Bristor, D., Hiller, M., Clarke, S. L., Schaar, B. T., Lowe, C. B., Wenger, A. M. and Bejerano, G. (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28(5): 495-501.
- Park, P. J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10(10): 669-680.
- Pepke, S., Wold, B. and Mortazavi, A. (2009). Computation for ChIP-seq and RNA-seq studies. Nat Methods 6(11 Suppl): S22-32.
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
- Renthal, W., Kumar, A., Xiao, G., Wilkinson, M., Covington, H. E., 3rd, Maze, I., Sikder, D., Robison, A. J., LaPlant, Q., Dietz, D. M., Russo, S. J., Vialou, V., Chakravarty, S., Kodadek, T. J., Stack, A., Kabbaj, M. and Nestler, E. J. (2009). Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 62(3): 335-348.
- Robison, A. J. and Nestler, E. J. (2011). Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12(11): 623-637.
- Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G. and Mesirov, J. P. (2011). Integrative genomics viewer. Nat Biotechnol 29(1): 24-26.
- Shao, N. Y., Loh, Y. H. and Shen, L. (2016). Region-analysis: a python program for genomic interval annotations. Github repository. Zenodo.
- Shen, L., Shao, N. Y., Liu, X., Maze, I., Feng, J. and Nestler, E. J. (2013). DiffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One 8(6): e65598.
- Shen, L., Shao, N., Liu, X. and Nestler, E. (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15: 284.
- Szalkowski, A. M. and Schmid, C. D. (2011). Rapid innovation in ChIP-seq peak-calling algorithms is outdistancing benchmarking efforts. Brief Bioinform 12(6): 626-633.
- Wilbanks, E. G. and Facciotti, M. T. (2010). Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One 5(7): e11471.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W. and Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9(9): R137.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Loh, Y. E., Feng, J., Nestler, E. and Shen, L. (2017). Bioinformatic Analysis for Profiling Drug-induced Chromatin Modification Landscapes in Mouse Brain Using ChlP-seq Data. Bio-protocol 7(3): e2123. DOI: 10.21769/BioProtoc.2123.
Category
Systems Biology > Epigenomics > Sequencing > ChIP-seq
Systems Biology > Epigenomics > Histone modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.